
Abstract
Intrauterine growth retardation (IUGR) has been linked to the development of type 2 diabetes in later life. The mechanisms underlying this phenomenon are unknown. Recent data suggest that some of the molecular defects underlying type 2 diabetes reside in the CNS. The enzyme carnitine palmitoyltransferase-1 (CPT1) regulates long-chain fatty acid (LCFA) entry into mitochondria, where LCFA undergo β-oxidation. Hypothalamic inhibition of CPT1 decreases food intake and suppresses endogenous glucose production. Our aim was to investigate the effects of uterine artery ligation, a procedure that mimics uteroplacental insufficiency, on the CNS expression of CPT1 and other key enzymes of LCFA metabolism. Bilateral uterine artery ligation was performed on d 19 of gestation in the pregnant rat; sham-operated pregnant rats served as controls. Hypothalamus, cerebellum, hippocampus, and cortex were dissected and analyzed at birth by real-time PCR. Nonesterified fatty acid (NEFA) serum levels were significantly higher in IUGR pups (p < 0.0001). In IUGR rats, the hypothalamic expression of CPT1 isoform C (p = 0.005) and acetyl-CoA carboxylase (ACC) isoforms alpha (p < 0.05) and beta (p = 0.005) were significantly decreased. The data presented here support the hypothesis that an abnormal intrauterine milieu can induce changes in hypothalamic lipid sensing.
Similar content being viewed by others


Main
Epidemiologic studies in man have shown that impaired intrauterine growth is associated with an increased incidence of insulin resistance, type 2 diabetes, and cardiovascular disease in the adult (1–5). These observations have led to the hypothesis that adult disease arises in utero, in part, as a result of changes in the development of key endocrine and metabolic pathways during suboptimal intrauterine conditions associated with impaired fetal growth. This hypothesis has been tested experimentally in a number of species, using a range of techniques to impair fetal growth. Inducing IUGR by placental insufficiency or by undernutrition, stress, or hormone treatment of the mother leads to endocrine and metabolic alterations in the adult offspring in several species (6). These findings have led to the “fetal origins” hypothesis, which suggests that an adverse intrauterine environment would program or imprint the development of fetal tissues, permanently determining responses producing later dysfunction and disease (7).
The hypothalamus is emerging as a critical site for the integration of nutritional, endocrine, and neural cues signaling the body's metabolic and nutritional status. Neurons in the hypothalamus (particularly in the arcuate nucleus) are primary targets of a number of key hormones and metabolic signals. Hypothalamic neurons, in turn, target several downstream sites to influence the coordinated autonomic, behavioral, and endocrine responses. These signals should normally activate a negative feedback loop between the availability of nutrients and their intake and metabolism (8–10). A major breakthrough in the understanding of mechanisms leading to type 2 diabetes has recently come from studies on the CNS-dependent regulation of glucose and fat metabolism, showing that some of the molecular defects underlying type 2 diabetes may reside in the CNS, supporting the concept that type 2 diabetes is, at least in part, a hypothalamic disorder (10–12).
Several lines of evidence indicate that lipid metabolism in neurons plays a pivotal role in mediating the hypothalamic responses to fuel availability. Consistent with this, intracerebroventricular (ICV) administration of FAS inhibitors reduces food intake, neuropeptide Y (NPY) expression, and blood glucose levels (13). The enzyme CPT1 regulates the entry of LCFA into mitochondria, the site where LCFA undergo β-oxidation. Hypothalamic inhibition of CPT1 decreases food intake and suppresses endogenous glucose production (14) via a neural pathway, which requires the activation of KATP channels localized in the arcuate nucleus and efferent vagal fibers innervating the liver (15,16). These findings indicate that changes in the rate of lipid oxidation in selective hypothalamic neurons signal nutrient availability to the hypothalamus, which in turn modulates the exogenous and endogenous flux of nutrients into the circulation (17).
Simmons and co-workers (18,19) developed an animal model of IUGR induced by bilateral uterine artery ligation. Blood flow to the fetus is not ablated but is reduced to a similar degree to that observed in human pregnancies complicated by uteroplacental insufficiency. In this model, the resulting uteroplacental insufficiency leads to growth retarded fetuses with a metabolic profile very similar to that of IUGR human fetuses (18–20). These animals exhibit mild peripheral insulin resistance and β-cell secretory defects very early in life but have adequate compensatory insulin secretion for several weeks. However, eventually, β-cell compensation fails, and overt diabetes occurs.
