
Abstract
Carnitine palmitoyltransferase 2 (CPTII) deficiency is among the most common inborn errors of mitochondrial fatty acid β-oxidation (FAO). Clinical phenotype varies in relation to the metabolic block, as assessed by studies of FAO in patient fibroblasts. Thus, fibroblasts from patients with mild manifestations have appreciable residual CPTII enzyme activity, in contrast to those from severely affected patients. In the present study, we hypothesized that the hypolipidemic drug bezafibrate, acting as an activator of the peroxisome proliferator-activated receptor α might stimulate FAO in CPTII-deficient cells. Data obtained show that bezafibrate treatment of mild-type CPTII-deficient cells resulted in a time- and dose- dependant increase in CPTII mRNA (from +47% to +66%) and residual enzyme activity (from +54% to 135%), and led to normalization of 3H-palmitate and 3H-myristate cellular oxidation rates. Bezafibrate did not correct FAO in fibroblasts from patients with severe phenotype. This study establishes for the first time that peroxisome proliferator-activated receptor activators, acting via stimulation of gene expression, can stimulate CPTII residual activity to a level sufficient to allow normal FAO flux in deficient human fibroblasts, and suggests that this approach should be tested in other inborn errors of mitochondrial β-oxidation.
Similar content being viewed by others

Main
CPTII (EC 2.3.1.21) deficiency (OMIM: 600650) is one of the most common inborn error of FAO (1, 2). CPTII plays a pivotal role in the transport of LCFA into the mitochondria. Indeed, LCFA, which cannot readily cross the mitochondrial membranes, are transferred from the cytosol to the mitochondrial matrix through the sequential action of CPTI and CPTII, located in the outer and inner mitochondrial membranes, respectively. CPTI and CPTII catalyze the trans-esterification of long-chain acyl-CoA into long-chain acylcarnitine in the cytosol, and the reverse reaction in the mitochondrial matrix, respectively. This acylcarnitine shuttle is completed by the action of the carnitine/acylcarnitine translocase. In the mitochondrial matrix, fatty acids are oxidized to acetyl-CoA through the four steps of intra-mitochondrial β-oxidation leading to ATP production.
CPTII deficiencies have been divided into three major clinical forms (1, 3). The mild form consists in recurrent attacks of rhabdomyolysis, usually triggered by prolonged exercise, fasting, or infections in teenagers. Life-threatening complications, e.g. acute renal failure and respiratory insufficiency, occasionally occur during episodes of rhabdomyolysis. The severe form of CPTII deficiency is characterized by acute liver failure with hypoketotic hypoglycemia and serious cardiac damages in the neonatal period or infancy. Finally, an intermediate juvenile form with hepatic, cardiac, and myopathic involvement has also been described. The clinical severity is usually related to the severity of the metabolic block (2). The severe neonatal form of CPTII deficiency is generally associated with extremely low levels of residual enzyme activity in the patient's fibroblasts, whereas a significant residual enzyme activity is detected in the milder form (1, 4). Owing to the possible occurrence of life-threatening events that can barely be prevented by classical dietary approaches (fat-restricted diet, medium-chain triglyceride supply), pharmacological agents able promoting mitochondrial FAO might be of help for management of CPTII-deficient patients.
Bezafibrate is one of the fibrate group of hypolipidemic drugs that have long been proven efficient in the treatment of hypertriglyceridemia in human and have protective effects on cardiovascular risks (5). Fibrates act as activators of a nuclear receptor, the PPARα that trans-activates several genes involved in lipid and lipoprotein metabolism (5, 6). Several lines of evidence suggest that fibrates up-regulate the expression of gene encoding mitochondrial enzymes (5, 6). For this reason, we hypothesized that bezafibrate could possibly improve LCFA oxidation in fibroblasts of CPTII-deficient patients by increasing residual CPTII enzyme activity. Here, we show that this compound restored mitochondrial FAO in cultured skin fibroblasts of CPTII-deficient patients.
