
Abstract
Currently, quantitative prediction of the impact of genetic polymorphism and drug-drug interactions mediated by cytochromes, based on in vivo data, is made by two separate methods and restricted to a single cytochrome. We propose a unified approach for describing the combined impact of drug-drug interactions and genetic polymorphism on drug exposure. It relies on in vivo data and uses the following three characteristic parameters: one for the victim drug, one for the interacting drug, and another for the genotype. These parameters are known for a wide range of drugs and genotypes. The metrics of interest are the ratio of victim drug area under the curve (AUC) in patients with genetic variants taking both drugs, to the AUC in patients with either variant or wild-type genotype taking the victim drug alone. The approach was evaluated by external validation, comparing predicted and observed AUC ratios found in the literature. Data were found for 22 substrates, 30 interacting drugs, and 38 substrate-interacting drug couples. The mean prediction error of AUC ratios was 0.02, and the mean prediction absolute error was 0.38 and 1.34, respectively. The model may be used to predict the variations in exposure resulting from a number of drug-drug–genotype combinations. The proposed approach will help (1) to identify comedications and population at risk, (2) to adapt dosing regimens, and (3) to prioritize the clinical pharmacokinetic studies to be done.



Similar content being viewed by others
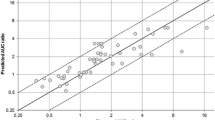
References
Zhang L, Zhang YD, Zhao P, Huang SM. Predicting drug-drug interactions: an FDA perspective. AAPS J. 2009;11(2):300–6.
Ohno Y, Hisaka A, Suzuki H. General framework for the quantitative prediction of CYP3A4-mediated oral drug interactions based on the AUC increase by coadministration of standard drugs. Clin Pharmacokinet. 2007;46(8):681–96.
Ohno Y, Hisaka A, Ueno M, Suzuki H. General framework for the prediction of oral drug interactions caused by CYP3A4 induction from in vivo information. Clin Pharmacokinet. 2008;47(10):669–80.
Tod M, Goutelle S, Clavel-Grabit F, Nicolas G, Charpiat B. Quantitative prediction of cytochrome P450 (CYP) 2D6-mediated drug interactions. Clin Pharmacokinet. 2011;50(8):519–30.
Tod M, Goutelle S, Gagnieu MC. Genotype-based quantitative prediction of drug exposure for drugs metabolized by CYP2D6. Clin Pharmacol Ther. 2011;90(4):582–7.
Goutelle S, Bourguignon L, Bleyzac N, Berry J, Clavel-Grabit F, Tod M, et al. In vivo quantitative prediction of the effect of gene polymorphisms and drug interactions on drug exposure for CYP2C19 substrates. AAPS J. 2013;15(2):415–26.
Castellan AC, Tod M, Gueyffier F, Audars M, Cambriels F, Kassaï B, et al. Quantitative prediction of the impact of drug interactions and genetic polymorphisms on cytochrome P450 2C9 substrate exposure. Clin Pharmacokinet. 2013;52(3):199–209.
Malhi H, Atac B, Daly AK, Gupta S. Warfarin and celecoxib interaction in the setting of cytochrome P450 (CYP2C9) polymorphism with bleeding complication. Postgrad Med J. 2004;80(940):107–9.
Puech R, Gagnieu MC, Planus C, Charpiat B, Boibieux A, Ferry T, et al. Extreme bradycardia due to multiple drug-drug interactions in a patient with HIV post-exposure prophylaxis containing lopinavir–ritonavir. Br J Clin Pharmacol. 2011;71(4):621–3.
Madadi P, Hildebrandt D, Gong IY, Schwarz UI, Ciszkowski C, Ross CJ, et al. Fatal hydrocodone overdose in a child: pharmacogenetics and drug interactions. Pediatrics. 2010;126(4):e986–9.
Marcucci C, Sandson NB, Thorn EM, Bourke DL. Unrecognized drug-drug interactions: a cause of intraoperative cardiac arrest. Anesth Analg. 2006;102(5):1569–72.
Yang J, Kjellsson M, Rostami-Hodjegan A, Tucker GT. The effects of dose staggering on metabolic drug-drug interactions. Eur J Pharm Sci. 2003;20(2):223–32.
Rostami-Hodjegan A, Tucker GT. Simulation and prediction of in vivo drug metabolism in human populations from in vitro data. Nat Rev Drug Discov. 2007;6(2):140–8.
