

Abstract
The role of mechanical properties in cancer disease and inflammation is still underinvestigated and even ignored in many oncological and immunological reviews. In particular, eight classical hallmarks of cancer have been proposed, but they still ignore the mechanics behind the processes that facilitate cancer progression. To define the malignant transformation of neoplasms and finally reveal the functional pathway that enables cancer cells to promote cancer progression, these classical hallmarks of cancer require the inclusion of specific mechanical properties of cancer cells and their microenvironment such as the extracellular matrix as well as embedded cells such as fibroblasts, macrophages or endothelial cells. Thus, this review will present current cancer research from a biophysical point of view and will therefore focus on novel physical aspects and biophysical methods to investigate the aggressiveness of cancer cells and the process of inflammation. As cancer or immune cells are embedded in a certain microenvironment such as the extracellular matrix, the mechanical properties of this microenvironment cannot be neglected, and alterations of the microenvironment may have an impact on the mechanical properties of the cancer or immune cells. Here, it is highlighted how biophysical approaches, both experimental and theoretical, have an impact on the classical hallmarks of cancer and inflammation. It is even pointed out how these biophysical approaches contribute to the understanding of the regulation of cancer disease and inflammatory responses after tissue injury through physical microenvironmental property sensing mechanisms. The recognized physical signals are transduced into biochemical signaling events that guide cellular responses, such as malignant tumor progression, after the transition of cancer cells from an epithelial to a mesenchymal phenotype or an inflammatory response due to tissue injury. Moreover, cell adaptation to mechanical alterations, in particular the understanding of mechano-coupling and mechano-regulating functions in cell invasion, appears as an important step in cancer progression and inflammatory response to injuries.
This may lead to novel insights into cancer disease and inflammatory diseases and will overcome classical views on cancer and inflammation. In addition, this review will discuss how the physics of cancer and inflammation can help to reveal whether cancer cells will invade connective tissue and metastasize or how leukocytes extravasate and migrate through the tissue.
In this review, the physical concepts of cancer progression, including the tissue basement membrane a cancer cell is crossing, its invasion and transendothelial migration as well as the basic physical concepts of inflammatory processes and the cellular responses to the mechanical stress of the microenvironment such as external forces and matrix stiffness, are presented and discussed. In conclusion, this review will finally show how physical measurements can improve classical approaches that investigate cancer and inflammatory diseases, and how these physical insights can be integrated into classical tumor biological approaches.
Export citation and abstract BibTeX RIS
1. Introduction
During the last twenty-five years, many aspects of the field of classical tumor biology have been investigated and, hence, in the year 2000, six 'classical' hallmarks were proposed, such as sustaining proliferative signaling, evading growth suppressors, activating invasion and metastasis, enabling replicative immortality, inducing angiogenesis and resisting cell death, in order to describe the process of cancer in a more detailed and precise mode (Hanahan and Weinberg 2000). How many regulatory circuits must a cancer-candidate cell deregulate to be cancerous? This question cannot be fully answered as the six hallmarks of the first set of regulatory circuits have been found to be insufficient in describing all types of cancer, and thus this first set was refined. Eleven years later, two additional hallmarks were introduced, namely the usage of abnormal metabolic pathways and the evasion of the immune system, to refine their number of principles for classification (Hanahan and Weinberg 2011). This important addition of the seventh and eighth hallmarks of cancer showed that the immune system was now regarded as being more important in malignant cancer progression.
Many molecules important for cancer cell motility and invasion, such as α6β4, αvβ3, αvβ5, α5β1, E-cadherin, Notch1-4 receptors, CXCR2 and CXCR4, have been reported to play a role in cancer disease (Gong et al [108], Bauer et al 2007, Mierke et al 2008a, 2008b, 2008c, Sawada et al 2008, Gilcrease et al 2009, Ricono et al 2009, Teicher and Fricker 2010). Despite all of these findings based on novel molecular or biochemical approaches such as genomics and proteomics, these novel approaches did not fundamentally change clinical outcomes in the field of cancer research. As the first enthusiastic hype passed, these approaches based on biological technologies have not yet reached the proposed goals, but they have led to significant insights into fundamental cancer biology, cancer diagnosis and prognosis. In particular, the classification of tumors, numerous marker proteins for certain cancer types and the detailed mapping of specific human cancer types have been started. The main criticism of these novel approaches remains, which is the variation of the gene and protein expression levels that are differently regulated during the progression of cancer, depending on the cancer disease stage. Thus it is still elusive how they contribute to or regulate the progression of cancer and to what extent. In more detail, genomic- and proteomic-based analysis methods ignore the spatial localization of the proteins in special compartments such as lipid rafts (Runz et al 2008), their state of activation and assembly, and finally their lifetime, turnover rate, modification rate and recycling rate (Veiga et al 1997, Garcia et al 1998, Caswell et al 2008, Gu et al 2011, Liu et al 2011).