METHODS
Patients.
All studies were approved by the Institutional Review Board and fibroblasts studies were conducted with informed consent. Patients 1–4 had a typical mild form of CPTII deficiency, with recurrent attacks of rhabdomyolysis, triggered by prolonged exercise and/or fasting and/or viral episodes (7). Patients 5 and 6 suffered from a severe neonatal form of CPTII deficiency with hypoketotic hypoglycemias, hepatomegaly, hypertrophic cardiomyopathy, and heart beat disorders; these two patients were unrelated (7). CPTII deficiency was ascertained by enzymatic assay on cultured skin fibroblasts (8) and CPTII mutations were identified in all six patients, as described in detail previously (7).
Material.
Bezafibrate was from Sigma Chemical (St. Louis, MO, U.S.A.). l-Aminocarnitine was kindly provided by D. Muscat (Sigma-Tau France, Ivry on the Seine, France). (9,10-3H) palmitic acid and (9,10-3H) myristic acid were from PerkinElmer Life Science Products (Boston, MA, U.S.A.).
Fibroblast culture.
Human skin fibroblasts from controls and CPTII-deficient patients were cultured as previously described (9). Briefly, fibroblasts were routinely cultured in Ham's F10 media with glutamine, 12% fetal bovine serum, 100 units/mL penicillin, and 0.1 mg/mL streptomycin. The cultures were incubated in a humidified CO2 incubator (5% CO2, 95% air) at 37°C. Bezafibrate was dissolved in DMSO. When ready for treatment, the media were removed and the cells were subsequently incubated with fresh media containing bezafibrate or the equivalent amount of DMSO (vehicle).
LCFA oxidation measurement in fibroblasts.
Fatty acid oxidation was measured by quantitating the production of 3H2O from (9,10-3H) palmitate or (9,10-3H) myristate as described previously (9). Briefly, the fibroblasts were trypsinized, counted, plated (6 × 104 cells per well in 24-well microplates) and allowed to grow for 4 d. Tritiated water release experiments were performed in triplicates. Cultured fibroblast layers were washed three times with Dulbecco's PBS. Then, 200 μL of PBS containing 100 μM 9,10(n)-3H) palmitic or 100 μM myristic acid (60 Ci/mmol, PerkinElmer Life Science Products) bound to fatty acid–free albumin were added per well; there was no bezafibrate in this assay medium. Incubation was carried out for 2 h at 37°C. In some experiments, 2 mM l-aminocarnitine (CPTII inhibitor) or 100 μM antimycin (NADH cytochrome c reductase inhibitor) were added to the incubation media. After incubation, the mixture was removed and added to 200 μL of cold 10% trichloroacetic acid. The tubes were centrifuged for 10 min at 2200 ×g at 4°C and aliquots of supernatants (350 μL) were removed, mixed with 55 μL of 6 N NaOH, and applied to ion-exchange resin. The columns were washed twice with 750 μL of water and the eluates were counted. Fibroblast proteins were determined by the Lowry method (10). Palmitate or myristate oxidation rates by fibroblasts were expressed as nanomole of 3H fatty acid (3H FA) oxidized per hour per milligram of cell protein (nmol 3H FA/h/mg protein).
CPT2 enzyme activity.
CPT2 activity was assessed by measuring the palmitoyl-l-(methyl-14C) carnitine formed from l-(methyl-14C) carnitine and palmitoyl-CoA, as previously described (8), with slight modifications. CPT2 activity was measured after solubilization of mitochondrial membranes by incubating fibroblasts at 4°C for 1 h in 0.5 M KCl 1% Tween 20, pH 7.2. The assay was performed at 30°C for 8 min with 200 μL of fibroblast extracts in 500 μL of a medium containing 100 mM Tris pH 7.2, 2.5 mM reduced glutathione, 0.4% Tween 20, 0.23 M KCl, 1 mM palmitoyl-CoA, and 2.5 mM l-(methyl-14C) carnitine (56 mCi/mmol, Amersham Pharmacia Biotech, Uppsala, Sweden). CPT2 activity was expressed as nanomole palmitoyl-l-(methyl-14C) carnitine formed per minute per milligram of cell protein (nmol 14C-PalCar/min/mg protein).