Guest EJ, Rowland-Yeo K, Rostami-Hodjegan A, Tucker GT, Houston JB, Galetin A. Assessment of algorithms for predicting drug-drug interactions via inhibition mechanisms: comparison of dynamic and static models. Br J Clin Pharmacol. 2011;71(1):72–87.
Hisaka A, Ohno Y, Yamamoto T, Suzuki H. Prediction of pharmacokinetic drug-drug interaction caused by changes in cytochrome P450 activity using in vivo information. Pharmacol Ther. 2010;125(2):230–48.
Abilify full product information. http://www.abilify.com/utilities/fullproductinfo.aspx.
Bertilsson L, Henthorn TK, Sanz E, Tybring G, Säwe J, Villén T. Importance of genetic factors in the regulation of diazepam metabolism: relationship to S-mephenytoin, but not debrisoquin, hydroxylation phenotype. Clin Pharmacol Ther. 1989;45(4):348–55.
Sohn DR, Kusaka M, Ishizaki T, Shin SG, Jang IJ, Shin JG, et al. Incidence of S-mephenytoin hydroxylation deficiency in a Korean population and the interphenotypic differences in diazepam pharmacokinetics. Clin Pharmacol Ther. 1992;52(2):160–9.
Qin XP, Xie HG, Wang W, He N, Huang SL, Xu ZH, et al. Effect of the gene dosage of CgammaP2C19 on diazepam metabolism in Chinese subjects. Clin Pharmacol Ther. 1999;66(6):642–6.
Ahonen J, Olkkola KT, Neuvonen PJ. The effect of the antimycotic itraconazole on the pharmacokinetics and pharmacodynamics of diazepam. Fundam Clin Pharmacol. 1996;10(3):314–8.
Park JY, Shon JH, Kim KA, Jung HJ, Shim JC, Yoon YR, et al. Combined effects of itraconazole and CYP2D6*10 genetic polymorphism on the pharmacokinetics and pharmacodynamics of haloperidol in healthy subjects. J Clin Psychopharmacol. 2006;26(2):135–42.
Kim YH, Cha IJ, Shim JC, Shin JG, Yoon YR, Kim YK, et al. Effect of rifampin on the plasma concentration and the clinical effect of haloperidol concomitantly administered to schizophrenic patients. J Clin Psychopharmacol. 1996;16(3):247–52.
Spaans E, van den Heuvel MW, Schnabel PG, Peeters PA, Chin-Kon-Sung UG, Colbers EP, et al. Concomitant use of mirtazapine and phenytoin: a drug-drug interaction study in healthy male subjects. Eur J Clin Pharmacol. 2002;58(6):423–9.
Samer CF, Daali Y, Wagner M, Hopfgartner G, Eap CB, Rebsamen MC, et al. Genetic polymorphisms and drug interactions modulating CYP2D6 and CYP3A activities have a major effect on oxycodone analgesic efficacy and safety. Br J Pharmacol. 2010;160(4):919–30.
Grönlund J, Saari TI, Hagelberg NM, Neuvonen PJ, Olkkola KT, Laine K. Exposure to oral oxycodone is increased by concomitant inhibition of CYP2D6 and 3A4 pathways, but not by inhibition of CYP2D6 alone. Br J Clin Pharmacol. 2010;70(1):78–87.
Schulz-Du Bois C, Schulz-Du Bois AC, Bewig B, Gerstner I, Aldenhoff JB, Cascorbi I, et al. Major increase of quetiapine steady-state plasma concentration following coadministration of clarithromycin: confirmation of the pharmacokinetic interaction potential of quetiapine. Pharmacopsychiatry. 2008;41(6):258–9.
Funck-Brentano C, Becquemont L, Kroemer HK, Bühl K, Knebel NG, Eichelbaum M, et al. Variable disposition kinetics and electrocardiographic effects of flecainide during repeated dosing in humans: contribution of genetic factors, dose-dependent clearance, and interaction with amiodarone. Clin Pharmacol Ther. 1994;55(3):256–69.
Becquemont L, Neuvonen M, Verstuyft C, Jaillon P, Letierce A, Neuvonen PJ, et al. Amiodarone interacts with simvastatin but not with pravastatin disposition kinetics. Clin Pharmacol Ther. 2007;81(5):679–84.
Furuta T, Ohashi K, Kobayashi K, Iida I, Yoshida H, Shirai N, et al. Effects of clarithromycin on the metabolism of omeprazole in relation to CYP2C19 genotype status in humans. Clin Pharmacol Ther. 1999;66(3):265–74.
