
Abstract
Chromatin structure has a crucial role in a diversity of physiological processes, including development, differentiation and stress responses, via regulation of transcription, DNA replication and DNA damage repair. Histone deacetylase (HDAC) inhibitors regulate chromatin structure and activate the DNA damage checkpoint pathway involving Ataxia-telangiectasia mutated (ATM). Herein, we investigated the impact of histone acetylation/deacetylation modification on the ATM-mediated transcriptional modulation to provide a better understanding of the transcriptional function of ATM. The prototype HDAC inhibitor trichostain A (TSA) reprograms expression of the myeloid cell leukemia-1 (MCL1) and Gadd45α genes via the ATM-mediated signal pathway. Transcription of MCL1 and Gadd45α is enhanced following TSA treatment in ATM+ cells, but not in isogenic ATM- or kinase-dead ATM expressing cells, in the ATM-activated E2F1 or BRCA1-dependent manner, respectively. These findings suggest that ATM and its kinase activity are essential for the TSA-induced regulation of gene expression. In summary, ATM controls the transcriptional upregulation of MCL1 and Gadd45α through the activation of the ATM-mediated signal pathway in response to HDAC inhibition. These findings are important in helping to design combinatory treatment schedules for anticancer radio- or chemo-therapy with HDAC inhibitors.
Similar content being viewed by others

Introduction
Functional DNA damage checkpoint pathways are required for maintaining genomic integrity by facilitating cellular responses to DNA damage. Ataxia-telangiectasia mutated (ATM) is a serine-threonine kinase that is activated by DNA damage (Kastan and Lim, 2000). The ATM checkpoint pathway leads to a set of diverse DNA damage responses including the activation of signal transduction, cell cycle regulation, DNA repair, transcription, and apoptosis, via the ATM-mediated phosphorylation of a number of proteins, including BRCA1/Rad51/BRCA2, Nbs1/Mre11/Rad50, Chk2, and p53/MDM2 (Kastan and Lim, 2000). Phosphorylation appears to be important for the optimal regulation of their activities during DNA damage responses. The surrogate ATM-mediated phosphorylation of p53 is required for its transcriptional function in cell cycle arrest and apoptosis (Polager and Ginsberg, 2009). Additionally, the transcription factors BRCA1 and CtIP mediate DNA damage responses via ATM-induced phosphorylations (Kastan and Lim, 2000; Lee et al., 2000). Although this evidence strongly suggests that ATM mediates a reprogramming of gene transcription, the function of ATM in regulating this process is not yet fully understood.
The acetylation/deacetylation of histones, which is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), plays an important role in the recognition and repair of DNA damage, as well as transcription and chromatin structure (Attikum and Gasser, 2009). Previous studies reported that exposure to UV light resulted in an increase in global histone acetylation levels (Smerdon et al., 1982) and that HAT and HDAC mutants are sensitive to DNA damaging stresses (Ferreiro et al., 2006). DNA damage induced an increase in global chromatin acetylation levels and appears to regulate gene expression and facilitate DNA repair by allowing transcription/repair proteins access to DNA (Pena et al., 2007). Recent studies reported that HDAC inhibitors (HDIs) induce the DNA damage checkpoint pathway (Bakkenist and Kastan, 2003; Lee, 2007a). Exposure of cells to chromatin-modifying drugs including HDIs induces rapid and diffuse phosphorylation of the ATM protein, suggesting that the activation of ATM may result from changes in chromatin structure (Bakkenist and Kastan, 2003; Jang et al., 2004a; Lee, 2007b). Also, ATM phosphorylates HDAC1 (Kim et al., 1999), and the interaction of ATM with chromatin is required for optimal ATM activation and thereby proper DNA damage responses (Kim et al., 2009); this supports the proposed interrelationship between chromatin structure and the activation of ATM. Decondensation of chromatin has been associated with A-T (Vergani et al., 1999). Aberrant HDAC activity has been detected in many cancer cells (Weichert, 2009). Taken together, these observations suggest that histone acetylation/deacetylation may regulate the ATM-mediated DNA damage responses, or that ATM may regulate epigenetic modification in many ways. However, the function of ATM in the epigenetic regulation of gene expression has not been fully elucidated.
