
Abstract
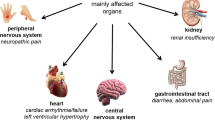
Fabry disease is a progressive and life-threatening glycolipid storage disorder affecting both males and females. The primary driver of the disease is the accumulation of glycolipids (globotriaosylceramide [GL-3]) in a variety of cell types, including vascular endothelial cells, a range of renal cell types, cardiomyocytes and neurons, which is caused by deficient activity of the lysosomal enzyme, α-galactosidase. The disease typically presents during childhood or adolescence. First manifestations reflect involvement of small nerve fibres of the peripheral and autonomic nervous systems. With age, severe complications involving the kidneys, heart and brain cause considerable morbidity and premature death.
Outside the US, enzyme replacement therapy (ERT) with agalsidase alfa 0.2 mg/kg every other week (EOW) and agalsidase beta 1.0mg/kg EOW is available for the treatment of patients with Fabry disease, while agalsidase beta 1.0mg/kg EOW is the only approved drug in the US.
To analyse the evidence for ERT, a systematic review of the literature was performed to identify prospectively designed randomized, controlled trials (RCTs) and open-label studies on the efficacy of agalsidase alfa and agalsidase beta. MEDLINE and EMBASE databases were searched; inclusion criteria for the systematic review were prospectively designed clinical studies evaluating ERT with quantifiable endpoints: double-blind and open-label studies were eligible. Exclusion criteria were review articles, case reports, case studies, letters to the editor and articles based on registry data (Fabry Outcome Survey or Fabry Registry). In addition, any studies with a retrospective design or data based on post hoc analyses were excluded. The evidence was reviewed with respect to the clinical benefits of ERT at the level of the end organ.
A total of 9 RCTs and 23 open-label studies were identified for inclusion. The efficacy of ERT in Fabry disease has been measured against a variety of endpoints, the majority of which were subclinical parameters rather than clinical outcomes. Plasma levels of GL-3 together with accumulation in the kidney, heart and skin were the most commonly studied endpoints, followed by renal endpoints of proteinuria and glomerular filtration rate, whereas cardiac and neurological endpoints were not commonly studied. To date, only one RCT with ERT defined hard clinical outcomes in the form of cardiac, renal or cerebrovascular events, or death as its primary endpoint.
The currently available data from prospective RCTs and open-label studies in patients with Fabry disease are more robust for ERT at a dose of 1 mg/kg EOW than a dose of 0.2mg/kg EOW, although the beneficial effects of ERT with either dose or preparation are variable.
















Similar content being viewed by others


References
Wanner C. Fabry disease model: a rational approach to the management of Fabry disease [published erratum appears in Clin Ther 2007 Oct; 29(10): 2268]. Clin Ther 2007; 29 Suppl. A: S2–5
Deegan PB, Baehner AF, Barba Romero MA, et al., European FOS Investigators. Natural history of Fabry disease in females in the Fabry Outcome Survey. J Med Genet 2006; 43(4): 347–52
Wang RY, Lelis A, Mirocha J, et al. Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet Med 2007 Jan; 9(1): 34–45
Wilcox WR, Oliveira JP, Hopkin RJ, et al., Fabry Registry. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab 2008 Feb; 93(2): 112–28
Ortiz A, Oliveira JP, Waldek S, et al., Fabry Registry. Nephropathy in males and females with Fabry disease: cross-sectional description of patients before treatment with enzyme replacement therapy. Nephrol Dial Transplant 2008 May; 23(5): 1600–7
Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest 2004 Mar; 34(3): 236–42
Eng CM, Fletcher J, Wilcox WR, et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis 2007 Apr; 30(2): 184–92
Cable WJ, Kolodny EH, Adams RD. Fabry disease: impaired autonomic function. Neurology 1982 May; 32(5): 498–502
Low M, Nicholls K, Tubridy N, et al. Neurology of Fabry disease. Intern Med J 2007 Jul; 37(7): 436–47
Møller AT, Jensen TS. Neurological manifestations in Fabry’s disease. Nat Clin Pract Neurol 2007 Feb; 3(2): 95–106
Ries M, Ramaswami U, Parini R, et al. The early clinical phenotype of Fabry disease: a study on 35 European chil dren and adolescents. Eur J Pediatr 2003 Nov; 162(11): 767–72
Ramaswami U, Whybra C, Parini R, et al., FOS European Investigators. Clinical manifestations of Fabry disease in children: data from the Fabry Outcome Survey. Acta Paediatr 2006 Jan; 95(1): 86–92
Gubler MC, Lenoir G, Grünfeld JP, et al. Early renal changes in hemizygous and heterozygous patients with Fabry’s disease. Kidney Int 1978 Mar; 13(3): 223–35
Sessa A, Meroni M, Battini G, et al. Renal pathological changes in Fabry disease. J Inherit Metab Dis 2001; 24 Suppl. 2: 66–70
Grünfeld JP, Lidove O, Joly D, et al. Renal disease in Fabry patients. J Inherit Metab Dis 2001; 24 Suppl. 2: 71–4
Branton M, Schiffmann R, Kopp JB. Natural history and treatment of renal involvement in Fabry disease. J Am Soc Nephrol 2002 Jun; 13 Suppl. 2: S139–43
Thadhani R, Wolf M, West ML, et al. Patients with Fabry disease on dialysis in the United States. Kidney Int 2002 Jan; 61(1): 249–55
Kampmann C, Baehner F, Whybra C, et al. Cardiac manifestations of Anderson-Fabry disease in heterozygous females. J Am Coll Cardiol 2002 Nov; 40(9): 1668–74
Linhart A, Kampmann C, Zamorano JL, et al., European FOS Investigators. Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. Eur Heart J 2007 May; 28(10): 1228–35
Mitsias P, Levine SR. Cerebrovascular complications of Fabry’s disease. Ann Neurol 1996 Jul; 40(1): 8–17
Fellgiebel A, Müller MJ, Ginsberg L. CNS manifestations of Fabry’s disease. Lancet Neurol 2006 Sep; 5(9): 791–5
Gold KF, Pastores GM, Botteman MF, et al. Quality of life of patients with Fabry disease. Qual Life Res 2002 Jun; 11(4): 317–27
Cole AL, Lee PJ, Hughes DA, et al. Depression in adults with Fabry disease: a common and under-diagnosed problem. J Inherit Metab Dis 2007 Nov; 30(6): 943–51
Vedder AC, Strijland A, vd Bergh Weerman MA, et al. Manifestations of Fabry disease in placental tissue. J Inherit Metab Dis 2006 Feb; 29(1): 106–11
Aerts JM, Groener JE, Kuiper S, et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci U S A 2008 Feb; 105(8): 2812–7
Blom D, Speijer D, Linthorst GE, et al. Recombinant enzyme therapy for Fabry disease: absence of editing of human alpha-galactosidase A mRNA. Am J Hum Genet 2003 Jan; 72(1): 23–31
Lee K, Jin X, Zhang K, et al. A biochemical and pharmacological comparison of enzyme replacement therapies for the glycolipid storage disorder Fabry disease. Glycobiology 2003 Apr; 13(4): 305–13
Sakuraba H, Murata-Ohsawa M, Kawashima I, et al. Comparison of the effects of agalsidase alfa and agalsidase beta on cultured human Fabry fibroblasts and Fabry mice. J Hum Genet 2006; 51(3): 180–8
Vedder AC, Breunig F, Donker-Koopman WE, et al. Treatment of Fabry disease with different dosing regimens of agalsidase: effects on antibody formation and GL-3. Mol Genet Metab 2008 Jul; 94(3): 319–25
Eng CM, Guffon N, Wilcox WR, et al., International Collaborative Fabry Disease Study Group. Safety and efficacy of recombinant human alpha-galactosidase A-replacement therapy in Fabry’s disease. N Engl J Med 2001 Jul; 345(1): 9–16
