
Abstract
The mechanisms underlying failure in sudden infant death syndrome may involve inadequate compensatory motor responses to a hypotensive challenge; the insult may result from a shock-like sequence, or from a ventilatory challenge that leads to a hypotensive event. Structures ordinarily not considered in mediating breathing or cardiovascular control, especially cerebellar-related structures, may play a critical role in compensatory responses, and underlie the position-dependent risk for SIDS. Dysfunction in affected brain areas appears to arise prenatally from a compromised fetal environment, with a nicotinic component contributing to the deficient mechanism.
Physiologic characteristics of infants who later succumb to SIDS, and cardiovascular events associated with the fatal scenario suggest a failure of interaction between somatomotor and autonomic control mechanisms in infants at risk for the syndrome. A failure of compensatory motor actions to overcome a profound hypotension, perhaps mediated by cerebellar mechanisms that regulate blood pressure, may underlie the fatal event.
Similar content being viewed by others

Main
Any determination of mechanisms underlying the sudden infant death syndrome (SIDS), the sudden and unexplained death of an infant occurring in the 1st year of life, must consider some of the known characteristics associated with the syndrome. These characteristics include enhanced risk with the prone sleeping position (1, 2), a substantially increased incidence following prenatal or postnatal tobacco exposure (3), a temporal association with sleep, periods of tachycardia before the fatal event (4), and an incidence confined principally between the 2nd and 4th months of life. Less well-documented associations, but findings repeatedly encountered in reports, are profuse sweating (5) and abnormally high core or environmental temperatures (6). Among the characteristics associated with the fatal event, a remarkable, often short-lasting bradycardia, accompanied by hypotension, sometimes in the presence of continued respiratory efforts, has been reported (7, 8).
The findings of tachycardia and profuse sweating before death implicate initial exaggerated sympathetic nervous system activity, while bradycardia and hypotension during the fatal event suggest that a subsequent sympathoinhibition, but parasympathetic nervous system recruitment accompany the fatal event; this sequence parallels the trend of events that are associated with shock from blood loss or deep pain (9). The possibility of a perfusion failure represents a departure from current interpretations of the mechanism of death as resulting primarily from a respiratory collapse. The sudden bradycardia also argues against particular arrhythmia failures, such as prolonged Q-T intervals (10) that normally result in death by ventricular fibrillation. Although a proportion of SIDS deaths may result from suffocation, ventricular fibrillation, CO2 intoxication, or hypoxia, a proportion may also result from a sympathoinhibition, precipitated by a shock or shock-like scenario. The significant element in this scenario is a profound loss of blood pressure, and an apparent inability to restore or induce compensatory responses to assist restoration of vascular tone. It may be the case that the initial hypotension may be triggered by a number of mechanisms other than the classic shock sequence of sympathoexcitation followed by sympathoinhibition. Marked blood pressure falls occur spontaneously during rapid eye movement sleep, for example, and hypotension follows sustained hypoxia.
The range of circumstances surrounding the fatal event in SIDS suggest a heterogeneous group of disorders, some of which appear to be of a respiratory origin, eg those associated with infant faces embedded in restricted air space of pillows or bedding (11). The airflow restriction may be of an internal obstructive nature, as indicated by increased incidence of obstructive apnea in infants who succumb (5). Other fatal events appear to be of cardiovascular nature, eg bradycardia and hypotension during the fatal event (8); the different scenarios suggest that infants may succumb through several failure modes, and that the capability to adequately recover from a challenge may be a more significant issue for survival than the particular mechanism of failure. Arousal mechanisms from sleep have been implicated as a common vehicle for recovery from compromising events (12), and are undoubtedly a significant issue in restoration of compensatory forebrain and other influences that assist recovery of vital processes. The concept of “arousal” should perhaps be extended to include restoration or activation of somatic muscle tone in addition to transition of sleep state. Recruitment of muscle tone is essential for respiratory challenges, eg head turning from an obstruction, enhanced tidal volumes to hypercapnia or hypoxia, or appropriate responses to a blood pressure loss. This last interaction implicates a role for somatic musculature on autonomic functioning (sympathetic control). The relationships derived from animal studies suggest that effective compensatory responses to loss of blood pressure involve somatomotor action, and especially the respiratory somatic musculature to restore blood pressure. Cats subjected to blood loss, for example, successfully respond to the hypotension and are able to restore blood pressure by enhanced inspiratory and expiratory efforts, tachypnea, and exaggerated extensor skeletal muscle action (13). Those successful compensatory motor responses are associated with substantial activity changes on the rostral ventral medullary surface, an area classically implicated in blood pressure regulation. However, the recovery efforts obviously also recruit motor areas that receive signals from central structures indicating low blood pressure, and activate muscles for recovery; the recovery response is particularly apparent for respiratory muscles (14).