In this study, we sought to determine the function of ATM during the regulation of gene transcription in response to HDAC inhibition and its underlying molecular mechanisms. To this end, we examined the transcription of the MCL1 and the Gadd45α gene that are increased by a surrogate HDI, trichostain A (TSA) in normal control cells (ATM+) but not isogenic A-T cells (ATM-). HDAC inhibition induced the expression of the MCL1 and Gadd45α mRNAs in an ATM-dependent manner. We better characterized this HDAC inhibition-responsive transcription using luciferase reporter constructs containing variations of the MCL1 promoter (either entire promoter, two truncated versions, or an E2F1 binding site-deleted variation) by testing whether TSA treatment led to an increase in the transcription of MCL1. The E2F1 binding site in the MCL1 promoter was required for the TSA-induced transcriptional responsiveness, and E2F1 was recruited to this region following TSA treatment. We also found that the phosphorylation of E2F1 by ATM was required for recruitment to the MCL1 promoter and for transcriptional modulation of this gene. Taken together, our findings indicate that ATM is involved in transcriptional regulation following epigenetic histone modification, and that the functions of ATM in DNA damage responses are vital to transcriptional modulation in response to HDAC inhibition.
Results
HDAC inhibition by TSA treatment regulates transcription of MCL1 in an ATM-dependent manner
Our previous reports demonstrate that MCL1 is a TSA-responsive gene whose transcription is mediated in an ATM-dependent manner by analyzing oligonucleotide microarrays (Jang et al., 2004a; Lee, 2007b). To understand the function of ATM during HDAC inhibition-induced transcriptional regulation and characterizes the molecular mechanisms underlying the ATM-mediated transcriptional modulation following HDAC inhibition, we tested whether HDAC inhibition by TSA treatment could modulate the transcription of MCL1 and whether the transcriptional upregulation of MCL1 gene is a result of ATM activity. To do this, we monitored the levels of MCL1 mRNA in ATM+ and ATM- cells following TSA-treatment. As shown in Figure 1A, the level of MCL1 mRNA was induced following TSA treatment in the ATM+ cells. In contrast, expression of MCL1 mRNA was not upregulated in the TSA-treated ATM- cells, indicating that the TSA-induced upregulation of MCL1 mRNA expression requires ATM. In addition, the basal level of the MCL1 mRNA in ATM+ cells was elevated, further suggesting that MCL1 transcription is regulated in an ATM-dependent manner.

Effects of HDAC inhibition (by TSA treatment) on the expression of MCL1 mRNA. (A) The expression levels of MCL1 mRNA in an isogenic ATM- cells and its ATM+ parental control cells treated with TSA for 24 h were determined using RT-PCR. GAPDH levels were used to confirm equal amounts of input RNA between samples. (B) The induction of MCL1 transcription by TSA in the TSA-treated (TSA) and non-treated (none) ATM- and ATM+ cells was analyzed by quantitative real-time RT-PCR. All reactions were normalized to GAPDH. (C) The relative expression levels of survivin mRNA in the TSA-treated and non-treated cells were monitored by quantitative real-time RT-PCR. (D) Expression of MCL1 transcripts in the presence and absence of TSA was analyzed by quantitative real-time RT-PCR in HCT116 and U87MG cells. (E) The expression levels of MCL1 mRNA in HCT116 cells expressing wild type (ATM-WT) or kinase-dead mutant type (ATM-KD) ATM were determined using real-time PCR.
To confirm that the HDAC inhibition-induced transcriptional upregulation of MCL1 is dependent on ATM, we measured the total levels of MCL1 transcript by real-time PCR analysis in the ATM+ and ATM- cells following TSA treatment. As expected, TSA induced MCL1 transcription in the ATM+ cells (Figure 1B). However, TSA had little effect on the MCL1 transcription in the ATM- cells (Figure 1B). In contrast, transcription of survivin, a gene which is TSA-responsive (Noh and Lee, 2003) but not ATM-regulated was reduced by TSA in both ATM+ and ATM- cells (Figure 1C), indicating that ATM may differentially, but not globally, regulate the transcription of histone modification-responsive genes. Similar expression levels of MCL1 mRNA were observed upon HDAC inhibition in HCT116, U87MG, and 293T cells (Figure 1D), suggesting that the HDAC inhibition-induced effect on MCL1 transcription is general.
Next, to determine whether ATM kinase activity is required for MCL1 expression, we monitored the level of MCL1 mRNA in HCT-116 cells in the presence or absence of TSA, following ectopic expression of wild type (ATM-WT) or mutant (ATM-KD) ATM (Figure 1E). ATM-WT induced the expression of the MCL1 mRNA upon TSA treatment. However, the increase in MCL1 mRNA expression was abolished in the ATM-KD-expressing cells in response to TSA treatment. Thus, we indicated that the TSA-induced MCL1 mRNA expression required ATM kinase activity.