Greenblatt DJ, Preskorn SH, Cotreau MM, Horst WD, Harmatz JS. Fluoxetine impairs clearance of alprazolam but not of clonazepam. Clin Pharmacol Ther. 1992;52(5):479–86.
Wang LS, Zhou G, Zhu B, Wu J, Wang JG, Abd El-Aty AM, et al. St John's wort induces both cytochrome P450 3A4-catalyzed sulfoxidation and 2C19-dependent hydroxylation of omeprazole. Clin Pharmacol Ther. 2004;75(3):191–7.
Purkins L, Wood N, Ghahramani P, Love ER, Eve MD, Fielding A. Coadministration of voriconazole and phenytoin: pharmacokinetic interaction, safety, and toleration. Br J Clin Pharmacol. 2003;56 Suppl 1:37–44.
Kanebratt KP, Diczfalusy U, Bäckström T, Sparve E, Bredberg E, Böttiger Y, et al. Cytochrome P450 induction by rifampicin in healthy subjects: determination using the Karolinska cocktail and the endogenous CYP3A4 marker 4beta-hydroxycholesterol. Clin Pharmacol Ther. 2008;84(5):589–94.
Kirby BJ, Collier AC, Kharasch ED, Whittington D, Thummel KE, Unadkat JD. Complex drug interactions of HIV protease inhibitors 1: inactivation, induction, and inhibition of cytochrome P450 3A by ritonavir or nelfinavir. Drug Metab Dispos. 2011;39(6):1070–8.
Liu P, Foster G, Gandelman K, LaBadie RR, Allison MJ, Gutierrez MJ, et al. Steady-state pharmacokinetic and safety profiles of voriconazole and ritonavir in healthy male subjects. Antimicrob Agents Chemother. 2007;51(10):3617–26.
Kirby BJ, Collier AC, Kharasch ED, Dixit V, Desai P, Whittington D, et al. Complex drug interactions of HIV protease inhibitors 2: in vivo induction and in vitro to in vivo correlation of induction of cytochrome P450 1A2, 2B6, and 2C9 by ritonavir or nelfinavir. Drug Metab Dispos. 2011;39(12):2329–37.
el-Yazigi A, Chaleby K, Gad A, Raines DA. Steady-state kinetics of fluoxetine and amitriptyline in patients treated with a combination of these drugs as compared with those treated with amitriptyline alone. J Clin Pharmacol. 1995;35(1):17–21.
Kubo M, Koue T, Inaba A, Takeda H, Maune H, Fukuda T, et al. Influence of itraconazole coadministration and CYP2D6 genotype on the pharmacokinetics of the new antipsychotic Aripiprazole. Drug Metab Pharmacokinet. 2005;20(1):55–64.
Azuma J, Hasunuma T, Kubo M, Miyatake M, Koue T, Higashi K, et al. The relationship between clinical pharmacokinetics of aripiprazole and CYP2D6 genetic polymorphism: effects of CYP enzyme inhibition by coadministration of paroxetine or fluvoxamine. Eur J Clin Pharmacol. 2012;68(1):29–37.
Kosuge K, Jun Y, Watanabe H, Kimura M, Nishimoto M, Ishizaki T, et al. Effects of CYP3A4 inhibition by diltiazem on pharmacokinetics and dynamics of diazepam in relation to CYP2C19 genotype status. Drug Metab Dispos. 2001;29(10):1284–9.
Saari TI, Laine K, Bertilsson L, Neuvonen PJ, Olkkola KT. Voriconazole and fluconazole increase the exposure to oral diazepam. Eur J Clin Pharmacol. 2007;63(10):941–9.
Lim KS, Cho JY, Jang IJ, Kim BH, Kim J, Jeon JY, et al. Pharmacokinetic interaction of flecainide and paroxetine in relation to the CYP2D6*10 allele in healthy Korean subjects. Br J Clin Pharmacol. 2008;66(5):660–6.
Birgersdotter UM, Wong W, Turgeon J, Roden DM. Stereoselective genetically-determined interaction between chronic flecainide and quinidine in patients with arrhythmias. Br J Clin Pharmacol. 1992;33(3):275–80.
Kumar V, Brundage RC, Oetting WS, Leppik IE, Tracy TS. Differential genotype dependent inhibition of CYP2C9 in humans. Drug Metab Dispos. 2008;36(7):1242–8.