We asked whether a change in hypothalamic lipid sensing is associated with IUGR and may account for the risk of developing insulin resistance and type 2 diabetes. To test this hypothesis, we examined the impact of uteroplacental insufficiency, obtained in Sprague-Dawley rats by bilateral uterine artery ligation, on the hypothalamic expression of CPT1 and other key enzymes of lipid metabolism.
METHODS
Animal model.
Time-dated Sprague-Dawley pregnant rats (Harlan Laboratories, Madison, WI) were individually housed under standard conditions and allowed free access to standard chow and water. On d 19 of gestation (term is 22 d), the maternal rats were anesthetized with intramuscular injections of xylazine (Sigma Chemical Co., St. Louis, MO), 8 mg/kg, and ketamine (Sigma Chemical Co.), 40 mg/kg, and the abdomen was opened along the midline. Suture was placed around both uterine arteries, then either tied or withdrawn before closing the abdomen (18,19). Dams recovered quickly from uterine artery ligation and sham procedures and resumed feeding the same day. After recovery, rats had ad libitum access to food and water. The pregnant rats were allowed to deliver spontaneously, and d 0 pups were weighed and killed by cervical dislocation. Tissues of interest were immediately harvested and frozen in liquid nitrogen and stored at –80°C. All procedures complied with Italian regulations for laboratory animal care, according to the guidelines and under supervision of the Animal Technology Station, Interdepartmental Service Center, Tor Vergata University, Rome, Italy.
RNA isolation and cDNA synthesis.
Total RNA was extracted using TriPure (Roche Diagnostics, Indianapolis, IN) according to the manufacturer's instructions and quantified in duplicate using UV absorbance at 260 nm. Gel electrophoresis confirmed the integrity of the samples. One microgram RNA, pretreated with RNase-free DNase (Invitrogen, Carlsbad, CA) was transcribed into the cDNA using the High-Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA) in a final volume of 50 μL following the manufacturer's protocol. To minimize variation in the reverse transcription reaction, all RNA samples from a single experimental setup were reverse transcribed simultaneously.
Real-time RT-PCR.
We explored the expression of key enzymes that regulate fatty acid metabolism in the hypothalamus (Fig. 1). Real-time RT-PCR was performed on an ABI PRISM 7000 Sequencer Detector (Applied Biosystems). PCR primers and TaqMan probes were used to amplify and detect CPT1A and CPT1B. ACCα, ACCβ, ACS, AMPK, FAS, malonyl-CoA decarboxylase (MCD), and the housekeeping gene 18S were commercially available (Assay-on-Demand Gene Expression Product; Applied Biosystems). CPT1C primers and TaqMan probe were designed using Primer Express Software (Applied Biosystems). Target probes were labeled with fluorescent dye FAM. Forward primer was 5′-ACGGCTTCCCGGAACAG, reverse primer was 5′-TGTGAGAGCCATGAAAACAAG, and probe was (5′-3′) AGGACAGACCAACAGTG (GenBank accession number XM_218625).

Model of neuronal integration of metabolic signals involved in regulation of glucose production. The hypothalamic lipid sensing integrates multiple metabolic homeostatic signals. Both glucose and FFA can influence the intracellular levels of LCFA-CoAs. Cellular oxidation of LCFA-CoAs is regulated by the levels of malonyl-CoA, a potent inhibitor of CPT1 activity. Glucose increases the levels of malonyl-CoA through glycolysis and increases LCFA-CoAs by inhibiting their transport into the mitochondria by CPT1. Circulating FFA are converted to LCFA-CoAs by the enzyme ACS. The enzyme malonyl-CoA decarboxylase lowers the levels of malonyl-CoA and prevents the accumulation of LCFA-CoAs by derepressing CPT1 activity and increasing LCFA-CoAs oxidation in the mitochondria. The genes tested in the study are underlined.