E2F1 is required for the HDAC inhibition-regulated transcription of MCL1
To better characterize the ATM-dependent transcriptional regulation of MCL1, we examined transcription from the MCL1 promoter following TSA treatment. A luciferase reporter joined to the MCL1 promoter (-294/+25 MCL1 promoter-pGL3) was generated and its transcriptional activity was measured in the presence or absence of TSA (Figure 2A). The reporter activity was enhanced approximately 38-fold following exposure to TSA in the ATM+ cells (Figure 2A). Importantly, TSA had no significant impact on the transcription of MCL1 in the ATM- cells (Figure 2A, ~4.3-fold), indicating that the transcriptional activity from the MCL1 promoter in response to TSA was regulated in part in an ATM-dependent fashion (~38-fold) and partially in an ATM-independent fashion (~4-fold). These observations are consistent with the RT-PCR described above for MCL1 (Figure 1).

The induction of MCL1 transcription following TSA treatment occurs in an ATM-dependent manner. (A) The luciferase activity of -294/+25 MCL1 promoter-driven reporter in ATM- and ATM+ cells was monitored in the presence and absence of TSA. The luciferase activity of the untreated cells transfected with the reporter was arbitrarily set to one. Results were obtained from three separate experiments, and standard deviations are shown (*P < 0.05). (B) The luciferase activity of truncated (-294/-140 and -140/+25) MCL1 promoters in ATM- and ATM+ cells was monitored in the presence and absence of TSA. The results were analyzed as described in (A). (C) The luciferase activity of promoters in which STAT (ΔSTAT) or E2F1 (ΔE2F1) binding elements were deleted, respectively, was monitored in 293T cells transfected with the indicated reporter constructs in the presence or absence of TSA. The results were analyzed as described in (A).
To understand the mechanism underlying the ATM-dependent transcriptional regulation of MCL1 in response to HDAC inhibition, we generated dissected promoters (-294/-140 and -140/+25) (Figure 2B) using the -294/+25 MCL1 promoter that contains binding elements for five transcription factors (Zhan et al., 1997). In order to identify which transcription factor(s) is/are involved in the regulation of MCL1 transcription in response to TSA the transcriptional activities from the two partial MCL1 promoters (-294/-140 and -140/+25) were analyzed in the presence and absence of TSA. As shown in Figure 2B, HDAC inhibition by TSA induced transcription from the -140/+25 MCL1 promoter containing binding sites of E2F1 and STAT by approximately 30-fold in the ATM+ cells, whereas transcription was not significantly affected by TSA in ATM- cells (~3.6-fold). These results were similar to that of -294/+25 MCL1 promoter. The transcriptional activity of the -294/-140 promoter containing binding sites for NFκB, SP1, and CREBP was only slightly induced upon TSA treatment in both ATM+ (~4.3-fold) and ATM- cells (~3.3-fold) (Figure 2B), suggesting that this promoter may have no ATM-regulated transcriptional binding sites. Taken together, it appears that the -140/+25 region of the MCL1 promoter plays a critical role in the ATM-dependent transcription of MCL1 in response to epigenetic histone modification.
Next, we sought to identify the transcription factor(s) that is/are involved in the HDAC inhibition-induced transcription of MCL1 in an ATM-dependent manner. To do this, we generated two reporters, in which either the E2F1 or STAT binding site was deleted, respectively (Figure 2C). In response to TSA treatment, the transcriptional activity of both deletion-promoters was dramatically decreased (Figure 2C) when compared to the wild type-MCL1 promoter. TSA had little effect on transcription from the E2F1 binding site-deletion promoter, suggesting that the E2F1 binding site within the MCL1 promoter is necessary for the TSA-induced transcription of MCL1. However, the transcription of the reporter gene in cells transfected with the STAT binding site-deletion promoter was activated approximately 9-fold following TSA treatment, indicating that deletion of the STAT binding site is not sufficient for abolishment of the TSA-induced MCL1 transcription but necessary for full induction. These results demonstrate that the E2F1 binding site plays a critical role in the regulation of transcription from the MCL1 promoter when induced by HDAC inhibition and that the STAT site may contribute to the TSA-induced transcription.
The recruitment of E2F1 to the MCL1 promoter is increased by HDAC inhibition
To further examine the role of E2F1 in the upregulation of MCL1 transcription in response to HDAC inhibition, we measured the recruitment of E2F1 to the MCL1 promoter following TSA treatment using the chromatin immunoprecipitation assay. Significantly, TSA induced the recruitment of E2F1 to the MCL1 promoter in the ATM+ cells (Figure 3A). But TSA had little effect on the recruitment of E2F1 into the promoter in the ATM- cells (Figure 3B). These results indicate that the recruitment of E2F1 to the MCL1 promoter was induced by TSA in an ATM-dependent manner. These findings correspond to the results from the promoter deletion-reporter experiments (Figures 2B, and 2C).