Park JY, Kim KA, Park PW, Park CW, Shin JG. Effect of rifampin on the pharmacokinetics and pharmacodynamics of gliclazide. Clin Pharmacol Ther. 2003;74(4):334–40.
Miura M, Tada H, Yasui-Furukori N, Uno T, Sugawara K, Tateishi T, et al. Enantioselective disposition of lansoprazole in relation to CYP2C19 genotypes in the presence of fluvoxamine. Br J Clin Pharmacol. 2005;60(1):61–8.
Fischer TL, Pieper JA, Graff DW, Rodgers JE, Fischer JD, Parnell KJ, et al. Evaluation of potential losartan-phenytoin drug interactions in healthy volunteers. Clin Pharmacol Ther. 2002;72(3):238–46.
Hamelin BA, Bouayad A, Méthot J, Jobin J, Desgagnés P, Poirier P, et al. Significant interaction between the nonprescription antihistamine diphenhydramine and the CYP2D6 substrate metoprolol in healthy men with high or low CYP2D6 activity. Clin Pharmacol Ther. 2000;67(5):466–77.
Damy T, Pousset F, Caplain H, Hulot JS, Lechat P. Pharmacokinetic and pharmacodynamic interactions between metoprolol and dronedarone in extensive and poor CYP2D6 metabolizers healthy subjects. Fundam Clin Pharmacol. 2004;18(1):113–23.
Werner D, Wuttke H, Fromm MF, Schaefer S, Eschenhagen T, Brune K, et al. Effect of amiodarone on the plasma levels of metoprolol. Am J Cardiol. 2004;94(10):1319–21.
Sharma A, Pibarot P, Pilote S, Dumesnil JG, Arsenault M, Bélanger PM, et al. Toward optimal treatment in women: the effect of sex on metoprolol–diphenhydramine interaction. J Clin Pharmacol. 2010;50(2):214–25.
Sitsen JM, Maris FA, Timmer CJ. Concomitant use of mirtazapine and cimetidine: a drug-drug interaction study in healthy male subjects. Eur J Clin Pharmacol. 2000;56(5):389–94.
Yu KS, Yim DS, Cho JY, Park SS, Park JY, Lee KH, et al. Effect of omeprazole on the pharmacokinetics of moclobemide according to the genetic polymorphism of CYP2C19. Clin Pharmacol Ther. 2001;69(4):266–73.
Chen BL, Chen Y, Tu JH, Li YL, Zhang W, Li Q, et al. Clopidogrel inhibits CYP2C19-dependent hydroxylation of omeprazole related to CYP2C19 genetic polymorphisms. J Clin Pharmacol. 2009;49(5):574–81.
Yasui-Furukori N, Takahata T, Nakagami T, Yoshiya G, Inoue Y, Kaneko S, et al. Different inhibitory effect of fluvoxamine on omeprazole metabolism between CYP2C19 genotypes. Br J Clin Pharmacol. 2004;57(4):487–94.
Cho JY, Yu KS, Jang IJ, Yang BH, Shin SG, Yim DS. Omeprazole hydroxylation is inhibited by a single dose of moclobemide in homozygotic EM genotype for CYP2C19. Br J Clin Pharmacol. 2002;53(4):393–7.
Böttiger Y, Tybring G, Götharson E, Bertilsson L. Inhibition of the sulfoxidation of omeprazole by ketoconazole in poor and extensive metabolizers of S-mephenytoin. Clin Pharmacol Ther. 1997;62(4):384–91.
Potkin SG, Thyrum PT, Alva G, Carreon D, Yeh C, Kalali A, et al. Effect of fluoxetine and imipramine on the pharmacokinetics and tolerability of the antipsychotic quetiapine. J Clin Psychopharmacol. 2002;22(2):174–82.
Uno T, Shimizu M, Yasui-Furukori N, Sugawara K, Tateishi T. Different effects of fluvoxamine on rabeprazole pharmacokinetics in relation to CYP2C19 genotype status. Br J Clin Pharmacol. 2006;61(3):309–14.
Spina E, Avenoso A, Scordo MG, Ancione M, Madia A, Gatti G, et al. Inhibition of risperidone metabolism by fluoxetine in patients with schizophrenia: a clinically relevant pharmacokinetic drug interaction. J Clin Psychopharmacol. 2002;22(4):419–23.
Mikus G, Schöwel V, Drzewinska M, Rengelshausen J, Ding R, Riedel KD, et al. Potent cytochrome P450 2C19 genotype-related interaction between voriconazole and the cytochrome P450 3A4 inhibitor ritonavir. Clin Pharmacol Ther. 2006;80(2):126–35.
