
TP53-Status-Dependent Oncogenic EZH2 Activity in Pancreatic Cancer

 , , , ,
, , , ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Mouse Strains and In Vivo Experiments
2.2. Primary PDAC Tissue, Primary PDAC Cells, and Gene-Panel-Sequencing
2.3. Cell Cultivation, Transfection, and Treatment
2.4. RNA Isolation and Quantitative Real-Time PCR (qRT-PCR)
2.5. Protein Harvesting and Western Blotting
2.6. Annexin/Propidium Iodide (PI) Staining
2.7. Immunofluorescence and Immunohistochemistry (IHC)
2.8. Chromatin Immunoprecipitation (ChIP) and RNA-Sequencing
2.9. Statistical Analysis
3. Results
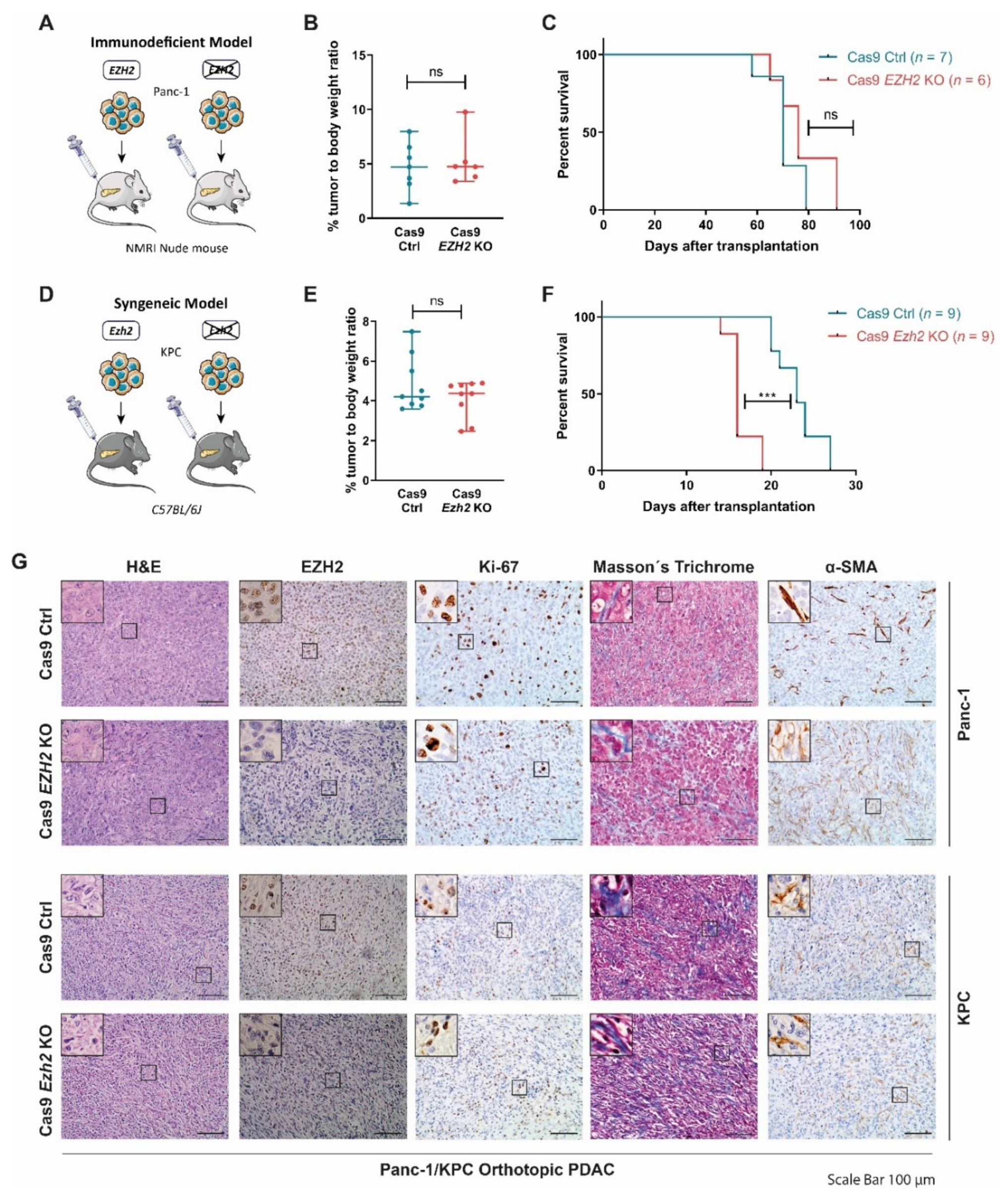
3.1. EZH2 Depletion Does Not Reduce Tumor Progression in Orthotopic PDAC Models
3.2. The TP53-Status Determines EZH2-Dependent Target Gene Regulation
3.3. The Induction of PDAC Cell Apoptosis upon EZH2 Depletion Is Restricted to p53wt Status
3.4. Loss of EZH2 Results in p53wt Stabilization in a CDKN2A-Dependent Manner
3.5. P53wt PDAC Evolving in the Absence of EZH2 Circumvents Induction of the CDKN2A-p53wt Axis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Volkel, P.; Dupret, B.; Le Bourhis, X.; Angrand, P.O. Diverse involvement of ezh2 in cancer epigenetics. Am. J. Transl. Res. 2015, 7, 175–193. [Google Scholar] [PubMed]
- Sparmann, A.; van Lohuizen, M. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer 2006, 6, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Chou, R.H.; Yu, Y.L.; Hung, M.C. The roles of ezh2 in cell lineage commitment. Am. J. Transl. Res. 2011, 3, 243–250. [Google Scholar] [PubMed]
- Bracken, A.P.; Dietrich, N.; Pasini, D.; Hansen, K.H.; Helin, K. Genome-wide mapping of polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006, 20, 1123–1136. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.A.; Varambally, S.; Arend, R.C. Histone methyltransferase ezh2: A therapeutic target for ovarian cancer. Mol. Cancer Ther. 2018, 17, 591–602. [Google Scholar] [CrossRef] [Green Version]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.; Otte, A.P.; Hayes, D.F.; et al. Ezh2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [Green Version]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein ezh2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Mallen-St Clair, J.; Soydaner-Azeloglu, R.; Lee, K.E.; Taylor, L.; Livanos, A.; Pylayeva-Gupta, Y.; Miller, G.; Margueron, R.; Reinberg, D.; Bar-Sagi, D. Ezh2 couples pancreatic regeneration to neoplastic progression. Genes Dev. 2012, 26, 439–444. [Google Scholar] [CrossRef] [Green Version]