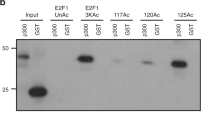
Recruitment of E2F1 to the MCL1 promoter. (A, B) Twenty four hours after transfection with a mammalian E2F1 expression construct, ATM+ (A) and ATM- cells (B) were treated with 0.33 µM TSA for 24 h. Next, ChIP assays were carried out using anti-E2F1 and acetyl-H4 antibodies. The endogenous MCL1 promoter DNA, which coimmunoprecipitated with the anti- E2F1 and acetyl-H4 antibodies, was detected using PCR. As a control, 0.01% of the total chromatin sample before immunoprecipitation (Input) was used for PCR amplification. All PCR were normalized to the input control. (C) 293T cells were transfected with wild type E2F1 or mutant E2F1-S31A in which the serine residue 31 was substituted with alanine. A ChIP assay was performed using an anti-E2F1 antibody. The results were analyzed as described in (A).
To better characterize the regulation of MCL1 transcription by E2F1 following HDAC inhibition in an ATM-dependent manner, we transfected 293T cells with expression constructs containing the wild type E2F1, and E2F1-S31A, a mutant that cannot be phosphorylated by ATM following DNA damage (Figure 3C). The recruitment of the exogenously expressed wild type E2F1 was induced by TSA, whereas that of the exogenously expressed E2F1-S31A was not altered by TSA. Together, our results support the notion that E2F1 is required for the induction of MCL1 expression in response to TSA, and that ATM is required for the HDAC inhibition-induced MCL1 transcription. In addition, our data indicate that the transcriptional regulation of MCL1 by HDAC inhibition is mediated by the ATM signaling pathway.
HDAC inhibition by TSA regulates transcription of another ATM target gene, Gadd45α in an ATM-dependent manner
Previously, we have identified another HDAC inhibition-regulated gene (Gadd45α), which is under the control of ATM (Lee, 2007a). The cell cycle regulator Gadd45α is a surrogate DNA damage responsive gene, which is upregulated by BRCA1 or p53 upon DNA damage (Lee et al., 2000). To determine whether ATM is involved in the regulation of Gadd45α transcription in response to HDAC inhibition, we studied Gadd45 transcription following TSA treatment. The transcriptional activity of a luciferase reporter hooked to Gadd45α promoter was monitored in the presence or absence of TSA (Figure 4). Gadd45α transcription was enhanced by approximately 10-fold following TSA treatment in the ATM+ cells (Figure 4A). In contrast, Gadd45α transcription was not significantly affected by TSA in the ATM- cells (Figure 4A, ~2.1-fold) when compared with its induction in the ATM+ cells, indicating that Gadd45α transcription in response to TSA was regulated primarily in an ATM-dependent manner. Also, we tested the HDAC inhibition-induced Gadd45α transcription in HeLa cells by analyzing transcription of the Gadd45α promoter-driven luciferase reporter (Figure 4B). Consistent with the results from ATM+ cells, Gadd45α transcription was induced by TSA treatment in HeLa cells (~5.2-fold). Ectopic expression of BRCA1, which is a transcriptional factor regulated by ATM, induced the Gadd45α transcription by approximately 2.6-fold in the absence of TSA, consistent with the previous report indicating that BRCA1 is a transcriptional activator for the Gadd45α gene (Park et al., 2008). Following TSA treatment, the Gadd45α transcription increased by approximately 16-fold in HeLa cells expressing BRCA1, suggesting that TSA-induced Gadd45α transcription can be mediated by ATM-BRCA1 pathway. These observations are consistent with the MCL1 transcription results, supporting the notion that ATM can regulate, and is required for transcriptional modulation in response to HDAC inhibition.

