
Abstract
Premature infants have higher cortisol precursor concentrations than term infants; however, many sick preterm infants have surprisingly low cortisol concentrations. Those who develop chronic lung disease (CLD) have lower cortisol values than those who recover. We hypothesized that some infants have a decreased ability to synthesize cortisol, leading to physiologic disruptions including amplified inflammatory responses, thereby resulting in CLD. We measured cortisol, 11-deoxycortisol, 17-hydroxyprogesterone, 17-hydroxypregnenolone, dehydroepiandrosterone sulfate, and ACTH in 40 extremely low birth weight infants enrolled in a study of low-dose hydrocortisone therapy to prevent CLD. Thirty-four infants survived and 15 developed CLD. Hydrocortisone therapy did not suppress ACTH or any measured steroid value. Before study (<48 h of life), 17-OH progesterone was higher in CLD infants, as was the ratio of 17-OH progesterone to 11-deoxycortisol. On d 15–19 (≥72 h after end of therapy), basal and stimulated cortisol concentrations were lower in CLD infants. In contrast, the basal ratio of 11-deoxycortisol to cortisol was higher in CLD infants, as were stimulated values of 17-OH progesterone and stimulated ratios of 17-OH progesterone to 11-deoxycortisol and 11-deoxycortisol to cortisol. Thus, infants who developed CLD had lower basal and stimulated cortisol values, but elevated cortisol precursors and precursor to product ratios, compared with infants who recovered. These data support the hypothesis that these immature infants have a decreased capacity to synthesize cortisol, which may lead to a relative adrenal insufficiency in the face of significant illness.
Similar content being viewed by others

Main
Premature infants have elevated concentrations of cortisol precursors compared with term infants, even to the point of requiring the development of different normal screening ranges for congenital adrenal hyperplasia (1–4). Cortisol concentrations, however, are not similarly elevated in these infants; instead, many sick premature infants have surprisingly low cortisol concentrations for their degree of illness (1, 2, 5, 6). Case reports and small studies have also reported clinical signs consistent with adrenal insufficiency in such infants, such as hypotension, which respond to hydrocortisone therapy (7–10).
Premature infants who develop CLD have been shown to have lower serum cortisol concentrations and decreased response to ACTH stimulation early in life (6, 11–13), compared with similar infants who recover without CLD. Because cortisol is central to the ability of the body to control inflammation, one consequence of cortisol insufficiency can be an amplified response to inflammatory stimuli (14–16). The earliest stages in the development of CLD are characterized by broad increases in lung inflammation (17, 18). We have reported that, in the first week of life, serum cortisol concentrations correlated inversely with concentrations of several cytokines and proteins in the airways of intubated premature infants (19).
From these findings, we developed the hypothesis that some premature infants have a decreased capacity to synthesize cortisol, resulting in a relative adrenal insufficiency in the face of stress or illness, a phenomenon that has been described in adult populations (20). This insufficiency could lead to a variety of physiologic disruptions, including amplified inflammatory responses, thereby resulting in the development of CLD. Such a decreased synthetic capacity could result from a specific enzyme deficiency; however, it would more likely represent a generalized immaturity of the definitive adrenal cortex.
Maturation of the fetal adrenal cortex may be regulated in part by decreasing exposure to maternal cortisol, reviewed by Pepe and Albrecht (21), as follows. Early in gestation, the fetal hypothalamic-pituitary-adrenal axis is exposed to negative feedback from maternal cortisol, which freely crosses through an immature placenta. Later in gestation, as placental metabolism matures, the enzyme 11β-hydroxysteroid dehydrogenase (11β-HSD) increasingly oxidizes maternal cortisol to its inactive metabolite, cortisone. Thus, negative feedback from maternal cortisol gradually decreases during the latter half of gestation, leading to increased fetal cortisol synthesis. When this normal maturational process is disrupted by extremely premature birth, the immature adrenal gland might lack the capacity to synthesize sufficient quantities of cortisol for the many stressors of extrauterine life, such as acute respiratory illness and mechanical ventilation.