Before performing real-time PCR, primers and probe concentration were determined to demonstrate their specificity and optimal reaction condition. 18S was used as an internal control for differences in cDNA loading. Before the use of 18S as a control, parallel serial dilutions of cDNA were quantified to prove the validity of using 18S as an internal control.
The real-time RT-PCR amplification was performed in tissues from 15 sham-operated and 15 IUGR rats. Experiments were performed in triplicate using 96-well tray and optical adhesive covers (Applied Biosystems) in a final reaction mixture of 20 μL containing 5 μL of undiluted cDNA. Real-time PCR was performed using Platinum Quantitative PCR SuperMix-UDG with ROX (Invitrogen). The cycling consisted of 2 min at 50°C, 2 min at 95°C followed by 45 cycles of 95°C for 15 s and 60°C for 45 s. Gene expression was presented using the 2–ΔΔCt method, first described by Livak [Livak K 1997 Relative quantitation of gene expression: ABI PRISM® 7700 Sequence Detection System. Applied Biosystems User Bulletin #2 (part no. 4303859)].
Determination of reaction efficiency was routinely used as an internal quality control for adequate assay performance. Crossing of threshold (Ct) values obtained for the target gene were normalized against each individual 18S value, which was run in the same well of the real-time RT-PCR run. Relative quantification of PCR products were then based on value differences between the target and 18S control using the comparative Ct method. The amount of target, normalized to an endogenous reference and relative to a calibrator, is given by the formula 2–ΔΔCT (fold change).
Plasma assays.
At birth, 30 sham-operated and 30 IUGR pups were decapitated, blood was collected and centrifuged at 1900 × g at 4°C for 10 min, and plasma was stored at –80°C. Glucose, insulin, and NEFA concentrations were measured.
Glucose was determined using a colorimetric commercial kit (Sigma Chemical Co.). Plasma insulin concentrations were measured in duplicate by a rat/mouse insulin ELISA kit, using rat insulin as the standard (Linco Research, St. Charles, MO) according to the manufacturer's instructions. The intraassay CV was 1.2–8.4%, the interassay CV was 6.0–17.9%, and the sensitivity limit was 0.2 ng/mL.
Quantification of NEFA was determined using a commercially available NEFA C test kit (Wako Chemicals, Osaka, Japan) as instructed by the manufacturer (reading absorbance is 550 nm). The intraassay CV was 1.1–2.7%. The linear range of the assay is 0–57.1 mg/dL.
Statistical analysis.
Real-time data were analyzed using the Statistical Package for Social Sciences (SPSS version 12.0, SPSS Inc., Chicago, IL). The available data consisted of dCt levels measured in rats. The aim of the study was to estimate and compare means of groups representing two different treatments.
The structure of the data suggested the use of a linear mixed-effects model (also called variance component ANOVA), to correctly represent the hierarchical structure of data (treatment/no treatment → mother → offspring → replicate), where the last three levels were considered as variance components and not as parameters of interest. This model handles correlated data and unequal variances, very common in situations such as repeated measurements or hierarchically nested experimental units.
For this reason, we started by fitting a variance components model to the data to estimate the effect of the treatment on the dCt level and to partition the variance between that arising due to differences among mothers and that due to differences among rat offspring nested into mothers. Furthermore, there was another source of variance due to the repeated measurements on each observation. In a linear mixed-effects model, responses from a subject are thought to be the sum (linear) of so-called fixed and random effects. In our model, treatment was considered the fixed effect, since it affects the population mean; mother, offspring, and repeated measurements effects were treated as random effects because they contributed only to the covariance structure of the data. Correlations between cases was introduced by the random effects. Formally, the outcome for the k-th replicate (measurement) of the j-th offspring from the i-th mother was modeled in the following way:
Xijk = μ0 + ΔTijk + MI + Oij + ɛijk i = 1…s, j = 1…ni, k = 1…Rij
where μ0 is the mean of the control group; Tijk is an indicator (yes/no = 1/0) of treatment, Δ represents the differential effect on the mean due to the treatment, and Mi, Oij, and ɛijk are the random effects of mother, offspring, and replicate, respectively, all assumed to be normally distributed with mean 0 and respective variances σm 2, σo 2, and σr 2.