The effects of TSA treatment on the expression of the Gadd45α mRNA. (A) Induction of Gadd45α transcription following TSA treatment occurred in an ATM-dependent manner. The activity of the Gadd45α promoter-driven reporter was analyzed in ATM- (-) and ATM+ (+) cells in the presence (+) or absence (-) of TSA. The results were analyzed as described in Figure 2. (B) The induction of Gadd45α transcription following TSA treatment in HeLa cells. HeLa cells were treated with (+) or without (-) TSA in the absence (-) or presence (+) of ectopically expressed BRCA1. The results were analyzed as described in (A). (C) The expression of the Gadd45α mRNA in 293T cells treated with or without TSA for 24 h was analyzed by RT-PCR. The abundance of GAPDH is shown and used as an internal control when determining MCL1 expression levels. (D) The recruitment of BRCA1 to the Gadd45α promoter was induced following treatment of 293T cells with TSA for 24 h. ChIP assays were carried out using an anti-BRCA1 antibody. The endogenous Gadd45α promoter DNA, which coimmunoprecipitated with the anti-BRCA1 antibody, was detected using PCR. The results were analyzed as described in Figure 3.
Next, we characterized the effect of HDAC inhibition on Gadd45α transcription by analyzing the level of Gadd45α mRNA in TSA-treated or non-treated 293T cells. Similar to the MCL1 mRNA expression results, the expression of Gadd45α mRNA was enhanced following HDAC inhibition by TSA (Figure 4C). Next, we investigated the recruitment of BRCA1, a key ATM kinase substrate to the Gadd45α promoter by performing chromatin immunoprecipitation experiments (Figure 4D). The recruitment of BRCA1 to the Gadd45α promoter was induced following TSA treatment and was accompanied by enhanced histone acetylation (Figure 4D). This increased recruitment of BRAC1 is consistent with the results from the E2F1 studies characterizing its recruitment to the MCL1 promoter in response to HDAC inhibition. Together with the MCL1 transcriptional modulation results, the observations from Gadd45α transcription support the hypothesis that ATM plays a role in transcriptional modulation in response to HDAC inhibition.
Discussion
DNA damage signal transduction pathways are activated following DNA damage and induce a diversity of important cellular responses. Among them, the ATM-mediated signal pathway is one of the most important elements during the induction of cellular DNA damage responses. Transcriptional modulations act as sequential intermediaries throughout the process of DNA damage responses, from the onset of the signaling events, to the final cellular responses that are displayed by downstream effectors. Although ATM plays pleiotropic roles in a variety of key cellular DNA damage responses, the precise functional interplay among the roles of ATM in transcriptional modulation and DNA damage responses following HDAC inhibition has not been fully elucidated. Previous results show that the alteration of chromatin remodeling can activate an ATM-mediated DNA damage signal pathway (Bakkenist and Kastan, 2003; Lee, 2007a) and can thereby regulate transcription in an ATM-dependent manner (Jang et al., 2004a; Lee, 2007b). Based on the above information, we sought to: i) determine if the inhibition of HDAC activity induces the ATM signal transduction pathway and regulates the transcription of ATM-target genes and ii) to characterize the molecular mechanisms that underlie the ATM-dependent transcriptional modulation in response to altered histone acetylation/deacetylation.
In this study, we have extended previous findings (Bakkenist and Kastan, 2003; Jang et al., 2004a; Lee, 2007a, 2007b) suggesting that the inhibition of HDAC activity induces the DNA damage checkpoint pathway and cellular DNA damage responses. We did this by characterizing the role of ATM in the transcriptional modulation of two ATM-regulated genes (MCL1 and Gadd45α) following HDAC inhibition (Jang et al., 2004a, 2007b) and by analyzing their gene expression patterns. Also, these two genes are known as DNA damage-inducible genes (Criswell et al., 2003; Jang et al., 2004a), supporting the notion that the induction of their transcription upon treatment with an HDAC inhibitor may be mediated through the DNA damage response. Our findings suggest that ATM regulates the transcription of the identified ATM target MCL1 and Gadd45α genes in response to HDAC inhibition (Figures 1 and 4). Furthermore, we have demonstrated that the HDAC inhibition-induced transcriptional modulation of MCL1 is regulated by ATM via phosphorylation of E2F1, and possibly mediated through the DNA damage signaling pathway. Taken together with previous reports demonstrating that the inhibition of HDAC induces chromosomal instability (Noh et al., 2009a), the cell cycle checkpoint at G2-M transition (Noh and Lee, 2003; Noh et al., 2009a, 2009b), cell death (Johnstone and Licht, 2003; Noh et al., 2005), and the ATM-mediated DNA damage signal pathway (Bakkenist and Kastan, 2003; Lee, 2007a), our results suggest that HDAC activity may be functionally implicated in DNA damage responses. Importantly, we found that ATM is required for the HDAC inhibition-induced transcription of MCL1 (Figures 1-3) and Gadd45α (Figure 4), supporting the functional interplay between HDAC activity and the ATM-controlled DNA damage responses. Also, our results indicate that epigenetic histone alteration can be recognized by the ATM-mediated signal transduction pathway, possibly by acting as a signal leading to the activation of ATM, an event which results in the initiation of a variety of ATM-mediated DNA responses.