Real-time quantitative PCR.
Total RNA was isolated from fibroblasts using the TriZol reagent according to the manufacturer's protocol (Invitrogen, Carlsbad, CA, U.S.A.), treated with DNAse I (Ambion, Austin, TX, U.S.A.) and quantified using the SYBR Green I kit from Roche Diagnostics (Mannheim, Germany). The real-time quantitative PCR (RTQ-PCR) was performed using a LightCycler instrument (Roche Diagnostics) according to the manufacturer's instructions. RTQ-PCR primers (Table 1) were designed using the sequences available in GenBank and spanned an intron/exon boundary. Gene expression levels were compared in fibroblasts treated with either vehicle or bezafibrate for 24 h. The amounts of CPTII and PPARα mRNA were normalized to the amount of β-actin mRNA measured by RTQ-PCR in each sample. The results of RTQ-PCR are given in arbitrary units and expressed as fold changes in mRNA levels in cells treated with bezafibrate, relative to vehicle-treated controls.
Expression of results and statistical analysis.
Data are means ± SE. Differences between vehicle- and bezafibrate-treated cells were analyzed by one-way ANOVA and the Fisher test or by paired t test; p < 0.05 was considered significant.
RESULTS
Effects of various concentrations of bezafibrate on fatty acid oxidation in CPTII-deficient fibroblasts.
The effects of bezafibrate (50–800 μM, for 3 d) were tested in mild-type CPTII deficient fibroblasts from patient 3, which exhibited a significantly lower FAO rate compared with control fibroblasts (1.8 ± 0.2 versus 3.3 ± 0.3 nanomol 3H FA/h/mg protein, p < 0.01) (Fig. 1). Bezafibrate induced a dose-dependant increase in 3H-palmitate oxidation rate. This increase was found statistically significant at a concentration of 100 μM bezafibrate or above, and the maximal stimulatory effect (observed at 800 μM; +66% compared with vehicle-treated cells) resulted in the restoration of normal palmitate oxidation in the CPTII-deficient fibroblasts.

Dose-response effect of bezafibrate on LCFA β-oxidation in CPTII-deficient fibroblasts. Fibroblasts of mild-type CPTII deficient patient were treated for 3 d with either vehicle (DMSO) or various concentrations of bezafibrate. 3H-palmitate oxidation measurements were performed in triplicate. Results are means ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001 compared with vehicle-treated control fibroblasts of three different cell lines.
Effects of bezafibrate on fatty acid oxidation in three CPTII-deficient fibroblasts.
The effects of bezafibrate (400 or 800 μM) were then tested on both 3H-palmitate and 3H-myristate oxidation in fibroblasts from controls and patients 1–3. In control cells, 400 or 800 μM bezafibrate induced similar stimulation of 3H-palmitate or 3H-myristate oxidation (+40%, p < 0.001 compared with vehicle-treated fibroblasts;Fig. 2). Bezafibrate also resulted in a marked increase of FAO in CPTII-deficient fibroblasts. Indeed, palmitate and myristate oxidation of fibroblasts from patients 1–3 treated by 400 μM bezafibrate was 1.7-, 1.8-, and 1.9-fold higher than vehicle-treated fibroblasts (p < 0.001), respectively. Increasing bezafibrate concentrations up to 800 μM resulted in a further increase in 3H-palmitate and 3H-myristate oxidation in fibroblasts from patients 1 and 3, that exhibited FAO rates similar to those measured in control fibroblasts treated in parallel, and significantly higher than untreated control fibroblasts FAO values. Overall, exposure of CPTII-deficient fibroblasts to 800 μM bezafibrate induced a 2.2- to 2.5-fold (p < 0.01) increase in both 3H-palmitate and 3H-myristate oxidation rates.