Five parameters were estimated: the three variances σm 2, σo 2, and σr 2 and the means μ0 and Δ. However, the main interest of this study was the estimate and the statistical significance of the parameter Δ. For plasma assays, statistical analysis was performed using unpaired two-tailed t test. Differences were considered statistically significant at p < 0.05.
RESULTS
Animal weights and metabolic profile.
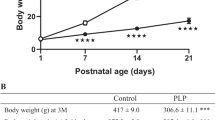
Birth weights of IUGR animals were significantly lower than those of controls (mean weight ± SD: 4.0 ± 0.57 versus 6.5 ± 0.32 g, p < 0.001). No significant differences were observed in blood glucose and insulin levels (Table 1). As one of the hallmarks of insulin resistance and type 2 diabetes is the increase of circulating FFA, we tested NEFA levels in sham and IUGR pups at d 0. NEFA levels were significantly higher in IUGR than sham animals (Table 1).
Expression of CPT1 isoforms in CNS.
Three CPT1 isoforms with various tissue distributions and encoded by distinct genes have been identified: liver (CPT1A) (21), muscle (CPT1B) (22), and brain (CPT1C) (23). Quantitative expression of CPT1A, CPT1B, and CPT1C was measured by quantitative RT-PCR in different CNS areas (hypothalamus, cortex, hippocampus, and cerebellum) of sham (n = 15) and IUGR rats (n = 15 from four different litters) at d 0. Statistical analysis showed that results were not affected by the different litters but only by the different treatments.
The expression of CPT1C resulted significantly lower (30% less) in the hypothalamus of IUGR pups, whereas was significantly increased in the hippocampus (Fig. 2). No change in CPT1C expression was found in the cortex and cerebellum. No significant difference was found in the expression of CPT1A and CPT1B in the different brain areas (Fig. 2).

Expression of CPT1 isoforms in different brain regions of sham-operated (n = 15) and IUGR rats (n = 15). CPT1A (white bars), CPT1B (shaded bars), and CPT1C (black bars). Transcripts were measured by real-time RT-PCR using appropriate primers and normalized to 18S mRNA. Data are expressed as fold change vs sham-operated group. *p = 0.01; **p < 0.05. Values are mean ± SEM.
Expression of ACCα, ACCβ, ACS, AMPK, FAS, and MCD in the hypothalamus.
LCFA act as nutrient abundance signals in the hypothalamus and function as signalling molecules informing the hypothalamus about the body's metabolic status (24). The expression of key enzymes that regulate hypothalamus lipid sensing was assessed (Fig. 1).
The expression of ACCα and ACCβ was significantly reduced (by 52% and 39%, respectively) in the hypothalamus of IUGR rats (Fig. 3). No significant variations in the hypothalamic expression of ACS, AMPK, FAS, and MCD were found in IUGR rats (Fig. 3). Although a tendency toward an increase of FAS expression and a reduction of AMPK expression was noted, these changes did not achieve statistical significance due to the high variability.

Expression of ACCα, ACCβ, MCD, FAS, AMPK, and ACS in hypothalamus of sham-operated (n = 15) and IUGR rats (n = 15). Transcripts were measured by real-time RT-PCR using appropriate primers and normalized to 18S mRNA. Data are expressed as fold change vs sham-operated group. *p < 0.05; **p = 0.005. Values are mean ± SEM.
DISCUSSION
The present study shows for the first time a down-regulation of the expression of hypothalamic CPT1C in an animal model of IUGR. CPT1 regulates LCFA entry into mitochondria, where it undergoes beta-oxidation. The most widely expressed form is the liver isoform, designated L-CPT1 or CPT1A. The isoform mostly expressed in muscle and adipose tissue is M-CPT1 or CPT1B. Finally, the most recently discovered form is the brain-specific isoform, named CPT1C. CPT1C protein sequence contains all the residues known to be important for both carnitine palmitoyltransferase activity and malonyl-CoA binding (23). Hypothalamic inhibition of CPT1 decreases food intake and suppresses endogenous glucose production (14), indicating that changes in the rate of lipid oxidation in selective hypothalamic neurons signal nutrient availability to the hypothalamus, which in turn modulates the exogenous and endogenous inputs of nutrients into the circulation.