Shi HY, Yan J, Zhu WH, Yang GP, Tan ZR, Wu WH, et al. Effects of erythromycin on voriconazole pharmacokinetics and association with CYP2C19 polymorphism. Eur J Clin Pharmacol. 2010;66(11):1131–6.
FDA Antiviral Drugs Advisory Committee. 2001. Briefing document for voriconazole (oral and intravenous formulations). http://www.fda.gov/ohrms/dockets/ac/01/briefing/3792b2_01_Pfizer.pdf.
Nivoix Y, Levêque D, Herbrecht R, Koffel JC, Beretz L, Ubeaud-Sequier G. The enzymatic basis of drug-drug interactions with systemic triazole antifungals. Clin Pharmacokinet. 2008;47(12):779–92.
Purkins L, Wood N, Ghahramani P, Greenhalgh K, Allen MJ, Kleinermans D. Pharmacokinetics and safety of voriconazole following intravenous to oral dose escalation regimens. Antimicrob Agents Chemother. 2002;46(8):2546–53.
Elsby R, Hilgendorf C, Fenner K. Understanding the critical disposition pathways of statins to assess drug-drug interaction risk during drug development: it’s not just about OATP1B1. Clin Pharmacol Ther. 2012;92(5):584–98.
Hisaka A, Ohno Y, Yamamoto T, Suzuki H. Theoretical considerations on quantitative prediction of drug-drug interactions. Drug Metab Pharmacokinet. 2010;25(1):48–61.
Mayhew BS, Jones DR, Hall SD. An in vitro model for predicting in vivo inhibition of cytochrome P450 3A4 by metabolic intermediate complex formation. Drug Metab Dispos. 2000;28(9):1031–7.
Obach RS, Walsky RL, Venkatakrishnan K. Mechanism-based inactivation of human cytochrome p450 enzymes and the prediction of drug-drug interactions. Drug Metab Dispos. 2007;35(2):246–55.
Acknowledgments
No sources of funding were used to conduct this study or prepare the manuscript.
Conflict of Interest
The authors declared no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Appendix: derivation of Eq. 11
Appendix: derivation of Eq. 11
In vitro, according to Hisaka (67), in case of reversible inhibition of a cytochrome, the ratio of victim drug intrinsic clearances is related to the inhibitor concentration, Iu, and the inhibition constant Ki as follows:
In case of mechanism-based inhibition, the ratio of intrinsic clearances depends on kinact, kdeg, and KI which are the maximum inactivation rate constant, degeneration constant, and inhibitor concentration when the rate constant of inactivation reaches half kinact, respectively (68):
In vivo, using the following assumptions: (1) the metabolic clearance of the victim drug is assumed to be close to total clearance, (2) metabolism is assumed to occur for a small part in the gut wall and for the main part in the liver, (3) hepatic clearance is related to intrinsic clearance by the well-stirred model, and (4) the kinetics of victim drug is linear, i.e., clearance is independent of time and dose, then the ratio of oral clearances is approximately equal to the ratio of intrinsic clearances. Replacing Iu by the time-averaged unbound concentration of interacting drug at the target site, Iu,av:
Defining the in vivo potency of an inhibitor as follows:
It comes
For a reversible inhibitor, and
For a mechanism-based inhibitor. Because kinact is much greater than kdeg (69), we have kinact/kdeg >> 1. Therefore,
Assuming linear kinetics of the inhibitor, its concentration Iu,av is proportional to its dose or dosing rate:
Hence,
for a reversible inhibitor, and
for a mechanism-based inhibitor. The final expression is the same for both types of inhibitor, but the expression of D50 is different.
Similarly, the inductive effect may be determined in vitro on hepatocyte cell cultures and modeled as (3) follows:
Where Emax is the maximal induction effect and I50 is the inducer unbound concentration resulting in a half maximal induction.
Defining the in vivo potency of an inducer as follows:
We have, by combining the last two equations:
Where Emax has been replaced by IXmax for consistency. Using I u,av = α.Dose, we find:
Rights and permissions
About this article
Cite this article
Tod, M., Nkoud-Mongo, C. & Gueyffier, F. Impact of Genetic Polymorphism on Drug-Drug Interactions Mediated by Cytochromes: A General Approach. AAPS J 15, 1242–1252 (2013). https://doi.org/10.1208/s12248-013-9530-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-013-9530-2