Palmitate and myristate oxidation are increased by bezafibrate in control and CPTII-deficient fibroblasts. Fibroblasts of three different CPTII-deficient patients and of one control were treated for 3 d with either vehicle (DMSO) or bezafibrate (400 or 800 μM). The experiments were performed in triplicate. Results are means ± SEM. ***p < 0.001 compared with the vehicle-treated cells of a given subject.
Time-course of bezafibrate effects on fatty acid oxidation.
Fibroblasts from patient 3 were cultured with 800 μM bezafibrate for 3, 6, 24, or 72 h. Figure 3 shows that bezafibrate induced a time-dependant stimulation of 3H-palmitate oxidation starting from 6 h, which reached a maximal level after 24 h, and then remained constant from 24 to 72 h. The overall increase in 3H-palmitate induced by 800 μM bezafibrate (2-fold) was similar to that observed in previous experiments.

Time course of bezafibrate treatment on palmitate oxidation. Fibroblasts from patient 3 were treated with 800 μM bezafibrate and 3H-palmitate oxidation experiments were performed at the different times indicated. For each time, three wells containing vehicle-treated cells were run in parallel. Points are means ± SEM of three wells. Absence of error bars indicates that SEM is smaller than symbol. **p < 0.01, ***p < 0.001 compared with the vehicle-treated cells at the same time.
Effects of mitochondrial inhibitors of fatty acid oxidation.
Experiments using specific inhibitors were then performed to ascertain that bezafibrate specifically impacted on mitochondrial FAO. Figure 4 shows that oxidation of 3H-palmitate in fibroblasts from patient 3 was completely abolished by l-aminocarnitine, a specific CPTII inhibitor. Similarly, there was no tritiated water release in the presence of antimycin, a respiratory chain inhibitor, demonstrating that palmitate is entirely oxidized by mitochondrial β-oxidation, without contribution of the peroxisomal pathway.

Palmitate oxidation in bezafibrate-treated cells is entirely mitochondrial. Cells from patient 3 were treated with vehicle or 800 μM bezafibrate for 3 d. In some wells, 2 mM l-aminocarnitine or 100 μM antimycin were added in the assay medium during 3H-palmitate oxidation. Experiments were run in triplicate. ***p < 0.001 compared with the vehicle-treated cells.Palmitate oxidation in bezafibrate-treated cells is entirely mitochondrial. Cells from patient 3 were treated with vehicle or 800 μM bezafibrate for 3 d. In some wells, 2 mM l-aminocarnitine or 100 μM antimycin were added in the assay medium during 3H-palmitate oxidation. Experiments were run in triplicate. ***p < 0.001 compared with the vehicle-treated cells.
Effects of bezafibrate on fatty acid oxidation in “mild” and “severe” CPTII-mutations.
The effects of bezafibrate were then tested on cultured skin fibroblasts of six patients carrying various CPTII mutations (Fig. 5). Patients 1–4 carried “mild” missense mutations in exons 1/3 (patient 1), exons 3/4 (patients 2 and 3), or exon 3 only (patient 4), whereas patients 5 and 6 carried a homozygous “severe” missense mutation in exon 4 of the CPTII gene (7). The residual FAO in vehicle-treated fibroblasts ranged from 50 to 70% of control values in patients 1–4, whereas it was <10% of controls in patients 5 and 6. After a 3-d exposure to 800 μM bezafibrate, fatty acid oxidation by fibroblasts from patients 1–4 was similar to controls, but remained unchanged in patients 5 and 6.