- Rao, Z.Y.; Cai, M.Y.; Yang, G.F.; He, L.R.; Mai, S.J.; Hua, W.F.; Liao, Y.J.; Deng, H.X.; Chen, Y.C.; Guan, X.Y.; et al. Ezh2 supports ovarian carcinoma cell invasion and/or metastasis via regulation of tgf-beta1 and is a predictor of outcome in ovarian carcinoma patients. Carcinogenesis 2010, 31, 1576–1583. [Google Scholar] [CrossRef]
- Smits, M.; Nilsson, J.; Mir, S.E.; van der Stoop, P.M.; Hulleman, E.; Niers, J.M.; de Witt Hamer, P.C.; Marquez, V.E.; Cloos, J.; Krichevsky, A.M.; et al. Mir-101 is down-regulated in glioblastoma resulting in ezh2-induced proliferation, migration, and angiogenesis. Oncotarget 2010, 1, 710–720. [Google Scholar] [CrossRef] [Green Version]
- Weikert, S.; Christoph, F.; Kollermann, J.; Muller, M.; Schrader, M.; Miller, K.; Krause, H. Expression levels of the ezh2 polycomb transcriptional repressor correlate with aggressiveness and invasive potential of bladder carcinomas. Int. J. Mol. Med. 2005, 16, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.; Forster, T.; Reutlinger, K.; Kopp, W.; Versemann, L.; Spitalieri, J.; Gaedcke, J.; Ströbel, P.; Singh, S.K.; Ellenrieder, V.; et al. Chromatin-independent interplay of nfatc1 and ezh2 in pancreatic cancer. Cells 2021, 10, 3463. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.; Steuber, B.; Kopp, W.; Kari, V.; Urbach, L.; Wang, X.; Kuffer, S.; Bohnenberger, H.; Spyropoulou, D.; Zhang, Z.; et al. Ezh2 regulates pancreatic cancer subtype identity and tumor progression via transcriptional repression of gata6. Cancer Res. 2020, 80, 4620–4632. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.M.; Neesse, A.; Dyck, M.L.; Steuber, B.; Koenig, A.O.; Lubeseder-Martellato, C.; Winter, T.; Forster, T.; Bohnenberger, H.; Kitz, J.; et al. Context-dependent epigenetic regulation of nuclear factor of activated t cells 1 in pancreatic plasticity. Gastroenterology 2017, 152, 1507–1520.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ougolkov, A.V.; Bilim, V.N.; Billadeau, D.D. Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase enhancer of zeste homologue 2. Clin. Cancer Res. 2008, 14, 6790–6796. [Google Scholar] [CrossRef] [Green Version]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Lomberk, G.; Blum, Y.; Nicolle, R.; Nair, A.; Gaonkar, K.S.; Marisa, L.; Mathison, A.; Sun, Z.; Yan, H.; Elarouci, N.; et al. Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes. Nat. Commun. 2018, 9, 1978. [Google Scholar] [CrossRef]
- Lomberk, G.; Dusetti, N.; Iovanna, J.; Urrutia, R. Emerging epigenomic landscapes of pancreatic cancer in the era of precision medicine. Nat. Commun. 2019, 10, 3875. [Google Scholar] [CrossRef]
- Roy, N.; Malik, S.; Villanueva, K.E.; Urano, A.; Lu, X.; Von Figura, G.; Seeley, E.S.; Dawson, D.W.; Collisson, E.A.; Hebrok, M. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev. 2015, 29, 658–671. [Google Scholar] [CrossRef] [Green Version]
- Von Figura, G.; Fukuda, A.; Roy, N.; Liku, M.E.; Morris Iv, J.P.; Kim, G.E.; Russ, H.A.; Firpo, M.A.; Mulvihill, S.J.; Dawson, D.W.; et al. The chromatin regulator brg1 suppresses formation of intraductal papillary mucinous neoplasm and pancreatic ductal adenocarcinoma. Nat. Cell Biol. 2014, 16, 255–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, M.; Klein, L.; Espinet, E.; Georgomanolis, T.; Wegwitz, F.; Li, X.; Urbach, L.; Danieli-Mackay, A.; Kuffer, S.; Bojarczuk, K.; et al. Tnf-alpha-producing macrophages determine subtype identity and prognosis via ap1 enhancer reprogramming in pancreatic cancer. Nat. Cancer 2021, 2, 1185–1203. [Google Scholar] [CrossRef] [PubMed]
- Hessmann, E.; Buchholz, S.M.; Demir, I.E.; Singh, S.K.; Gress, T.M.; Ellenrieder, V.; Neesse, A. Microenvironmental determinants of pancreatic cancer. Physiol. Rev. 2020, 100, 1707–1751. [Google Scholar] [CrossRef] [PubMed]
- Hessmann, E.; Johnsen, S.A.; Siveke, J.T.; Ellenrieder, V. Epigenetic treatment of pancreatic cancer: Is there a therapeutic perspective on the horizon? Gut 2017, 66, 168–179. [Google Scholar] [CrossRef] [Green Version]
- Food and Drug Administration. Fda Approves first Treatment Option Specifically for Patients with Epithelioid Sarcoma, a Rare Soft Tissue Cancer. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-option-specifically-patients-epithelioid-sarcoma-rare-soft-tissue (accessed on 20 May 2022).