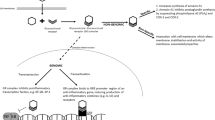
If such a functional immaturity of the adrenal gland contributes to the development of CLD in these infants, such infants should show accumulation of cortisol precursors (Fig. 1), compared with infants who recover without CLD. To examine this hypothesis, we measured serum cortisol and its precursors 11-deoxycortisol, 17-hydroxyprogesterone, and 17-hydroxypregnenolone, as well as DHEAS as a measure of fetal zone adrenal activity, in a cohort of extremely low birth weight infants enrolled in pilot study of hydrocortisone therapy for prevention of CLD.

Synthetic pathway for cortisol. As shown, 17-OH pregnenolone is also an intermediary in the synthetic pathway for dehydroepiandrosterone (DHEA). HSD = hydroxysteroid dehydrogenase (22).
METHODS
Patient population and procedures.
The infants studied were extremely low birth weight infants (500–999 g birth weight) who were enrolled in a prospective, randomized, double-blind, placebo-controlled pilot study of hydrocortisone to prevent CLD. This study was approved by the institutional review boards and conducted at two centers: the Hershey Medical Center of The Pennsylvania State University (June 1996 through May 1998) and Pennsylvania Hospital of the University of Pennsylvania (June 1997 through May 1998). Infants were eligible if they were appropriate for gestational age, between 12 and 48 h old at study entry, and mechanically ventilated. Exclusion criteria included maternal diabetes, congenital sepsis, and small for gestational age infants.
Patients were enrolled after parental consent was obtained. Patients were randomized to receive either placebo or hydrocortisone (Solu-Cortef plain, Pharmacia & Upjohn, Kalamazoo, MI, U.S.A.) at a dose of 1.0 mg/kg/d divided every 12 h (∼8–10 mg/m2/d) for 9 d, followed by 0.5 mg/kg/d for 3 d. All other care was provided at the direction of the attending physician. CLD was defined as a continuing requirement for supplemental oxygen at 36 wk postmenstrual age to maintain an arterial oxygen saturation >90%, measured by pulse oximeter.
Blood specimens were obtained once before study, at 12–48 h of age, and once on d 5–7 of life (day of birth = d 0). Three days after the therapy had been completed (d 15–19), response to ACTH challenge was evaluated. After a baseline specimen was obtained, 3.5 μg/kg i.v. corticotropin (Cortrosyn, Organon Inc., West Orange, NJ, U.S.A.) was administered. Because infants do not display circadian rhythms of cortisol secretion at this age, no specific time of day was mandated for sample collection (22). After 30 min, another blood specimen was obtained. No infant was tested while receiving open-label corticosteroid (dexamethasone) for treatment of continuing lung disease. ACTH testing was deferred until at least 3 d after such therapy was stopped.
A corticotropin dose of 3.5μg/kg was chosen for the ACTH stimulation test to compare these results with previous work investigating the relationship of cortisol response to ACTH during the first week of life to the subsequent development of bronchopulmonary dysplasia (11). Although lower doses of corticotropin have recently been used in infants (6, 12, 23), in this study we wanted to compare maximal responses between these groups. Similarly, the sampling time of 30 min after ACTH administration was chosen for consistency with previous work. This time point has recently been shown to be appropriate in premature infants, using a standard ACTH dose of 250 μg/1.73 m2(23).
Laboratory procedures.
Blood specimens were collected in tubes containing EDTA; plasma was separated and frozen at −70°C until analysis. All analyses were performed in duplicate, by RIA. Cortisol was measured using an assay that had cross-reactivity with 11-deoxycortisol of 6.3% and with other naturally occurring steroids of <2% (GammaCoat, INCSTAR, Stillwater, MN, U.S.A.). Inter- and intra-assay coefficients of variation (CV) were 9.2% and 7.0%, respectively.