Mild but not severe forms of CPTII deficiency are corrected by bezafibrate treatment. (A) Control or CPTII-deficient patient fibroblasts were treated with either vehicle or 800 μM bezafibrate and 3H-palmitate oxidation was measured in triplicate. Results are means ± SEM. ***p < 0.001 compared with the corresponding vehicle-treated cells. (B) Genomic organization of the human CPTII gene. The exons are numbered from 1 to 5 and the position of the various mutations are indicated.
Effects of bezafibrate on CPTII activity.
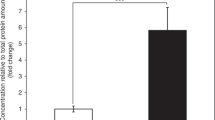
In control fibroblasts, CPTII enzyme activity was raised from 0.77 to 1.03 nmol 14C-PalCar/min/mg protein (+34%, p < 0.001), in response to bezafibrate (800 μM for 3 d). Exposure of mild-type CPTII-deficient cells to bezafibrate resulted in a significant increase in enzyme activity, ranging from +54 to +135% compared with vehicle-treated cells. Yet, these CPTII activities measured in treated patient cells (0.32 ± 0.01 nmol 14C-PalCar/min/mg protein, n = 3) remained significantly lower than control values (Fig. 6).

Bezafibrate increases residual CPTII activity. Control and mild-type CPTII-deficient fibroblasts were treated for 3 d with bezafibrate or vehicle, and the cells were harvested for CPTII measurement as described in “Methods.” Results are means ± SEM of three determinations. ***p < 0.001 compared with the vehicle-treated cells.
Effects of bezafibrate on CPTII and PPARα transcript levels.
Quantitative PCR analysis showed that bezafibrate induced largely comparable increases in CPTII transcripts in control and patient cells (1.47- to 1.66-fold), and the differences between bezafibrate-treated and vehicle-treated cells were highly significant (p < 0.001;Table 2). Furthermore, bezafibrate stimulated PPARα gene expression 1.39- to 1.94-fold (p < 0.01) in control and patients fibroblasts, respectively.
DISCUSSION
The present study reports on the increase of fatty acid oxidation by bezafibrate in cultured skin fibroblasts of CPTII-deficient patients. The stimulatory effects of bezafibrate were time- and dose-dependant and led to a full restoration of LCFA oxidation in mild-type CPTII deficiency. Because the effects of bezafibrate were related to an increased residual enzyme activity, they were observed in “mild-type” CPTII deficiency (with significant residual enzyme levels) but not in “severe” CPTII deficiency (with extremely low residual enzyme activity). Thus, correction of FAO flux by bezafibrate involved stimulation of mutated CPTII enzyme expression, rather than indirect compensatory mechanisms.
It is worth noting that restoration of FAO by bezafibrate was not associated with full restoration of CPTII activity. These findings are in keeping with previous studies showing that CPTII enzyme activity is in great excess in normal human cells (1, 11). Accordingly, drastic reduction of FAO observed in the severe form of the disease, is only noted when residual CPTII activity is <10% of control value (1, 11). Interestingly, heterozygous parents of severely affected patients, exhibiting about 50% CPTII activity, have normal β-oxidation flux in fibroblasts, and are asymptomatic (11). This is consistent with the present data, which suggest that the threshold of residual CPTII activity needed to support maximal fatty acid oxidation flux is around 40% of normal enzyme activity.
Quantitative PCR showed that bezafibrate induced a significant increase in CPTII mRNA. Bezafibrate has been previously shown to stimulate gene expression of β-oxidation enzymes after specific binding to its nuclear receptor, PPARα(5, 6). The underlying mechanism involves binding of activated PPARα on specific DNA sequences called PPAR response elements (PPRE), leading to stimulation of gene transcription (5). PPRE have been identified on the 5′-flanking region of human CPTI muscular isoform (CPTI-β) (12, 13), which plays a key role in the transport of long-chain acyl-CoA within the mitochondria, and of human medium-chain acyl-CoA dehydrogenase (MCAD) (14), which catalyzes the initial step of mitochondrial medium-chain fatty acid β-oxidation. Accordingly, regulation of CPTI-β and MCAD gene expression levels by the PPAR signaling pathway has been shown to occur in rodents (12, 14–16) and humans as well (17). PPARα null mice exhibit low steady-state levels of CPTII mRNA (18), and administration of PPARα agonists fails to stimulate CPTII gene expression in these knockout mice (19). Furthermore, our results are consistent with the recent identification of a PPRE in the 5′-flanking region of the human CPTII gene (20), and suggest that control of CPTII gene by PPARα operates in human skin fibroblasts.