Neural mechanisms responsible for compensatory responses to a hypercapnic, hypoxic, or bradycardic/sympathoinhibitory challenge may involve brain sites not normally considered as “respiratory,” “arousal,” or “cardiovascular” areas. A principal focus in respiratory control studies has been to determine brain areas responsible for central pattern generation underlying regular breathing rhythm, and that attention has usually been directed to the medulla. Issues of blood pressure control similarly focus on medullary reflex loops. However, a major interest for SIDS research is determining which brain structures are recruited to restore breathing when pattern generators fail, ie from an apneic episode, or which neural sites compensate for blood pressure falls incompatible with survival; those structures may lie outside normally considered medullary sites. Recent evidence from functional magnetic resonance studies in normal subjects (15) and in children afflicted with congenital central hypoventilation syndrome (16), as well as electrophysiological recording and lesion studies in animals, implicate portions of the cerebellum, especially the cerebellar fastigial nucleus, in modulating appropriate responses to O2 and blood pressure challenges. Bilateral lesions of the fastigial nucleus result in a fatal progression after blood pressure lowering (17, 18), or uncorrected influences from hypercapnia on breathing (19). Regions within the cerebellum are often associated with an “error correction” role, ie the correction of appropriate motor output after sensing of aberrant afferent signals. That “error correction” is classically associated with motor performance, and tested clinically with a motor task, such as finger pointing. Comparable regulatory roles for the cerebellum apparently extend to blood pressure and breathing control (15).
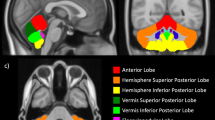
Cerebellar compensatory reactions to hypotension or hypertension are likely mediated through afferent activity from the inferior olivary nucleus via climbing fibers to Purkinje cells and the fastigial nucleus of the cerebellum, and output to vestibular sympathetic pathways, as well as somatomotor regions. Portions of the inferior olive, a major input relay to the cerebellum, show significant gliosis in infants who have succumbed to SIDS, and isolated cases of inferior olive hypoplasia are associated with profound respiratory dysfunction (20). More recent findings indicate deficiencies in SIDS victims in muscarinic and kainate receptors within the ventral medullary surface (21, 22) and serotonergic receptors in medullary sites including the inferior olive and caudal medullary raphe regions associated with hypotension and sympathoinhibition (23, 24). The inferior olive also shows increased c fos expression to a variety of manipulations that trigger vasodepression (Bandler and Keay, Department of Anatomy and Histology, University of Sydney, NSW, Australia, personal communication, 1999). Delayed maturation of cerebellar regions has been observed in SIDS victims (25). Vestibular contributions to blood pressure regulation are well-known, since body positioning requires rapidly acting compensation of regional blood pressure to assist appropriate perfusion, frequently operating in a “feed-forward” or anticipatory fashion (26, 27). Deficiencies in such regulation are frequently observed clinically, for example, in rapid rising from a supine to vertical position.
A cerebellar/vestibular role for compensatory responses to cardiovascular or breathing challenges may participate in the position-dependent risk factor for SIDS. Afferent position information from the position-sensitive receptors travel by way of the vestibular nuclei and inferior olive to the cerebellum, which then projects to reticular and rostral ventrolateral medullary sympathetic areas via vestibular nuclei (27). Such vestibular contributions may be the significant factor underlying the substantial prone versus supine position differences in cardiovascular and breathing control in low birth weight infants (28); the blood pressure response to head-up tilt is greatly reduced in full-term infants in the prone position as well (29). Cerebellar-mediated vestibular input from rocking also reduces obstructive apneic events (30).