DHEAS, 11-deoxycortisol, 17-OH progesterone, and 17-OH pregnenolone were also measured by RIA (ImmuChem, ICN Biomedicals, Costa Mesa, CA, U.S.A.). Inter- and intra-assay CV were, respectively, 6.1% and 8.8% for DHEAS, 12.6% and 4.1% for 11-deoxycortisol, 13.3% and 9.3% for 17-OH progesterone, and 9.3% and 7.1% for 17-OH-pregnenolone. Samples obtained on d 5–7 of life were also analyzed for ACTH using RIA (ImmuChem®). For this analysis, the interassay CV was 10.7% and the intra-assay CV was 6.0%.
For DHEAS, cross-reactivity with steroids other than DHEA, androsterone, and androstenedione was <1%. For 11-deoxycortisol and 17-OH progesterone, cross-reactivities for all other reported steroids were <3%. For 17-OH pregnenolone, cross-reactivity with 17-OH progesterone was 32%; for all other steroids reported, cross-reactivity was <1%.
Cortisol precursors were measured after toluene extraction as modified from Saedi et al.(3). Briefly, 100 μL of standard, control, and sample serum were added to 1 mL of toluene in polypropylene tubes, shaken for 2 min, and allowed to stand for 10 min. One hundred microliters of organic phase was transferred into glass tubes, dried under nitrogen, and analyzed.
Statistical analysis.
Population differences were analyzed with t teSt. Because hormone values were generally not normally distributed, these data were analyzed after log transformation; however, data are presented in the text as geometric mean, with 25th –75th percentile values, for ease of reading. Cortisol and precursors are presented as nanomoles per liter. To convert cortisol to micrograms per deciliter, divide by 27.6. To convert precursors to nanograms per milliliter, divide 11-deoxycortisol values by 2.86 and divide 17-OH progesterone and 17-OH pregnenolone by 3.03. DHEAS is presented as micromoles per liter; to convert to micrograms per milliliter, divide by 2.56.
Analysis was performed using multiple regression analysis (NCSS 2000, Numbercruncher Statistical Systems, Salt Lake City, UT, U.S.A.), with the individual hormone concentration as the dependent variable. Initial independent factors included study center, maternal ethnic group, exposure to prenatal steroids, gestational age, chorioamnionitis, and CLD, with stepwise removal of nonsignificant factors.
Before these analyses, the effect of prenatal glucocorticoid exposure on hormone concentrations was evaluated. Because the majority of infants (85%) received prenatal betamethasone, only three samples were available from nonexposed infants for baseline and d 5–7 analyses, and two samples for poststudy analyses. These data were not apparently different from those from infants who received glucocorticoids, and were included in the final analyses. Similarly, the time interval between maternal betamethasone administration and delivery did not appear to affect steroid values. Inclusion of data from infants not exposed to prenatal betamethasone did not change the p value of any analysis in relationship to the designated significance value of 0.05.
RESULTS
Population characteristics of the patients enrolled in this study are shown in Table 1. Forty patients were enrolled in the study; 34 survived to 36 wk postconceptional age. Because the outcome variable for this study was CLD, only those infants who survived to 36 wk postmenstrual age were included in the analyses. Patients developing CLD were smaller than those who recovered, as might be expected. Gestational ages were similar, possibly because of the narrow gestational age range of patients enrolled. The clinical outcomes and effects of hydrocortisone supplementation have been reported elsewhere (24).
Hydrocortisone therapy.