HDAC activity is deregulated in many cancer cells (Johnstone and Licht, 2003). Currently, HDIs have been effective for inhibiting cancer cell proliferation (Noh et al., 2003, 2005, 2009a, 2009b) and thus have emerged as encouraging drugs or sensitizers for the treatment of cancer (Jang et al., 2004b). In this regard, a proper understanding of how HDAC activity influences the cellular response to DNA damage is desirable to address questions whether and how epigenetic alteration is involved in carcinogenesis accompanied by genomic instability, which is promoted by defective DNA damage responses. In addition, it is required to facilitate the effective use of epigenetic modulators such as HDAC or DNA methyl transferase inhibitors as anticancer drugs or chemo- and radio-sensitizers.
When cells encounter environmental or cellular stresses, changes in transcriptional regulation act as an important adaptive mechanism, leading to stress-responsive phenotypes. Several transcription factors are known to be responsive to DNA-damaging stresses, including p53, NF-κB, and the Sp1-related retinoblastoma control proteins, and BRCA1 (Kastan and Lim, 2000). Most previous observations underscore the role of ATM-p53 pathway-mediated transcription in DNA damage responses, and suggest that other ATM-transcription factor pathways may be involved. In this study, we found that impaired ATM function altered the transcriptional reprogramming in response to HDAC inhibition, suggesting that ATM plays a role in: i) regulating the transcription of a diversity of genes and ii) shaping cellular phenotypes in response to a variety of stresses.
MCL1 can promote cell viability, although these effects may be more short-term than the effects of BCL2 (Akgul et al., 2000). Also, a previous report demonstrating that the expression of MCL1 is found to be increased upon exposure of ML-1 cells to various types of DNA damaging agents, including ionizing radiation, ultraviolet radiation, and alkylating drugs (Akgul et al., 2000), supports our speculation that HDAC inhibition can act as a kind of DNA damaging signal and thereby enhances MCL1 expression. Interestingly, our data showed that the expression of the pro-apoptotic cell-cycle regulator Gadd45α was activated in an ATM-dependent fashion, and that this effect occurred concomitantly with a marked increase in the expression of MCL1 in ATM+ cells following HDAC inhibition. These functionally contradictory results can be explained as temporal responses throughout the ATM-mediated DNA damage responses. At the onset of the DNA damage response, the ATM-dependent increase of Gadd45α may play a role in the induction of a cell cycle checkpoint leading to arrest and thereby allowing for DNA (or epigenetic) repair. The ATM-dependent induction of MCL1 may play a role in rescuing cured (or repaired) cells and conferring survival, possibly leading to chemo- or radio-resistance, and tumor recurrence. As such, these paradoxical functions of ATM in transcriptional reprogramming partially account for its roles in tumor suppression and therapeutic resistance.
The ATM protein can function within diverse epigenetic modification responses in addition to DNA damage responses through its ability to transduce stress signals as well as reprogram gene expression profiles. As demonstrated here, ATM can direct the expression of its target genes and can result in cellular phenotypes in response to epigenetic histone modification. Thus, our study adds to our current understanding of the transcription-mediated role of ATM. In addition, this work may provide valuable molecular information for understanding functional crosstalk between genetics and epigenetics and improving clinical applications and courses of chemotherapy and radiotherapy for cancers.
Methods
Cell culture
AT22IJE pEBS7 (ATM-) and AT22IJE pEBS7-YZ5 (ATM+) cells (gifts from Y. Shiloh) were cultured in DMEM supplemented with hygromycin B (Hyclone, Logan, UT, USA). The 293T, HeLa, and HCT 116 cells were grown in DMEM supplemented with penicillin/streptomycin.
RNA extraction and RT-PCR
Total cellular RNA was isolated using the TRIzol reagent (Life Technologies, Inc., Grand Island, NY) according to the manufacturer's instructions. The primer sequences are as follows: GAPDH (forward 5'-GTCAACGGATTTGGTCTGTATT-3', reverse 5'-AGTCTTCTGGGTGGCAGTGAT-3'), MCL1 (forward 5'-CTTCCGTAATTAGGAACCTG-3', reverse 5'-CTTGCATATAATGAAGTGAA-3'), BRCA1 (forward 5'-TGACTTTGGAGGAATTCTCGGC-3', reverse 5'-ATGAATGTGGATTCGTCACCAGCACGCAGT-3'), and survin (forward 5'-CTACATTCAAGAACTGGCCC-3', reverse 5'-CAATCCATGGCAGCCAGCTG-3').