The intracellular increase in LCFA induces a sensing mechanism in hypothalamic neurons, ultimately reducing liver glucose production (8). An increased rate of glucose production is a major determinant of fasting hyperglycemia in type 2 diabetes (25). Circulating levels of LCFA, which are often elevated in obese and diabetic individuals, have been suggested as an important causative link between obesity and type 2 diabetes (26–29). LCFA are potent stimulators of hepatic gluconeogenesis (30–34). Despite these effects on gluconeogenesis, changes in circulating LCFA levels often do not modify glucose production because of concomitant and opposite changes in the rate of glycogenolysis (35). The central action of circulating LCFA is required to counteract LCFA-induced stimulation of gluconeogenesis and to prevent an increase in glucose production, thereby providing an extrahepatic site for hepatic autoregulation.
The present finding of reduced CPT1C expression in the hypothalamus of IUGR animals is consistent with our previous observation of reduced blood glucose levels in children born small for gestational age (36). Reduced CPT1C activity might in fact lead to the accumulation of LCFA, which in turn inhibit food intake and endogenous glucose production. However, no alterations in blood glucose and insulin levels were observed in IUGR rats, as previously reported (20). The finding of normal expression of CPT1A and CPT1B isoforms in CNS suggests that these two isoforms may compensate for CPT1C decrease.
The finding of elevated NEFA concentrations in IUGR rats is consistent with a previous report showing significant elevations of NEFA in IUGR Sprague-Dawley rats before the onset of diabetes (37). FFA have been implicated in the pathogenesis of insulin resistance and type 2 diabetes (33), and the increase of NEFA levels in IUGR pups may be related to the risk of developing diabetes in adulthood.
The up-regulation of CPT1C expression in the hippocampus is a novel finding. Rat hippocampus contains high concentrations of NPY (38), a key regulator of feeding behavior. It is tempting to speculate that hippocampus may be another regulatory site of food intake and lipid metabolism, potentially affected by uteroplacental insufficiency.
The other major finding of the present study was the reduced expression of ACCα and ACCβ in the hypothalamus of IUGR rats. ACC catalyzes the formation of malonyl-CoA, an essential substrate for fatty acid synthesis in lipogenic tissues and a key regulatory molecule in muscle, brain, and other tissues (39). Cellular levels of malonyl-CoA repress CPT1 activity and decrease LCFA-CoAs oxidation. MCD converts malonyl-CoA to acetyl-CoA. Recent evidence underscores the role played by malonyl-CoA in the hypothalamic arcuate nucleus as a major modulator of intracellular LCFA-CoA levels (40,41). MCD overexpression in arcuate nucleus leads to reduced accumulation of LCFA-CoAs, increased food intake, and augmented endogenous glucose production (41). These results suggest that neuronal levels of malonyl-CoA act as neural sensors of fuel availability and regulators of energy balance and glucose homeostasis. The reduced expression of ACC in the hypothalamus of IUGR animals might lead to decreased formation of malonyl-CoA, eventually increasing LCFA utilization and, consequently, reducing LCFA cellular levels.
Overall, our findings are intriguing, showing reduced expression of two enzymes that physiologically lead to opposite final effects. Reduced CPT1C may in fact lead to the accumulation of LCFAs; on the contrary, decreased ACC may reduce intracellular LCFA. This observation suggests the existence of a negative feedback loop between the two enzymatic systems, finely controlling cellular concentrations of LCFA and ultimately regulating food intake and endogenous glucose production.
Our study was limited at birth and investigated gene expression only. Therefore, present data do not permit us to know whether the alterations in gene expression observed at birth are transient or permanent, or whether they compensate each other or in the long-term may determine hypothalamic dysregulation, eventually leading to insulin resistance and type 2 diabetes in adulthood, as previously described in the same animal model (20).
In conclusion, our data show that uteroplacental insufficiency which leads to IUGR affects the expression of hypothalamic lipid sensing genes. The detailed knowledge of central lipid sensing mechanisms may lead to the design of specific modulators potentially helpful in the prevention and therapy of metabolic alterations associated to IUGR.