Infants receiving hydrocortisone therapy had significantly higher cortisol concentrations while receiving therapy (d 5–7 sample: geometric mean [25th –75th percentile] = 617 [338–1163] nmol/L, n = 16, versus 190 [119–341] nmol/L for placebo-treated infants, n = 17;p < 0.001). In the infants receiving hydrocortisone, there was a strong correlation between the time interval after the hydrocortisone dose and the serum cortisol concentration, as would be expected (F = 41.68, p < 0.001). However, comparing only those values obtained >6 h after a dose was given, hydrocortisone-treated infants still had higher values than placebo-treated infants (n = 8, 491 [319–737] nmol/L;F = 166.5, p < 0.001 versus placebo group). With this exception, there were no other significant differences in hormone concentrations between infants treated with hydrocortisone and those receiving placebo, even while infants were receiving hydrocortisone therapy. Of particular interest, there was no suppression of baseline or stimulated cortisol concentrations when tested, ≥72 h after cessation of therapy. [Note: Because of the clinical significance of this information, these two cortisol data points were also reported with the clinical outcomes of the study infants (24).]
ACTH concentrations.
ACTH values were measured on d 5–7, while patients were receiving hydrocortisone or placebo. Values were similar between hydrocortisone-treated and placebo infants (geometric mean [25% to 75%] = 10.3 [9.2–11.3]versus 10.3 [8.3–13.4] pmol/L). Values were also similar between infants developing CLD (10.0 [8.3–12.1]) and those recovering without CLD (10.5 [8.4–12.4]).
Postnatal dexamethasone therapy.
Nine infants received dexamethasone before their final blood samples and ACTH testing. All were tested >72 h after cessation of dexamethasone therapy, and no infant had received >3 d of dexamethasone therapy before testing. Although this group of infants had lower baseline and stimulated cortisol concentrations than infants not exposed to dexamethasone, this difference was entirely accounted for by the effect of the factor CLD. Within the patient population developing CLD, basal cortisol values were similar between infants exposed to dexamethasone (n = 5; 206 [164–270] nmol/L) and those infants not exposed (n = 7; 194 [149–246] nmol/L). Stimulated cortisol values were 433 [279–573] nmol/L for infants exposed to dexamethasone versus 411 [365–456] nmol/L for infants not treated with dexamethasone. Similarly, there were no significant differences in other hormone values between infants treated with dexamethasone before ACTH testing versus those not treated, when the factor CLD was included in the analyses. (Two infants could not be tested because of continuing dexamethasone therapy, and one patient was missed.)
CLD.
At all time points, DHEAS concentrations were similar between those infants developing CLD and those recovering without CLD. At study entry, values were 22.4 (14.9–33.9) μmol/L; on d 15–19 of life, concentrations were 4.8 (2.8–7.4) μmol/L before ACTH stimulation and 6.3 (3.7–10.5) μmol/L after stimulation.
Cortisol and precursor hormone values before study entry and after completion of study are shown in Figure 2, A–F, divided into those infants developing CLD and those recovering without CLD. Cortisol values were similar between groups at study entry. Basal cortisol concentrations at study entry were significantly higher than values obtained after completion of study, by paired t test (519 [346–861] nmol/L versus 262 [175–366] nmol/L, n = 29;p < 0.003). At study entry, 17-OH progesterone concentrations were significantly higher in infants who subsequently developed CLD, as was the ratio of 17-OH progesterone to 11-deoxycortisol (Fig. 2). Gestational age was not a significant factor in these regressions.

Concentrations of (A) cortisol, (B) 11-deoxycortisol, (C) 17-OH progesterone, (D) 17-OH pregnenolone, (E) the ratio of 11-deoxycortisol (x1000)/cortisol, and (F) the ratio of 17-OH progesterone/11-deoxycortisol. For ease of reading, values are presented as nanomoles per liter (mean ± SEM); however, data were analyzed after log transformation. Data are grouped into patients developing CLD (closed squares) and those recovering without CLD (open squares); *denote significant differences between those groups (p < 0.05). Data are shown before study entry (between 12 and 48 h of life; CLD, n = 13; no CLD, n = 18), and 3 d after cessation of therapy with hydrocortisone or placebo (between d 15 and 19 of life; CLD, n = 12; no CLD, n = 19). Gestational age was initially included in all analyses; however, it was a significant factor only in the analysis for stimulated cortisol value, and was therefore removed from the other analyses. Data from patients treated with hydrocortisone and placebo are grouped together because values were not significantly different between the groups. Significant differences were seen as follows: (A) Cortisol: pre-ACTH, F = 6.60, p = 0.02; post-ACTH, F = 8.22, p < 0.01; (C) 17-OH progesterone: at study entry, F = 6.00, p = 0.02; post-ACTH, F = 5.99, p = 0.02; (E) 11-deoxycortisol (×1000)/cortisol: pre-ACTH, F = 14.83, p < 0.001; post-ACTH, F = 13.77, p < 0.001; (F) 17-OH progesterone/11-deoxycortisol: at study entry, F = 4.81, p < 0.04; post-ACTH, F = 8.05, p < 0.01.