Preparation of MCL1 reporter gene constructs
The MCL1 promoter (-294~+25) (Zhan et al., 1997) was cloned into the luciferase reporter plasmid pGL3 (Promega, Madison, WI) to generate the -294/+25 MCL1-pGL3 luciferase reporter construct. This construct was created by amplifying the MCL1 promoter with PCR and introducing a restriction enzyme site (HindIII) at position +25. Next, the amplicon was digested with XhoI and HindIII and subcloned into pGL3. The -140/+25 and -294/-140 constructs were generated in a similar manner by digestion with PvuII, and subcloning the PvuII/HindIII and XhoI/PvuII fragments into pGL3, respectively.
Reporter assay
ATM- and ATM+ cells were transfected with a MCL1- or Gadd45α (Park et al., 2008)-luciferase reporter and pRL-CMV (Promega) to control for transfection efficiency, in quadruplicate. One day after transfection, cells were incubated for another 24 h with or without 0.33 µM TSA before being harvested and assayed using the Dual Luciferase Reporter Assay (Promega). The luciferase activity was standardized against the transfection efficiency for each sample. Values are presented as mean ± SEM, from 3 separate experiments (*p < 0.05 compared with reporter alone).
Real time PCR
All reactions were performed in triplicate using the SYBR Green PCR Master Mix kit (Applied Biosystems) and an ABI PRISM 7000 Sequence Detector (Applied Biosystems). Primers were used as described above in the RT-PCR section.
Chromatin immunoprecipitation (ChIP) assay
About 1 × 106 293T, ATM-, and ATM+ cells were transfected with E2F1 and E2F1-S31A. The ChIP was performed using anti-E2F1 (NEOMARKERS), anti-BRCA1 (Santa Cruz Biotechnology), and anti-acetyl H4 (Upstate) antibodies following treatment of the transfected cells with 0.33 or 1 µM TSA for 24 h according to the manufacturer's instructions (Upstate). The endogenous MCL1 and Gadd45α promoter DNA, which coimmunoprecipitated with the indicated antibody, respectively, was detected using PCR.
Abbreviations
- ATM:
-
Ataxia-telangiectasia mutated
- ChIP:
-
chromatin immunoprecipitation
- HDAC:
-
histone deacetylase
- MCL1:
-
myeloid cell leukemia-1
- TSA:
-
trichostatin A
References
Akgul C, Turner PC, White MR, Edwards SW . Functional analysis of the human MCL1 gene . Cell Mol Life Sci 2000 ; 57 : 684 - 691
van Attikum H, Gasser SM . Crosstalk getween histone modifications during the DNA damage response . Trends in Cell Biol 2009 ; 19 : 207 - 217
Bakkenist CJ, Kastan MB . DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation . Nature 2003 ; 421 : 499 - 506
Criswell T, Leskov K, Miyamoto S, Luo G, Boothman DA . Transcription factors activated in mammalian cells after clinically relevant doses of ionizing radiation . Oncogene 2003 ; 22 : 5813 - 5827
Ferreiro JA, Powell NG, Karabetsou N, Mellor J, Waters R . Roles for Gen5p and Ada2p in transcription and nucleotide excision repair at the Saccharomyces cerevisiae Met16 gene . Nucleic Acid Res 2006 ; 34 : 976 - 985
Jang ER, Lee JH, Lim DS, Lee JS . Analysis of ataxia-telangiectasia mutated (ATM)- and Nijmegen breakage syndrome (NBS)-regulated gene expression patterns . J Cancer Res Clin Oncol 2004a ; 130 : 225 - 234
Jang ER, Lim SJ, Lee ES, Jeong G, Kim TY, Bang YJ, Lee JS . The HDAC inhibitor TSA sensitizes ER α-negative breast cancer cells to tamoxifen . Oncogene 2004b ; 23 : 1724 - 1736
Johnstone RW, Licht JD . HDAC inhibitors in cancer therapy: is transcription the primary target ? Cancer Cell 2003 ; 4 : 13 - 18
Kastan MB, Lim DS . The many substrates and functions of ATM . Nat Rev Mol Cell Biol 2000 ; 1 : 179 - 186
Kim GD, Choi YH, Dimtchev A, Jeong SJ, Dritschilo A, Jung M . Sensing of ionizing radiation-induced DNA damage by ATM through interaction with histone deacetylase . J Biol Chem 1999 ; 274 : 31127 - 31130