A substantial number of cerebellar developmental abnormalities have been identified, including intracerebellar hemorrhage (31); some of these aberrations are associated with marked respiratory disturbances; other case reports describe significant cardiovascular disturbances following midline cerebellar stimulation in humans (32). If cerebellar abnormalities are present in SIDS victims, the resulting physiologic characteristics are subtle, since cardiac and respiratory patterns that differentiate future SIDS victims from controls can only be identified with careful attention to partitioning breathing and cardiac patterns by sleep state and time-of-night (33). However, the pronounced influences on vital functions from cerebellar syndromes emphasize the potential capacity for more-modest cerebellar deficiencies to disturb physiologic functions. Animal data suggest that cerebellar mechanisms are particularly recruited during extreme challenges, rather than during routine regulation of breathing and blood pressure (17, 19).
The epidemiologic evidence for SIDS suggests a prominent role for prenatal nicotine exposure and low maternal hematocrit (34). Recent animal evidence demonstrates the importance of adrenal catecholamine release on autoresuscitation from hypoxia, and that prenatal nicotine exposure compromises autoresuscitation, possibly by interference with cardiac conductance changes associated with altered adrenal catecholamine outflow (35–37). The potential role of loss of protective vagal influences on cardiac conduction disturbances has been outlined earlier (38).
However, prenatal or postnatal central damage could also affect compensatory recovery mechanisms. Cerebellar Purkinje cells receive afferents from the climbing fibers of the inferior olive and are especially sensitive to neurotoxic damage from agents such as harmaline (39), perhaps because of the unique limited geometry of the afferent system. It appears that the peculiar arrangement of Purkinje fibers poses a risk for excitotoxic injury from a variety of challenges, including hypoxic exposure, nicotine, or harmaline. Harmaline exposure results in aberrant responses to blood pressure challenges that are comparable to lesions of the fastigial nucleus of the cerebellum. In rats, it appears that such disturbances are time-dependent, with recovery by alternate pathways several weeks after damage (40). It may be the case that repetitive hypoxic insults occurring postnatally, or fetal damage to this blood pressure/breathing regulatory system from nicotine exposure establishes a less-than-optimal system for responding to ventilatory or blood pressure challenges; the time-dependent response capability may be an issue, because of the narrow window of SIDS risk (between the second and fourth months of life).
Abbreviations
- SIDS:
-
sudden infant death syndrome
- RVLM:
-
rostral ventral lateral medulla
- fast. nuc.:
-
fastigial nucleus
- lat. retic. form.:
-
lateral reticular formation
- inf. olive:
-
inferior olive
- IML:
-
intermediolateral
References
Fleming PJ, Gilbert RE, Azaz Y, Berry PJ, Rudd PT, Stewart A, Hall E 1990 The interaction between bedding and sleeping position in sudden infant death syndrome: a population-based case-control study. BMJ 301: 85–89.
Ponsonby AL, Dwyer T, Gibbons LE, Cochrane JA, Wang YA 1993 Factors potentiating the risk of sudden infant death syndrome associated with the prone position. N Engl J Med 329: 377–382.
Blair PS, Fleming PJ, Bensley D, Smith I, Bacon C, Taylor E, Berry J, Golding J, Tripp J 1996 Smoking and the sudden infant death syndrome: results from 1993–5 case-control study for confidential inquiry into stillbirths and deaths in infancy. BMJ 313: 195–198.
Schechtman VL, Harper RM, Kluge KA, Wilson AJ, Hoffman HJ, Southall DP 1988 Cardiac and respiratory patterns in normal infants and victims of the sudden infant death syndrome. Sleep 11: 413–424.
Kahn A, Groswasser J, Rebuffat E, Sottiaux M, Blum D, Foerster M, Franco P, Bochner A, Alexander M, Bachy A, Richard P, Verghote M, Le Polain D, Wayenberg JL 1992 Sleep and cardiorespiratory characteristics of infant victims of sudden death: a prospective case-control study. Sleep 15: 287–292.