After completion of the study (d 15–19 of life), basal and stimulated cortisol concentrations were significantly lower in infants who subsequently developed CLD compared with infants who recovered without CLD (Fig. 2A). In contrast, stimulated concentrations of 17-OH progesterone were higher in infants who developed CLD, as were precursor to product ratios, described in the legend for Figure 2. Gestational age was a marginally significant factor only in the analysis of stimulated cortisol values (p = 0.07).
Concentrations of 17-OH pregnenolone were not significantly different between the two groups at any time point tested. Because of the high cross-reactivity of this assay with 17-OH progesterone, the ratio of these two hormones was not evaluated. The apparent concentrations of 17-OH pregnenolone were undoubtedly affected by this high cross-reactivity; however, because concentrations of 17-OH progesterone were higher in infants developing CLD, the cross-reactivity should have amplified, rather than obscured, differences between the groups.
DISCUSSION
In this study, we demonstrated that small premature infants who develop CLD have elevated concentrations of cortisol precursors compared with infants who recover without CLD. Infants developing CLD also had lower cortisol concentrations and a lower response to ACTH in the third week of life compared with infants who recovered, a finding consistent with previous reports during the first week of life (6, 11–13). These findings are compatible with immaturity of the adrenal axis in these infants, who have immaturity of most organ systems. The fact that these infants were also enrolled in a randomized, placebo-controlled interventional trial of low-dose hydrocortisone therapy to prevent CLD, in which hydrocortisone-treated infants showed significantly improved respiratory outcomes (24), supports the hypothesis that cortisol deficiency is one etiologic factor in the pathogenesis of CLD.
It may be difficult to define a “normal” response to ACTH stimulation in extremely premature infants, a population that is by definition abnormal. However, in a recent report of adrenal function in infants <30 wk gestation (6), the authors found that stimulated cortisol values in relatively well infants (nonventilated infants and infants with low illness severity scores) exceeded the cut-off values generally used to define adrenal insufficiency in adults (>500–550 nmol/L, or >18–20 μg/dL) (20). In our study, 74% of infants who recovered without CLD but only 25% of those developing CLD achieved stimulated cortisol values >500 nmol/L (p < 0.01). These data suggest that previously established normal values for other populations might also be applicable to this population. Further study of ACTH dose, timing of blood sampling, and outcomes are required to determine whether these values can be applied to extremely premature infants, and whether the high prevalence of lower values in this population reflects a high incidence of adrenal insufficiency.
We found no difference in ACTH concentrations between infants receiving hydrocortisone and those receiving placebo, nor did we see a difference between infants developing CLD and those recovering. This could represent immature pituitary regulation, with autonomous ACTH secretion neither increasing with stress nor decreasing in response to negative feedback from exogenous hydrocortisone. However, previous studies have shown that ACTH concentrations increase in response to exogenous CRH in extremely low birth weight infants (25, 26). The values we obtained in our study were similar to the CRH-stimulated ACTH values reported in those studies (25, 26). These higher values could simply result from different laboratory assays; however, they might also indicate continuing pituitary stimulation, unsuppressed by the relatively low dose of hydrocortisone (HC). Deciding between these possibilities would require further study of both basal and stimulated ACTH values in this population.