Thurberg BL, Byers HR, Granter SR, et al. Monitoring the 3-year efficacy of enzyme replacement therapy in Fabry disease by repeated skin biopsies. J Invest Dermatol 2004 Apr; 122(4): 900–8
Bierer G, Balfe D, Wilcox WR, et al. Improvement in serial cardiopulmonary exercise testing following enzyme replacement therapy in Fabry disease. J Inherit Metab Dis 2006 Aug; 29(4): 572–9
Banikazemi M, Bultas J, Waldek S, et al., Fabry Disease Clinical Trial Study Group. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med 2007 Jan; 146(2): 77–86
Schiffmann R, Kopp JB, Austin 3rd HA, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 2001 Jun; 285(21): 2743–9
Moore DF, Altarescu G, Herscovitch P, et al. Enzyme replacement reverses abnormal cerebrovascular responses in Fabry disease. BMC Neurol 2002 Jun; 2: 4
Hajioff D, Goodwin S, Quiney R, et al. Hearing improvement in patients with Fabry disease treated with agalsidase alfa. Acta Paediatr Suppl 2003 Dec; 92(443): 28–30
Schiffmann R, Hauer P, Freeman B, et al. Enzyme replacement therapy and intraepidermal innervation density in Fabry disease. Muscle Nerve 2006 Jul; 34(1): 53–6
Hughes DA, Elliott PM, Shah J, et al. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson-Fabry disease: a randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Heart 2008 Feb; 94(2): 153–8
Eng CM, Banikazemi M, Gordon RE, et al. A phase 1/2 clinical trial of enzyme replacement in Fabry disease: pharmacokinetic, substrate clearance, and safety studies. Am J Hum Genet 2001 Mar; 68(3): 711–22
Thurberg BL, Rennke H, Colvin RB, et al. Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int 2002 Dec; 62(6): 1933–46
Weidemann F, Niemann M, Breunig F, et al. Long-term effects of enzyme replacement therapy on Fabry cardiomyopathy: evidence for a better outcome with early treatment. Circulation 2009 Feb; 119(4): 524–9
Hilz MJ, Brys M, Marthol H, et al. Enzyme replacement therapy improves function of C-, Aq-, and Ab-nerve fibers in Fabry neuropathy. Neurology 2004 Apr; 62(7): 1066–72
Wilcox WR, Banikazemi M, Guffon N, et al., International Fabry Disease Study Group. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Genet 2004 Jul; 75(1): 65–74
Eto Y, Ohashi T, Utsunomiya Y, et al. Enzyme replacement therapy in Japanese Fabry disease patients: the results of a phase 2 bridging study. J Inherit Metab Dis 2005; 28(4): 575–83
Pisani A, Spinelli L, Sabbatini M, et al. Enzyme replacement therapy in Fabry disease patients undergoing dialysis: effects on quality of life and organ involvement. Am J Kidney Dis 2005 Jul; 46(1): 120–7
Beer M, Weidemann F, Breunig F, et al. Impact of enzyme replacement therapy on cardiac morphology and function and late enhancement in Fabry’s cardiomyopathy. Am J Cardiol 2006 May; 97(10): 1515–8
Breunig F, Weidemann F, Strotmann J, et al. Clinical benefit of enzyme replacement therapy in Fabry disease. Kidney Int 2006 Apr; 69(7): 1216–21
Kalliokoski RJ, Kantola I, Kalliokoski KK, et al. The effect of 12-month enzyme replacement therapy on myocardial perfusion in patients with Fabry disease. J Inherit Metab Dis 2006 Feb; 29(1): 112–8
Germain DP, Waldek S, Banikazemi M, et al. Sustained, long-term renal stabilization after 54 months of agalsidase beta therapy in patients with Fabry disease. J Am Soc Nephrol 2007 May; 18(5): 1547–57
Schiffmann R, Floeter MK, Dambrosia JM, et al. Enzyme replacement therapy improves peripheral nerve and sweat function in Fabry disease. Muscle Nerve 2003 Dec; 28(6): 703–10
Schiffmann R, Ries M, Timmons M, et al. Long-term therapy with agalsidase alfa for Fabry disease: safety and effects on renal function in a home infusion setting. Nephrol Dial Transplant 2006 Feb; 21(2): 345–54