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53r172h and krasg12d cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, S.; Chen, N.M.; Siveke, J.T.; Konig, A.; Zhang, J.S.; Singh, S.K.; Wolf, E.; Bartkuhn, M.; Esposito, I.; Hessmann, E.; et al. Inflammation-induced nfatc1-stat3 transcription complex promotes pancreatic cancer initiation by krasg12d. Cancer Discov. 2014, 4, 688–701. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.M.; Singh, G.; Koenig, A.; Liou, G.Y.; Storz, P.; Zhang, J.S.; Regul, L.; Nagarajan, S.; Kuhnemuth, B.; Johnsen, S.A.; et al. Nfatc1 links egfr signaling to induction of sox9 transcription and acinar-ductal transdifferentiation in the pancreas. Gastroenterology 2015, 148, 1024–1034.e9. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Chen, N.M.; Hessmann, E.; Siveke, J.; Lahmann, M.; Singh, G.; Voelker, N.; Vogt, S.; Esposito, I.; Schmidt, A.; et al. Antithetical nfatc1-sox2 and p53-mir200 signaling networks govern pancreatic cancer cell plasticity. EMBO J. 2015, 34, 517–530. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Hasselluhn, M.C.; Schmidt, G.E.; Ellenrieder, V.; Johnsen, S.A.; Hessmann, E. Aberrant nfatc1 signaling counteracts tgfβ-mediated growth arrest and apoptosis induction in pancreatic cancer progression. Cell Death Dis. 2019, 10, 446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgart, S.; Glesel, E.; Singh, G.; Chen, N.M.; Reutlinger, K.; Zhang, J.; Billadeau, D.D.; Fernandez-Zapico, M.E.; Gress, T.M.; Singh, S.K.; et al. Restricted heterochromatin formation links nfatc2 repressor activity with growth promotion in pancreatic cancer. Gastroenterology 2012, 142, 388–398.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Raghavachari, N.; Garcia-Reyero, N. Gene Expression Analysis; Humana New York: New York, NY, USA, 2018. [Google Scholar]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Huber, M.; Brehm, C.U.; Gress, T.M.; Buchholz, M.; Alashkar Alhamwe, B.; von Strandmann, E.P.; Slater, E.P.; Bartsch, J.W.; Bauer, C.; Lauth, M. The immune microenvironment in pancreatic cancer. Int. J. Mol. Sci. 2020, 21, 7307. [Google Scholar] [CrossRef]
- Karamitopoulou, E. Tumour microenvironment of pancreatic cancer: Immune landscape is dictated by molecular and histopathological features. Br. J. Cancer 2019, 121, 5–14. [Google Scholar] [CrossRef]
- Murakami, T.; Hiroshima, Y.; Matsuyama, R.; Homma, Y.; Hoffman, R.M.; Endo, I. Role of the tumor microenvironment in pancreatic cancer. Ann. Gastroenterol. Surg. 2019, 3, 130–137. [Google Scholar] [CrossRef]
- Erkan, M.; Michalski, C.W.; Rieder, S.; Reiser-Erkan, C.; Abiatari, I.; Kolb, A.; Giese, N.A.; Esposito, I.; Friess, H.; Kleeff, J. The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 2008, 6, 1155–1161. [Google Scholar] [CrossRef]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of ezh2 activates stat3 signaling via stat3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremer, S.C.B.; Conradi, L.C.; Mechie, N.C.; Amanzada, A.; Mavropoulou, E.; Kitz, J.; Ghadimi, M.; Ellenrieder, V.; Ströbel, P.; Hessmann, E.; et al. Enhancer of zeste homolog 2 in colorectal cancer development and progression. Digestion 2021, 102, 227–235. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Couldwell, W.T.; Hinton, D.R.; He, S.; Chen, T.C.; Sebat, I.; Weiss, M.H.; Law, R.E. Protein kinase c inhibitors induce apoptosis in human malignant glioma cell lines. FEBS Lett. 1994, 345, 43–46. [Google Scholar] [CrossRef] [Green Version]
- Oliner, J.D.; Kinzler, K.W.; Meltzer, P.S.; George, D.L.; Vogelstein, B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992, 358, 80–83. [Google Scholar] [CrossRef]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Lukashchuk, N.; Vousden, K.H. Ubiquitination and degradation of mutant p53. Mol. Cell. Biol. 2007, 27, 8284–8295. [Google Scholar] [CrossRef] [Green Version]
- Frum, R.A.; Grossman, S.R. Mechanisms of mutant p53 stabilization in cancer. Sub Cell. Biochem. 2014, 85, 187–197. [Google Scholar]