The pattern of steroid concentrations seen in the present study was consistent with decreased activity of both the 21- and the 11β-hydroxylase enzymes. Concentrations of 17-OH pregnenolone did not differ significantly between the two groups. This is consistent with a previous report showing no increase in the ratio of 17-OH pregnenolone to 17-OH progesterone in sick preterm infants, in which the authors suggested no reduction of 3β-HSD activity (2). However, it would be surprising to find much activity of this enzyme early in life in extremely premature infants. Previous evaluation of the ontogeny of 3β-HSD in the human fetal adrenal gland has shown very little immunoreactive 3β-HSD at this time in gestation (27). An alternative explanation may be that 17-OH pregnenolone is also an intermediate compound in the synthetic pathway for DHEA and DHEAS (Fig. 1). This pathway is active in the fetal zone of the adrenal gland (22), and may account for the lack of accumulation of 17-OH pregnenolone.
The accumulation of cortisol precursors, and the increased ratios of precursor to product (Fig. 2, E and F), additionally support the hypothesis that these infants have a decreased capacity to synthesize cortisol, resulting in a relative adrenal insufficiency in the face of acute stress or illness. Previous investigators have documented higher concentrations of cortisol precursors in premature infants compared with term infants, and have described even higher precursor concentrations in sick premature infants compared with well babies (1, 2, 6). One previous abstract has linked an elevated ratio of 11-deoxycortisol to cortisol with subsequent development of bronchopulmonary dysplasia (28), and a new report documents elevated 17-OH progesterone concentrations in such infants (6).
A number of case reports and small studies have suggested that some premature infants with low cortisol concentrations show clinical signs of adrenal insufficiency responsive to glucocorticoid therapy (7–10). Other investigators have found very low cortisol concentrations in otherwise well premature infants, and have suggested that such concentrations are not necessarily harmful, and do not require treatment (3). Both of these scenarios may well be true; that is, low concentrations of cortisol may be adequate for a well infant but insufficient for an infant experiencing significant illness. This would be consistent with the concept of relative adrenal insufficiency, which has been described in adults (20).
The existence of a relative adrenal insufficiency has been suggested in adult populations in the setting of acute stress. As reviewed by Lamberts et al.(20), cortisol concentrations regarded as normal in well subjects may be inappropriately low for patients who are acutely ill. In such patients, a poor response to ACTH may predict increased mortality (20). These authors also review the experience reported by the University Hospital of Glasgow, Scotland, where the inadvertent introduction of a drug that inhibited 11β-hydroxylase was associated with decreased cortisol concentrations and increased 11-deoxycortisol concentrations, as well as increased mortality among patients with multiple injuries (20). Thus, in other populations, relative adrenal insufficiency has been postulated to play a part in adverse outcome.
The actions of cortisol are so numerous that a deficiency of endogenous glucocorticoid could affect many aspects of lung development and lung injury, including growth and differentiation (29), surfactant production (29, 30), microvascular integrity (31), and development of antioxidant enzymes (30). Of particular interest in this population is the relationship of cortisol to inflammation, because the development of CLD has been convincingly linked to increased lung inflammation in the first days of life (17, 18). Cortisol is central to the ability of the body to dampen the response to inflammatory stimuli; in its absence, inflammatory responses are amplified (14) and repair processes disordered (32). We have previously documented an inverse correlation between cortisol concentrations and markers of inflammation and protein leak in the lungs of mechanically ventilated preterm infants (19).
This study supports previous reports that many small premature infants show clinical and biochemical evidence of relative adrenal insufficiency early in life (5–13). The effect of low-dose hydrocortisone therapy seen in this pilot study—reduction of CLD without apparent suppression of endogenous cortisol production after therapy (24) —provides an encouraging basis for further studies of glucocorticoid replacement therapy for relative adrenal insufficiency of prematurity in these extremely immature infants.