Fleming PJ, Levine MR, Azaz Y, Wigfield R 1993 The development of thermoregulation and interactions with the control of respiration in infants: possible relationship to sudden infant death. Acta Paediatr Scand Suppl 389: 57–59.
Meny RG, Carroll JL, Carbone MT, Kelly DH 1994 Cardiorespiratory recordings from infants dying suddenly and unexpectedly at home. Pediatrics 93: 43–49.
Ledwidge M, Fox G, Matthews T 1998 Neurocardiogenic syncope: a model for SIDS. Arch Dis Child 78: 481–483.
Harper RM, Bandler R 1998 Finding the failure mechanism in the Sudden Infant Death Syndrome. Nat Med 4: 157–158.
Schwartz PJ, Stramba-Badiale M, Segantini A, Austoni P, Bosi G, Giorgetti R, Grancini F, Marni ED, Perticone F, Rosti D, Salice P 1998 Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med 338: 1709–1714.
Kemp JS, Thach BT 1991 Sudden death in infants sleeping on polystyrene-filled cushions. N Engl J Med 324: 1858–1864.
Hunt C 1989 Impaired arousal from sleep: relationship to sudden infant death syndrome. [Review]. J Perinatol 9: 184–187.
Harper RM, Richard CA, Rector DM 1999 Physiological and ventral medullary surface activity during hypovolemia. Neuroscience 94: 579–586.
Ohtake PJ, Jennings DB 1992 Ventilation is stimulated by small reductions in arterial pressure in the awake dog. J Appl Physiol 73: 1549–1557.
Harper RM, Bandler R, Spriggs D, Alger JR 2000 Lateralized and widespread brain activation during transient blood pressure elevation revealed by magnetic resonance imaging. J Comp Neurol 417: 195–204.
Harper RM, Spriggs D, Saeed MM, Alger JR, Woo MA, Woo MS, Gozal D, Keens TG 1999 Functional magnetic resonance imaging during hypoxia challenges in congenital central hypoventilation syndrome (CCHS) reveals lateralized neural responses. Soc Neurosci Abstr 25: 280
Lutherer LO, Lutherer BC, Dormer KJ, Janssen HF, Barnes CD 1983 Bilateral lesions of the fastigial nucleus prevent the recovery of blood pressure following hypotension induced by hemorrhage or administration of endotoxin. Brain Res 269: 251–257.
Chen CH, Williams JL, Lutherer LO 1994 Cerebellar lesions alter autonomic responses to transient isovolaemic changes in arterial pressure in anaesthetized cats. Clin Auton Res 4: 263–272.
Xu F, Owen J, Frazier DT 1994 Cerebellar modulation of ventilatory response to progressive hypercapnia. J Appl Physiol 77: 1073–1080.
Cortez SC, Kinney HC 1996 Brainstem tegmental necrosis and olivary hypoplasia: a lethal entity associated with congenital apnea. J Neuropathol Exp Neurol 55: 841–849.
Kinney HC, Filiano JJ, Sleeper L, Mandell F, Valdes-Dapena M, White WF 1995 Decreased muscarinic receptor binding in the arcuate nucleus in sudden infant death syndrome. Science 269: 1446–1450.
Panigrahy A, Filiano JJ, Sleeper LA, Mandell F, Valdes-Dapena M, Krous HF, Rava LA, White WF, Kinney HC 1997 Decreased kainate receptor binding in the arcuate nucleus of the sudden infant death syndrome. J Neuropathol Exp Neurol 56: 1253–1261.
Panigrahy A, Filiano JJ, Sleeper LA, Mandell F, Valdes-Dapena M, Krous HF, Rava LA, Foley E, White WF, Kinney HC 2000 Decreased serotonergic receptor binding in rhombic-lipped derived regions of the medulla oblongata in the sudden infant death syndrome. J Neuropathol Exp Neurol (in press)
Coleman MJ, Dampney RA 1995 Powerful depressed sympathoinhibitory effects evoked from neurons in the caudal raphe pallidus and obscurus. Am J Physiol 268: R1295–R1302.
Cruz-Sánchez FF, Lucena J, Ascaso C, Tolosa E, Quintò L, Rossi M 1997 Cerebellar cortex delayed maturation in sudden infant death syndrome. J Neuropathol Exp Neurol 56: 340–346.