- Zhu, Q.; Wani, G.; Yao, J.; Patnaik, S.; Wang, Q.E.; El-Mahdy, M.A.; Prætorius-Ibba, M.; Wani, A.A. The ubiquitin–proteasome system regulates p53-mediated transcription at p21waf1 promoter. Oncogene 2007, 26, 4199–4208. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xiong, Y.; Yarbrough, W.G. Arf promotes mdm2 degradation and stabilizes p53: Arf-ink4a locus deletion impairs both the rb and p53 tumor suppression pathways. Cell 1998, 92, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Comet, I.; Riising, E.M.; Leblanc, B.; Helin, K. Maintaining cell identity: Prc2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 2016, 16, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Pinton, G.; Wang, Z.; Balzano, C.; Missaglia, S.; Tavian, D.; Boldorini, R.; Fennell, D.A.; Griffin, M.; Moro, L. Cdkn2a determines mesothelioma cell fate to ezh2 inhibition. Front. Oncol. 2021, 11, 2505. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, M.; Uchimaru, K. Targeting ezh2 in cancer therapy. Curr. Opin. Oncol. 2017, 29, 375–381. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, M.; Wang, X.; Hamdan, F.H.; Rapp, J.; Eggert, J.; Kosinsky, R.L.; Wegwitz, F.; Kutschat, A.P.; Younesi, F.S.; Gaedcke, J.; et al. Arid1a facilitates kras signaling-regulated enhancer activity in an ap1-dependent manner in colorectal cancer cells. Clin. Epigenetics 2019, 11, 92. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Friedland, S.C.; Guo, B.; O’Dell, M.R.; Alexander, W.B.; Whitney-Miller, C.L.; Agostini-Vulaj, D.; Huber, A.R.; Myers, J.R.; Ashton, J.M.; et al. Arid1a, a swi/snf subunit, is critical to acinar cell homeostasis and regeneration and is a barrier to transformation and epithelial-mesenchymal transition in the pancreas. Gut 2019, 68, 1245–1258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, C.; Yu, S.; Jia, C.; Yan, J.; Lu, Z.; Chen, J. Loss of arid1a expression correlates with tumor differentiation and tumor progression stage in pancreatic ductal adenocarcinoma. Technol. Cancer Res. Treat. 2018, 17, 1533034618754475. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. Ezh2 oncogenic activity in castration-resistant prostate cancer cells is polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef] [Green Version]
- Behrens, C.; Solis, L.M.; Lin, H.; Yuan, P.; Tang, X.; Kadara, H.; Riquelme, E.; Galindo, H.; Moran, C.A.; Kalhor, N.; et al. Ezh2 protein expression associates with the early pathogenesis, tumor progression, and prognosis of non-small cell lung carcinoma. Clin. Cancer Res. 2013, 19, 6556–6565. [Google Scholar] [CrossRef] [Green Version]
- Ntziachristos, P.; Tsirigos, A.; Van Vlierberghe, P.; Nedjic, J.; Trimarchi, T.; Flaherty, M.S.; Ferres-Marco, D.; da Ros, V.; Tang, Z.; Siegle, J.; et al. Genetic inactivation of the polycomb repressive complex 2 in t cell acute lymphoblastic leukemia. Nat. Med. 2012, 18, 298–301. [Google Scholar] [CrossRef]
- Vanharanta, S.; Shu, W.; Brenet, F.; Hakimi, A.A.; Heguy, A.; Viale, A.; Reuter, V.E.; Hsieh, J.J.; Scandura, J.M.; Massagué, J. Epigenetic expansion of vhl-hif signal output drives multiorgan metastasis in renal cancer. Nat. Med. 2013, 19, 50–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgart, S.; Ellenrieder, V.; Fernandez-Zapico, M.E. Oncogenic transcription factors: Cornerstones of inflammation-linked pancreatic carcinogenesis. Gut 2013, 62, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Labuschagne, C.F.; Zani, F.; Vousden, K.H. Control of metabolism by p53—Cancer and beyond. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Siolas, D.; Vucic, E.; Kurz, E.; Hajdu, C.; Bar-Sagi, D. Gain-of-function p53(r172h) mutation drives accumulation of neutrophils in pancreatic tumors, promoting resistance to immunotherapy. Cell Rep 2021, 36, 109578. [Google Scholar] [CrossRef] [PubMed]