Abbreviations
- CLD:
-
chronic lung disease
- DHEAS:
-
dehydroepiandrosterone sulfate
- 3β-HSD:
-
3β-hydroxysteroid dehydrogenase
- CRH:
-
corticotropin releasing hormone
References
Lee MM, Rajagopalan L, Berg GJ, Moshang T 1989 Serum adrenal steroid concentrations in premature infants. J Clin Endocrinol Metab 69: 1133–1136
Hingre RV, Gross SJ, Hingre KS, Mayes DM, Richman RA 1994 Adrenal steroidogenesis in very low birth weight preterm infants. J Clin Endocrinol Metab 78: 266–270
Saedi SA, Dean H, Dent W, Cronin C 1995 Reference ranges for serum cortisol and 17-hydroxyprogesterone levels in preterm infants. J Pediatr 126: 985–987
Allen DB, Hoffman GL, Fitzpatrick P, Laessig R, Maby S, Slyper A 1997 Improved precision of newborn screening for congenital adrenal hyperplasia using weight-adjusted criteria for 17-hydroxyprogesterone levels. J Pediatr 130: 128–133
Scott SM, Watterberg KL 1995 Effect of gestational age, postnatal age, and illness on plasma cortisol concentrations in premature infants. Pediatr Res 37: 112–116
Huysman MWA, Hokken-Koelega ACS, DeRidder MAJ, Sauer PJJ 2000 Adrenal function in sick very preterm infants. Pediatr Res 48: 629–633
Ward RM, Kimura RE, Rich-Denson C 1991 Addisonian crisis in extremely premature neonates. Clin Res 39: 11A( abstr)
Colasurdo MA, Hanna CE, Gilhooly JT, Reynolds JW 1989 Hydrocortisone replacement in extremely premature infants with cortisol insufficiency. Clin Res 37: 180A( abstr)
Helbock HJ, Insoft RM, Conte FA 1993 Glucocorticoid-responsive hypotension in extremely low birth weight newborns. Pediatrics 92: 715–717
Bourchier D, Weston PJ 1997 Randomised trial of dopamine compared with hydrocortisone for the treatment of hypotensive very low birth weight infants. Arch Dis Child 76: F174–F178
Watterberg KL, Scott SM 1995 Evidence of early adrenal insufficiency in babies who develop bronchopulmonary dysplasia. Pediatrics 95: 120–125
Korte C, Styne D, Merritt TA, Mayes D, Wertz A, Helbock HJ 1996 Adrenocortical function in the very low birth weight infant: improved testing sensitivity and association with neonatal outcome. J Pediatr 128: 257–263
Banks BA, Merrill JD, Cnaan A, Stouffer N, Ballard RA, Ballard PL, North American TRH Trial Collaborators 1999 Weak association between plasma cortisol and chronic lung disease in preterm infants. Pediatr Res 45: 183A( abstr)
Goujon E, Parnet P, Laye S, Combe C, Dantzer R 1996 Adrenalectomy enhances pro-inflammatory cytokines gene expression, in the spleen, pituitary and brain of mice in response to lipopolysaccharide. Brain Res Mol Brain Res 36: 53–62
Farsky SP, Sannomiya P, Garcia-Leme J 1995 Secreted glucocorticoids regulate leukocyte-endothelial interactions in inflammation. A direct vital microscopic study. J Leukoc Biol 57: 379–386
Chrousos GP 1995 The hypothalamic-pituitary adrenal axis and immune-mediated inflammation. N Engl J Med 332: 1351–1362
Pierce MR, Bancalari E 1995 The role of inflammation in the pathogenesis of bronchopulmonary dysplasia. Pediatr Pulmonol 19: 371–378
Ozdemir A, Brown MA, Morgan WJ 1997 Markers and mediators of inflammation in neonatal lung disease. Pediatr Pulmonol 23: 292–306
Watterberg KL, Scott SM, Backstrom C, Gifford KL, Cook KL 2000 Links between early adrenal function and respiratory outcome in preterm infants: airway inflammation and patient ductus arteriosus. Pediatrics 105: 320–324
Lamberts SWJ, Bruining HA, de Jong FH 1997 Corticosteroid therapy in severe illness. N Engl J Med 337: 1285–1292