Abbreviations
- ACC:
-
acetyl-CoA carboxylase
- ACS:
-
acyl-CoA synthase
- AMPK:
-
AMP-activated protein kinase
- CPT1:
-
carnitine palmitoyltransferase-1
- FAS:
-
fatty acid synthase
- LCFA:
-
long-chain fatty acid
- MCD:
-
malonyl-CoA decarboxylase
- NEFA:
-
nonesterified fatty acids
References
Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ 1989 Weight in infancy and death from ischaemic heart disease. Lancet 2: 577–580
Barker DJ, Osmond C, Golding J, Kuh D, Wadsworth ME 1989 Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 298: 564–567
Barker DJ 1995 Fetal origins of coronary disease. BMJ 311: 171–174
Eriksson JG, Forsen T, Tuomilehto J, Osmond C, Barker DJ 2001 Early growth and coronary heart disease in later life: a longitudinal study. BMJ 322: 949–953
Geremia C, Cianfarani S 2004 Insulin sensitivity in children born small for gestational age (SGA). Rev Diabetic Stud 1: 58–65
Fowden AL, Giussani DA, Forhead AJ 2005 Endocrine and metabolic programming during intrauterine development. Early Hum Dev 81: 723–734
Purdy LP, Metzger BE 1996 Influences of intrauterine metabolic environment on adult disease: what may we infer from size at birth?. Diabetologia 39: 1126–1130
Obici S, Feng Z, Morgan K, Stein D, Karkanias G, Rossetti L 2002 Central administration of oleic acid inhibits glucose production and food intake. Diabetes 51: 271–275
Obici S, Zhang BB, Karkanias G, Rossetti L 2002 Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med 8: 1376–1382
Puglianiello A, Cianfarani S 2006 Central control of glucose homeostasis. Rev Diabetic Stud 3: 54–60
Elmquist JK, Marcus JN 2003 Rethinking the central causes of diabetes. Nat Med 9: 645–647
Prodi E, Obici S 2006 The brain as a molecular target for diabetic therapy. Endocrinology 147: 2664–2669
Loftus TM, Jaworsky DE, Frehywot GL, Townsend CA, Ronnett GV, Lane MD, Kuhajda FP 2000 Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science 288: 2379–2381
Obici S, Feng Z, Arduini A, Conti R, Rossetti L 2003 Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nat Med 9: 756–761
Pocai A, Obici S, Schwartz GJ, Rossetti L 2005 A brain-liver circuit regulates glucose homeostasis. Cell Metab 1: 53–61
Lam TK, Pocai A, Gutierrez-Juarez R, Obici S, Bryan J, Aguilar-Bryan L, Schwartz GJ, Rossetti L 2005 Hypothalamic sensing of circulating fatty acids is required for glucose homeostasis. Nat Med 11: 320–327
Pocai A, Lam TK, Obici S, Gutierrez-Juarez R, Muse ED, Arduini A, Rossetti L 2006 Restoration of hypothalamic lipid sensing normalizes energy and glucose homeostasis in overfed rats. J Clin Invest 116: 1081–1091
Simmons RA, Gounis AS, Bangalore SA, Ogata ES 1992 Intrauterine growth retardation: fetal glucose transport is diminished in lung but spared in brain. Pediatr Res 31: 59–63
Unterman T, Lascon R, Gotway M, Oehler D, Gounis A, Simmons RA, Ogata ES 1990 Circulating levels of insulin-like growth factor binding protein-1 (IGFBP-1) and hepatic mRNA are increased in the small for gestational age fetal rat. Endocrinology 127: 2035–2037
Simmons RA, Templeton LJ, Gertz SJ 2001 Intrauterine growth retardation leads to the development of type 2 diabetes in rat. Diabetes 50: 2279–2286
Britton CH, Schulz RA, Zhang B, Esser V, Foster DW, McGarry JD 1995 Human liver mitochondrial carnitine palmitoyltransferase I: characterization of its cDNA and chromosomal localization and partial analysis of the gene. Proc Natl Acad Sci U S A 92: 1984–1988
Yamazaki N, Shinohara Y, Shima A, Yamanaka Y, Terada H 1996 Isolation and characterization of cDNA and genomic clones encoding human muscle type carnitine palmitoyltransferase I. Biochim Biophys Acta 1307: 157–161