Doba N, Reis DJ 1974 Role of the cerebellum and vestibular apparatus in regulation of orthostatic reflexes in the cat. Circ Res 34: 9–18.
Yates BJ 1996 Vestibular influences on the autonomic nervous system. In: Highstein, SM, Cohen B, Buttner-Ennever JA (eds) New Directions in Vestibular Research. Ann NY Acad Sci 458–470.
Sahni R, Schulze KF, Kashyap S, Ohira-Kist K, Myers MM, Fifer WP 1999 Body position, sleep states, and cardiorespiratory activity in developing low birth weight infants. Early Hum Dev 54: 197–206.
Chong A, Murphy N, Matthews T 2000 Effect of prone sleeping on circulatory control in infants. Arch Dis Child 82: 253–256.
Groswasser J, Sottiaux M, Rebuffat E, Simon T, Vandeweyer M, Kelmanson I, Blum D, Kahn A 1995 Reduction in obstructive breathing events during body rocking: a controlled polygraphic study in preterm and full-term infants. Pediatrics 96: 64–68.
Martin R, Roessmann U, Fanaroff A 1976 Massive intracerebellar hemorrhage in low-birth-weight infants. J Pediatr 89: 290–293.
Elisevich K, Redekop G 1991 The fastigial pressor response. J Neurosurg 74: 147–151.
Harper RM, Schechtman VL 1995 Physiological measurements as predictive tests for SIDS. In: Rognum TO (ed) Sudden Infant Death Syndrome: New Trends in the Nineties. Scandinavian University Press, Oslo, 314–319.
Bulterys MG, Greenland S, Kraus JF 1990 Chronic fetal hypoxia and sudden infant death syndrome: interaction between maternal smoking and low hematocrit during pregnancy. Pediatrics 86: 535–540.
Slotkin TA, Lappi SE, McCook EC, Lorber BA, Seidler FJ 1995 Loss of neonatal hypoxia tolerance after prenatal nicotine exposure: implications for sudden infant death syndrome. Brain Res Bull 38: 69–75.
Yuan SZ, Runold M, Lagercrantz H 1997 Adrenalectomy reduces the ability of newborn rats to gasp and survive anoxia. Acta Physiol Scand 159: 285–292.
Fewell JE, Smith FG 1998 Perinatal nicotine exposure impairs ability of newborn rats to autoresuscitate from apnea during hypoxia. J Appl Physiol 85: 2066–2074.
Gootman PM, Gootman N, Sica AL 1996 A neuro-cardiac theory for sudden infant death syndrome: role of the autonomic nervous system. J Sudden Infant Death Syndrome Infant Mortality 1: 169–182.
O'Hearn E, Molliver ME 1997 The olivocerebellar projection mediates ibogaine-induced degeneration of Purkinje cells: A model of indirect trans-synaptic excitotoxicity. J Neurosci 17: 8828–8841.
Lutherer LO, Purdom AR 1999 Reversal of early functional deficits resulting from harmaline-induced cerebellar lesions. FASEB J LB77.
Author information
Authors and Affiliations
Additional information
Supported by RO1-HD-22506, RO1-HD-22695, and RO3-HD-36228 from the National Institute of Child, Health and Human Development.
Rights and permissions
About this article
Cite this article
Harper, R. Sudden Infant Death Syndrome: A Failure of Compensatory Cerebellar Mechanisms?. Pediatr Res 48, 140–142 (2000). https://doi.org/10.1203/00006450-200008000-00004
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200008000-00004
This article is cited by
-
Assessment of tobacco smoke effects on neonatal cardiorespiratory control using a semi-automated processing approach
Medical & Biological Engineering & Computing (2018)
-
Real-time detection, classification, and quantification of apneic episodes using miniature surface motion sensors in rats
Pediatric Research (2015)
-
The role of spiking and bursting pacemakers in the neuronal control of breathing
Journal of Biological Physics (2011)
-
Leptomeningeal neurons are a common finding in infants and are increased in sudden infant death syndrome
Acta Neuropathologica (2009)
-
Sudden infant death while awake
Forensic Science, Medicine, and Pathology (2008)