Pepe GJ, Albrecht ED 1995 Actions of placental and fetal adrenal steroid hormones in primate pregnancy. Endocr Rev 16: 608–648
Winter JSD 1992 Fetal and neonatal adrenocortical physiology. In: Polin RA, Fox WW (eds) Fetal and Neonatal Physiology. W.B. Saunders, Philadelphia, pp 1829–1841
Karlsson R, Kallio J, Toppari J, Kero P 1999 Timing of peak serum cortisol values in preterm infants in low-dose and the standard ACTH tests. Pediatr Res 45: 367–369
Watterberg KL, Gerdes JS, Gifford KL, Lin H-M 1999 Prophylaxis against adrenal insufficiency to prevent chronic lung disease in premature infants. Pediatrics 104: 1258–1263
Ng PC, Wong GWK, Lam CWK, Lee CH, Wong MY, Fok TF, Wong W, Chan DC 1997 Pituitary-adrenal response in preterm very low birth weight infants after treatment with antenatal corticosteroids. J Clin Endocrinol Metab 82: 3548–3552
Hanna CE, Keith LD, Colasurdo MA, Buffkin DC, Laird MR, Mandel SH, Cook DM, LaFranchi SH, Reynolds JW 1993 Hypothalamic pituitary adrenal function in the extremely low birth weight infant. J Clin Endocrinol Metab 76: 384–387
Parker CR, Faye-Peterson O, Stankovic AK, Mason JI, Grizzle WE 1995 Immunohistochemical evaluation of the cellular localization and ontogeny of 3 beta-hydroxysteroid dehydrogenase/delta 5–4 isomerase in the human fetal adrenal gland. Endocr Res 21: 69–80
Guttentag S, Rubin LP, Richardson D, et al 1991 The glucocorticoid pathway in ill and well extremely low birth weight infants. Pediatr Res 29: 77A( abstr)
Gross I 1990 Regulation of fetal lung maturation. Am J Physiol 259: L337–L344
Frank L, Lewis PL, Sosenko IRS 1985 Dexamethasone stimulation of fetal rat lung antioxidant enzyme activity in parallel with surfactant stimulation. Pediatrics 75: 569–574
Williams TJ, Yarwood H 1990 Effect of glucocorticosteroids on microvascular permeability. Am Rev Respir Dis 141: S39–S43
Rannels DE, Stockstill B, Mercer RR, Crapo JD 1991 Cellular changes in the lungs of adrenalectomized rats following left pneumonectomy. Am J Respir Cell Mol Biol 5: 351–362
Acknowledgements
The authors thank Drs. C. Richard Parker Jr. and John D. Johnson for their valuable critical reviews of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported, in part, by grant MCJ-420633 from the Maternal and Child Health Bureau (Title V, Social Security Act), Health Resources and Services Administration, Department of Health and Human Services.
Rights and permissions
About this article
Cite this article
Watterberg, K., Gerdes, J. & Cook, K. Impaired Glucocorticoid Synthesis in Premature Infants Developing Chronic Lung Disease. Pediatr Res 50, 190–195 (2001). https://doi.org/10.1203/00006450-200108000-00005
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200108000-00005
This article is cited by
-
Glucocorticoid signature of preterm infants developing bronchopulmonary dysplasia
Pediatric Research (2023)
-
Impact of postnatal steroids on peripheral avascular retina and severity of retinopathy of prematurity
Pediatric Research (2023)
-
Hydrocortisone and bronchopulmonary dysplasia: variables associated with response in premature infants
Journal of Perinatology (2020)
-
Assessment of adrenal function at birth using adrenal glucocorticoid precursor to product ratios to predict short-term neonatal outcomes
Pediatric Research (2020)
-
Medical morbidities and DNA methylation of NR3C1 in preterm infants
Pediatric Research (2017)