Kim YC, Gerlitz G, Furusawa T, Catez F, Nussenzweig A, Oh KS, Kraemer KH, Siloh Y, Bustin M . Activation of ATM depends on chromatin interactions occurring before induction of DNA damage . Nat Cell Biol 2009 ; 11 : 92 - 96
Lee JS, Collins KM, Brown AL, Lee CH, Chung JH . hCds1-mediated phosphorylation of BRAC1 regulates the DNA damage response . Nature 2000 ; 404 : 201 - 204
Lee JS . Activation of ATM-dependent of DNA damage signal pathway by a histone deacetylase inhibitor, Trichostain A . Cancer Res Treat 2007a ; 39 : 125 - 130
Lee JS . Functional link between DNA damage responses and transcriptional regulation by ATM in response to a histone deacetylase inhibitor TSA . Cancer Res Treat 2007b ; 39 : 116 - 124
Noh EJ, Lee JS . Functional interplay between modulation of HDAC activity and its regulatory role in G2-M transition . Biochem Biophys Res Commun 2003 ; 310 : 267 - 273
Noh EJ, Jang ER, Jeong G, Lee YM, Min CK, Lee JS . MBD3 mediates cancer-selective cytotoxicity by HDAC inhibitors via differential transcriptional reprogramming in lung cancer cells . Cancer Res 2005 ; 65 : 11400 - 11410
Noh EJ, Lim DS, Jeong G, Lee JS . An HDAC inhibitor, TSA, induces a delay at G2-M transition, slippage of spindle checkpoint, and cell death in a transcription-dependent manner . Biochem Biophys Res Commun 2009a ; 378 : 326 - 331
Noh EJ, Lim DS, Lee JS . A novel role for MBD3, a component of the HDAC complex, in regulation of cell-cycle progression and cells death . Biochem Biophys Res Commun 2009b ; 378 : 332 - 337
Park MA, Seok YJ, Jeong G, Lee JS . SUMO1 negatively regulates BRCA1-mediated transcription, via modulation of promoter occupancy . Nucleic Acids Res 2008 ; 36 : 263 - 283
Pena AN, Pereira-Smith OM . The role of the MORF/MRG family of genes in cell growth, differentiation, DNA repair, and thereby aging . Ann N Y Acad Sci 2007 ; 1100 : 299 - 305
Polager S, Ginsberg D . p53 and E2f: partners in life and death . Nat Rev Cancer 2009 ; 9 : 738 - 748
Smerdon MJ, Lan SY, Calza RE, Reeves F . Sodium butyrate stimulates DNA repair in UV-irradiated normal and xeroderma pigmentosum human fibroblast . J Biol Chem 1982 ; 257 : 13441 - 13447
Vergani L, Fugazza G, Chessa L, Nicolini C . Changes of chromatin condensation in one patient with ataxia telangiectasia disorder: a structural study . J Cell Biochem 1999 ; 75 : 578 - 586
Weichert W . HDAC expression and clinical prognosis in human malignancies . Cancer Lett 2009 ; 280 : 168 - 176
Zhan Q, Bieszczad CK, Bae I, Fornace AJ, Craig RW . Induction of BCL2 family member MCL1 as an early response to DNA damage . Oncogene 1997 ; 14 : 1031 - 1039
Acknowledgements
This work is supported by Korea Research Foundation (KRF E00050), Korea Science and Engineering Foundation (2009-0078170) and Health Industry Development Institute (A080542).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Experimental & Molecular Medicine website
Supplementary information
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Jang, E., Choi, J., Park, M. et al. ATM modulates transcription in response to histone deacetylase inhibition as part of its DNA damage response. Exp Mol Med 42, 195–204 (2010). https://doi.org/10.3858/emm.2010.42.3.020
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2010.42.3.020
Keywords
This article is cited by
-
MLK4 regulates DNA damage response and promotes triple-negative breast cancer chemoresistance
Cell Death & Disease (2021)
-
Development and validation of the TGx-HDACi transcriptomic biomarker to detect histone deacetylase inhibitors in human TK6 cells
Archives of Toxicology (2021)
-
Loss of the spectraplakin gene Short stop induces a DNA damage response in Drosophila epithelia
Scientific Reports (2020)
-
PARP inhibitor veliparib and HDAC inhibitor SAHA synergistically co-target the UHRF1/BRCA1 DNA damage repair complex in prostate cancer cells
Journal of Experimental & Clinical Cancer Research (2018)
-
New factors in mammalian DNA repair—the chromatin connection
Oncogene (2017)