- Klemke, L.; Fehlau, C.F.; Winkler, N.; Toboll, F.; Singh, S.K.; Moll, U.M.; Schulz-Heddergott, R. The gain-of-function p53 r248w mutant promotes migration by stat3 deregulation in human pancreatic cancer cells. Front. Oncol. 2021, 11, 642603. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Stark, N.; Edmunds, S.J.; Li, J.; Conradi, L.C.; Bohnenberger, H.; Ceteci, F.; Greten, F.R.; Dobbelstein, M.; Moll, U.M. Therapeutic ablation of gain-of-function mutant p53 in colorectal cancer inhibits stat3-mediated tumor growth and invasion. Cancer Cell 2018, 34, 298–314.e297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serresi, M.; Gargiulo, G.; Proost, N.; Siteur, B.; Cesaroni, M.; Koppens, M.; Xie, H.; Sutherland, K.D.; Hulsman, D.; Citterio, E.; et al. Polycomb repressive complex 2 is a barrier to kras-driven inflammation and epithelial-mesenchymal transition in non-small-cell lung cancer. Cancer Cell 2016, 29, 17–31. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yu, X.; Gong, W.; Liu, X.; Park, K.S.; Ma, A.; Tsai, Y.H.; Shen, Y.; Onikubo, T.; Pi, W.C.; et al. Ezh2 noncanonically binds cmyc and p300 through a cryptic transactivation domain to mediate gene activation and promote oncogenesis. Nat. Cell Biol. 2022, 24, 384–399. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Li, Z.; Wu, Z.; Aau, M.; Guan, P.; Karuturi, R.K.; Liou, Y.C.; Yu, Q. Context-specific regulation of nf-kappab target gene expression by ezh2 in breast cancers. Mol. Cell 2011, 43, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Wienken, M.; Dickmanns, A.; Nemajerova, A.; Kramer, D.; Najafova, Z.; Weiss, M.; Karpiuk, O.; Kassem, M.; Zhang, Y.; Lozano, G.; et al. Mdm2 associates with polycomb repressor complex 2 and enhances stemness-promoting chromatin modifications independent of p53. Mol. Cell 2016, 61, 68–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Ding, L.; Wang, D.; Ye, Z.; He, Y.; Ma, L.; Zhu, R.; Pan, Y.; Wu, Q.; Pang, K.; et al. Ezh2 cooperates with gain-of-function p53 mutants to promote cancer growth and metastasis. EMBO J. 2019, 38, e99599. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, M.; Wang, D.; Hou, P.; Chen, X.; Chu, S.; Chai, D.; Zheng, J.; Bai, J. Post-translational modifications of ezh2 in cancer. Cell Biosci. 2020, 10, 143. [Google Scholar] [CrossRef]
- Ko, H.W.; Lee, H.H.; Huo, L.; Xia, W.; Yang, C.C.; Hsu, J.L.; Li, L.Y.; Lai, C.C.; Chan, L.C.; Cheng, C.C.; et al. Gsk3beta inactivation promotes the oncogenic functions of ezh2 and enhances methylation of h3k27 in human breast cancers. Oncotarget 2016, 7, 57131–57144. [Google Scholar] [CrossRef] [Green Version]
- Knutson, S.K.; Kawano, S.; Minoshima, Y.; Warholic, N.M.; Huang, K.C.; Xiao, Y.; Kadowaki, T.; Uesugi, M.; Kuznetsov, G.; Kumar, N.; et al. Selective inhibition of ezh2 by epz-6438 leads to potent antitumor activity in ezh2-mutant non-hodgkin lymphoma. Mol. Cancer Ther. 2014, 13, 842–854. [Google Scholar] [CrossRef] [Green Version]
- Ma, A.; Stratikopoulos, E.; Park, K.S.; Wei, J.; Martin, T.C.; Yang, X.; Schwarz, M.; Leshchenko, V.; Rialdi, A.; Dale, B.; et al. Discovery of a first-in-class ezh2 selective degrader. Nat. Chem. Biol. 2020, 16, 214–222. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Versemann, L.; Patil, S.; Steuber, B.; Zhang, Z.; Kopp, W.; Krawczyk, H.E.; Kaulfuß, S.; Wollnik, B.; Ströbel, P.; Neesse, A.; et al. TP53-Status-Dependent Oncogenic EZH2 Activity in Pancreatic Cancer. Cancers 2022, 14, 3451. https://doi.org/10.3390/cancers14143451
Versemann L, Patil S, Steuber B, Zhang Z, Kopp W, Krawczyk HE, Kaulfuß S, Wollnik B, Ströbel P, Neesse A, et al. TP53-Status-Dependent Oncogenic EZH2 Activity in Pancreatic Cancer. Cancers. 2022; 14(14):3451. https://doi.org/10.3390/cancers14143451
Chicago/Turabian StyleVersemann, Lennart, Shilpa Patil, Benjamin Steuber, Zhe Zhang, Waltraut Kopp, Hannah Elisa Krawczyk, Silke Kaulfuß, Bernd Wollnik, Philipp Ströbel, Albrecht Neesse, and et al. 2022. "TP53-Status-Dependent Oncogenic EZH2 Activity in Pancreatic Cancer" Cancers 14, no. 14: 3451. https://doi.org/10.3390/cancers14143451