Price N, van der Leij F, Jackson V, Corstorphine C, Thomson R, Sorensen A, Zammit V 2002 A novel brain-expressed protein related to carnitine palmitoyltransferase I. Genomics 80: 433–442
Lopez M, Tovar S, Vazquez MJ, Nogueiras R, Senaris R, Dieguez C 2005 Sensing the fat: fatty acid metabolism in the hypothalamus and the melanocortin system. Peptides 26: 1753–1758
Taylor SI 1999 Deconstructing type 2 diabetes. Cell 97: 9–12
McGarry JD 1992 What if Minkowski had been ageusic? An alternative angle on diabetes. Science 258: 766–770
Boden G 1997 Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes 46: 3–10
Shulman GI 2000 Cellular mechanisms of insulin resistance. J Clin Invest 106: 171–176
McGarry JD 2002 Banting Lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 51: 7–18
Boden G, Chen X, Ruiz J, White JV, Rossetti L 1994 Mechanisms of fatty acid induced inhibition of glucose uptake. J Clin Invest 93: 2438–2446
Rebrin K, Steil GM, Mittelman SD, Bergman RN 1996 Causal linkage between insulin suppression of lipolysis and suppression of liver glucose output in dogs. J Clin Invest 98: 741–749
Lam TK, van de Werve G, Giacca A 2003 Free fatty acids increase basal hepatic glucose production and induce hepatic insulin resistance at different sites. Am J Physiol Endocrinol Metab 284: E281–E290
Lam TK, Carpentier A, Lewis GF, van de Werve G, Fantus IG, Giacca A 2003 Mechanisms of the free fatty acid-induced increase in hepatic glucose production. Am J Physiol Endocrinol Metab 284: E863–E873
Williamson JR, Browning ET, Scholz R 1969 Control mechanisms of gluconeogenesis and ketogenesis. I. Effects of oleate on gluconeogenesis in perfused rat liver. J Biol Chem 244: 4607–4616
Chen X, Iqbal N, Boden G 1999 The effects of free fatty acids on gluconeogenesis and glycogenolysis in normal subjects. J Clin Invest 103: 365–372
Cianfarani S, Maiorana A, Geremia C, Scire G, Spadoni GL, Germani D 2003 Blood glucose concentrations are reduced in children born small for gestational age (SGA), and thyroid-stimulating hormone levels are increased in SGA with blunted postnatal catch-up growth. J Clin Endocrinol Metab 88: 2699–2705
Selak MA, Storey BT, Peterside I, Simmons RA 2003 Impaired oxidative phosphorylation in skeletal muscle of intrauterine growth-retarded rats. Am J Physiol Endocrinol Metab 285: E130–E137
Stenfors C, Hellman U, Silberring J 1997 Characterization of endogenous neuropeptide Y in rat hippocampus and its metabolism by nanospray mass spectrometry. J Biol Chem 272: 5747–5751
Brownsey RW, Boone AN, Elliott JE, Kulpa JE, Lee WM 2006 Regulation of acetyl-CoA carboxylase. Biochem Soc Trans 34: 223–227
Hu Z, Cha SH, Chohnan S, Lane MD 2003 Hypothalamic malonyl-CoA as a mediator of feeding behavior. Proc Natl Acad Sci U S A 100: 12624–12629
He W, Lam TK, Obici S, Rossetti L 2006 Molecular disruption of hypothalamic nutrient sensing induces obesity. Nat Neurosci 9: 227–233
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported in part by Ministero dell'Istruzione dell'Università e della Ricerca (MIUR), Grant COFIN 40%-2003064547, and by Novo Nordisk Italy.
Rights and permissions
About this article
Cite this article
Puglianiello, A., Germani, D., Antignani, S. et al. Changes in the Expression of Hypothalamic Lipid Sensing Genes in Rat Model of Intrauterine Growth Retardation (IUGR). Pediatr Res 61, 433–437 (2007). https://doi.org/10.1203/pdr.0b013e3180332d4e
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/pdr.0b013e3180332d4e
This article is cited by
-
Effect of intrauterine growth retardation on liver and long-term metabolic risk
International Journal of Obesity (2012)
-
Uteroplacental insufficiency down regulates insulin receptor and affects expression of key enzymes of long-chain fatty acid (LCFA) metabolism in skeletal muscle at birth
Cardiovascular Diabetology (2008)