Clarke JT, West ML, Bultas J, et al. The pharmacology of multiple regimens of agalsidase alfa enzyme replacement therapy for Fabry disease. Genet Med 2007 Aug; 9(8): 504–9
Schiffmann R, Askari H, Timmons M, et al. Weekly enzyme replacement therapy may slow decline of renal function in patients with Fabry disease who are on long-term biweekly dosing. J Am Soc Nephrol 2007 May; 18(5): 1576–83
Vedder AC, Linthorst GE, Houge G, et al. Treatment of Fabry disease: outcome of a comparative trial with agalsidase alfa or beta at a dose of 0.2mg/kg. PloS ONE 2007 Jul; 2(7): e598
Baehner F, Kampmann C, Whybra C, et al. Enzyme replacement therapy in heterozygous females with Fabry disease: results of a phase IIIB study. J Inherit Metab Dis 2003; 26(7): 617–27
Wraith JE, Tylki-Szymanska A, Guffon N, et al. Safety and efficacy of enzyme replacement therapy with agalsidase beta: an international, open-label study in pediatric patients with Fabry disease. J Pediatr 2008 Apr; 152(4): 563–70
Ries M, Clarke JT, Whybra C, et al. Enzyme-replacement therapy with agalsidase alfa in children with Fabry disease. Pediatrics 2006 Sep; 118(3): 924–32
Ramaswami U, Wendt S, Pintos-Moreli G, et al. Enzyme replacement therapy with agalsidase alfa in children with Fabry disease. Acta Paediatr 2007 Jan; 96(1): 122–7
Ries M, Clarke JT, Whybra C, et al. Enzyme replacement in Fabry disease: pharmacokinetics and pharmacodynamics of agalsidase alfa in children and adolescents. J Clin Pharmacol 2007 Oct; 47(10): 1222–30
Schiffmann R, Murray GJ, Treco D, et al. Infusion of alpha-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proc Natl Acad Sci U S A 2000 Jan; 97(1): 365–70
Ohashi T, Sakuma M, Kitagawa T, et al. Influence of antibody formation on reduction of globotriaosylceramide (GL-3) in urine from Fabry patients during agalsidase beta therapy. Mol Genet Metab 2007 Nov; 92(3): 271–3
Linthorst GE, Hollak CE, Donker-Koopman WE, et al. Enzyme therapy for Fabry disease: neutralizing antibodies toward agalsidase alpha and beta. Kidney Int 2004 Oct; 66(4): 1589–95
Mengel E, Baron K, Kalkum G, et al. Is there a neutralizing effect of antibodies against agalsidase alpha and agalsidase beta? Acta Paediatr 2007; 96(s455): 108
Vedder AC, Cox-Brinkman J, Hollak CE, et al. Plasma chitotriosidase in male Fabry patients: a marker for monitoring lipid-laden macrophages and their correction by enzyme replacement therapy. Mol Genet Metab 2006 Nov; 89(3): 239–44
Mignani R, Feriozzi S, Pisani A, et al. Agalsidase therapy in patients with Fabry disease on renal replacement therapy: a nationwide study in Italy. Nephrol Dial Transplant 2008 May; 23(5): 1628–35
Acknowledgements
The authors received writing/editorial support in preparation of this article, funded by Genzyme Europe BV. Hester van Lier of Excerpta Medica provided writing support. The authors maintained full and independent responsibility for the content of this paper.
R.M. Schaefer certifies that there is no actual or potential conflict of interest in relation to this article, except that he received fees for lectures and consulting from both Genzyme and Shire. A. Tylki-Szymańska received fees for lectures from Genzyme and Shire. M.J. Hilz received funding support for studies, honoraria for scientific presentations, and compensation as a consultant from Genzyme Inc., Cambridge, MA, USA.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Schaefer, R.M., Tylki-Szymańska, A. & Hilz, M.J. Enzyme Replacement Therapy for Fabry Disease. Drugs 69, 2179–2205 (2009). https://doi.org/10.2165/11318300-000000000-00000
Published:
Issue Date:
DOI: https://doi.org/10.2165/11318300-000000000-00000