In conclusion, this study brings the first data indicating that bezafibrate can correct FAO in CPTII-deficient human cells. Correction by bezafibrate was obtained similarly in cell lines harboring four different “mild” CPTII genotypes. It would be interesting to extend this study to other genotypes responsible for mild CPT2 deficiency (7), and eventually to assess the putative effect of intragenic polymorphisms (21) on the response to bezafibrate, for a given CPT2 genotype.
Finally, several other β-oxidation defects are known to present with significant cell residual enzyme activity (2), and expression of most corresponding genes has been shown to be PPAR regulated (5, 6). Screening possible effects of PPAR agonists could thus provide a new and valuable way to develop cell therapy models for these orphan diseases and assess their potential applications for clinical use.
Abbreviations
- FAO:
-
mitochondrial fatty acid β-oxidation
- CPTII:
-
carnitine palmitoyltransferase 2
- CPTI:
-
carnitine palmitoyltransferase 1
- PPARα:
-
peroxisome proliferator-activated receptor α
- LCFA:
-
long-chain fatty acid
References
Bonnefont JP, Demaugre F, Prip-Buus C, Saudubray JM, Brivet M, Abadi N, Thuillier L 1999 Carnitine palmitoyltransferase deficiencies. Mol Genet Metab 68: 424–440
Gregersen N, Andresen BS, Corydon MJ, Corydon TJ, Olsen RK, Bolund L, Bross P 2001 Mutation analysis in mitochondrial fatty acid oxidation defects: exemplified by acyl-CoA dehydrogenase deficiencies, with special focus on genotype-phenotype relationship. Hum Mutat 18: 169–189
Brivet M, Boutron A, Slama A, Costa C, Thuillier L, Demaugre F, Rabier D, Saudubray JM, Bonnefont JP 1999 Defects in activation and transport of fatty acids. J Inherit Metab Dis 22: 428–441
Bonnefont JP, Taroni F, Cavadini P, Cepanec C, Brivet M, Saudubray JM, Leroux JP, Demaugre F 1996 Molecular analysis of carnitine palmitoyltransferase II deficiency with hepatocardiomuscular expression. Am J Hum Genet 58: 971–978
Berger J, Moller DE 2002 The mechanisms of action of PPARs. Annu Rev Med 53: 409–435
Vamecq J, Latruffe N 1999 Medical significance of peroxisome proliferator-activated receptors. Lancet 354: 141–148
Thuillier L, Rostane H, Droin V, Demaugre F, Brivet M, Khadom N, Prip-Buus C, Gobin S, Saudubray JM, Bonnefont JP 2003 Correlation between genotype, metabolic data, and clinical presentation in carnitine palmitoyltransferase 2 deficiency. Hum Mutat 5: 493–501
Demaugre F, Bonnefont JP, Colonna M, Cepanec C, Leroux JP, Saudubray JM 1991 Infantile form of carnitine palmitoyltransferase II deficiency with hepatomuscular symptoms and sudden death. Physiopathological approach to carnitine palmitoyltransferase II deficiencies. J Clin Invest 87: 859–864
Manning NJ, Olpin SE, Pollitt RJ, Webley J 1990 A comparison of [9,10–3H]palmitic and [9,10–3H]myristic acids for the detection of defects of fatty acid oxidation in intact cultured fibroblasts. J Inherit Metab Dis 13: 58–68
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ 1951 Protein measurement with the folin phenol reagent. J Biol Chem 193: 265–275
Eaton S 2002 Control of mitochondrial beta-oxidation flux. Prog Lipid Res 41: 197–239
Brandt JM, Djouadi F, Kelly DP 1998 Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator-activated receptor alpha. J Biol Chem 273: 23786–23792
Mascaro C, Acosta E, Ortiz JA, Marrero PF, Hegardt FG, Haro D 1998 Control of human muscle-type carnitine palmitoyltransferase I gene transcription by peroxisome proliferator-activated receptor. J Biol Chem 273: 8560–8563
Gulick T, Cresci S, Caira T, Moore DD, Kelly DP 1994 The peroxisome proliferator-activated receptor regulates mitochondrial fatty acid oxidative enzyme gene expression. Proc Natl Acad Sci U S A 91: 11012–11016
Ouali F, Djouadi F, Merlet-Benichou C, Bastin J 1998 Dietary lipids regulate beta-oxidation enzyme gene expression in the developing rat kidney. Am J Physiol 275: F777–F784
Yu GS, Lu YC, Gulick T 1998 Co-regulation of tissue-specific alternative human carnitine palmitoyltransferase Ibeta gene promoters by fatty acid enzyme substrate. J Biol Chem 273: 32901–32909
Muoio DM, Way JM, Tanner CJ, Winegar DA, Kliewer SA, Houmard JA, Kraus WE, Dohm GL 2002 Peroxisome proliferator-activated receptor-alpha regulates fatty acid utilization in primary human skeletal muscle cells. Diabetes 51: 901–909
Watanabe K, Fujii H, Takahashi T, Kodama M, Aizawa Y, Ohta Y, Ono T, Hasegawa G, Naito M, Nakajima T, Kamijo Y, Gonzalez FJ, Aoyama T 2000 Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor alpha associated with age-dependent cardiac toxicity. J Biol Chem 275: 22293–22299
Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, Gonzalez FJ 1998 Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J Biol Chem 273: 5678–5684
Barrero MJ, Camarero N, Marrero PF, Haro D 2003 Control of human carnitine palmitoyltransferase II gene transcription by peroxisome proliferator-activated receptor through a partially conserved PPRE. Biochem J 369: 721–729
Taroni F, Verderio E, Fiorucci S, Cavadini P, Finocchiaro G, Uziel G, Lamantea E, Gellera C, DiDonato S 1992 Molecular characterization of inherited carnitine palmitoyl transferase II deficiency. Proc Natl Acad Sci U S A 89: 8429–8433
Acknowledgements
The authors thank Mrs. D. Muscat (Sigma-Tau France) for providing us with l-aminocarnitine. We also thank Laurie Laugier for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported, in part, by a grant from the Association Française contre les Myopathies.
Rights and permissions
About this article
Cite this article
Djouadi, F., Bonnefont, JP., Thuillier, L. et al. Correction of Fatty Acid Oxidation in Carnitine Palmitoyl Transferase 2–Deficient Cultured Skin Fibroblasts by Bezafibrate. Pediatr Res 54, 446–451 (2003). https://doi.org/10.1203/01.PDR.0000083001.91588.BB
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000083001.91588.BB
This article is cited by
-
Mitochondrial membrane transporters and metabolic switch in heart failure
Heart Failure Reviews (2019)
-
Carnitine palmitoyl transferase 2 polymorphism may be associated with enterovirus 71 severe infection in a Chinese population
Archives of Virology (2016)
-
Mitochondrial trifunctional protein deficiency in human cultured fibroblasts: effects of bezafibrate
Journal of Inherited Metabolic Disease (2016)
-
Genetic variations underlying self-reported physical functioning: a review
Quality of Life Research (2015)
-
Stilbenes and resveratrol metabolites improve mitochondrial fatty acid oxidation defects in human fibroblasts
Orphanet Journal of Rare Diseases (2014)