As current classical biological and biochemical approaches have not yet captured the full complexity of the cancer disease and have failed, particularly, to give more insight into the malignant progression of cancer disease, cancer research–adapted classical physical approaches and novel biophysical methods have been developed to be suitable for usage in the field of cancer research. Until now, these new directions of physical-based cancer research have pronouncedly changed the field of current cancer research and have broken down the classical biological and biochemical view on cancer disease. As cancer disease is associated with inflammation, the physical view has also started to be adapted to inflammatory diseases. Here, the focus is on inflammation related to cancer or after tissue injury. In addition to solid tumors, also 'soft' leukemias will be addressed to compare them with cancer cells of epithelial origin.
As there is still a hallmark missing that includes the mechanical properties of cancer cells, this review will focus on the effect of the physical properties of cancer cells, their physical microenvironment such as the extracellular matrix, and on neighboring cells such as endothelial cells, cancer-associated macrophages and fibroblasts. All these processes or circuits will be presented from a physical point of view and, thus, break down the classical eight-hallmarks-based view on cancer.
1.1. Overview of the biological background on cell transmigration and invasion
Whether a neoplasm is benign or malign depends on several parameters that are influenced and regulated in complex networks depending on the environmental conditions (regulatory units). Until now, this question could not be answered successfully and not even in a basic way as many of the parameters have not yet been described and even the regulation of these parameters has not been fully discovered yet.
The malignant progression of cancer involves the process of cancer metastasis, and hence is the worst-case scenario in cancer disease as it is responsible for the main cause of cancer deaths. The process of metastasis includes many consecutive steps, each of which is regulated in a precise way, including many positive or negative regulatory units. The onset of metastasis starts even in the primary tumor, where some cancer cells gain the ability to be altered in a special fashion, and thus they become able to weaken their cell–cell adhesions, remodel their cell–matrix adhesions, get a highly migratory phenotype and subsequently may be able to migrate through the other basal cells of the primary tumor and the tumor basal membrane and invade into the tumor microenvironment (figure 1). As the microenvironment changes the ability of protrusion formation, it has been suggested that breaks in the basement membrane can facilitate the invasion and dissemination of cancer cells upon direct contact with collagen I (Nguygen-Ngoc et al 2012). From a broader point of view, cancer metastasis starts with the spreading of cancer cells from the primary tumor, which migrate into the local tumor microenvironment in a certain way that is dependent on genetic and molecular parameters that are mutated or deregulated in a special way. This highly invasive subgroup of cancer cells transmigrate into blood or lymph vessels (intravasation), get transported through the vessel flow to the target region, adhere to the endothelial cell lining of the vessels, grow within the vessel (Al-Mehdi et al 2000, Elzarrad et al 2008) and form a secondary tumor inside the vessel, or they possibly transmigrate through the endothelial vessel lining (extravasation) into the extracellular matrix of connective tissue. At their target region, this cancer cell subtype migrates further into the targeted tissue, grows and forms a secondary tumor, which means that the tumor metastasizes (figure 2). In addition, cancer cells can escape from this metastasis pathway for a certain time by staying in the vascular niche as dormant cancer cells (Ghajar et al 2013). In particular, glycoprotein interactions of circulating cancer cells and the endothelial vessel linings regulate cancer cell adhesion during hydrodynamic shear stress through adapting the glycomechanics of relevant glycoproteins such as Muc-1 (Geng et al 2012, Mitchell and King 2014). How cancer cells manage to get out of the primary tumor is currently under investigation. In addition, what role the other non-metastatic cancer cells in a primary tumor play seems to be elusive. How the target region of cancer metastasis is defined for a particular cancer type is purely hypothetical, as it is supposed that the aggressive cancer cells of the primary tumor choose mechanically comparable regions of the human body (regarding stiffness) as the targeted tissue. This hypothesis is not yet proven by an experimental approach. Why cancer cells meet blood or lymph vessels is unclear. However, it has been suggested that cancer cells sense endothelial cells probably through a gradient of substances released by endothelial cells, such as chemokines, cytokines or extracellular matrix proteins such as endothelial fibronectin. Several tumor endothelial associated markers have been identified (Al-Mehdi et al 2000), but still there seems to be no common path used by all the different kinds on cancer cell types. What role immunoregulatory cells such as macrophages and T-lymphocytes in the primary tumor play seems to be elusive and should be investigated more thoroughly.
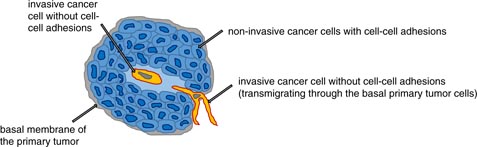
Figure 1. The initial step for the selection of an invasive cancer cell subtype. An invasive cancer cell loses its cell–cell adhesion and migrates through the primary tumor and the basement membrane into the connective tissue microenvironment.
Download figure:
Standard image High-resolution image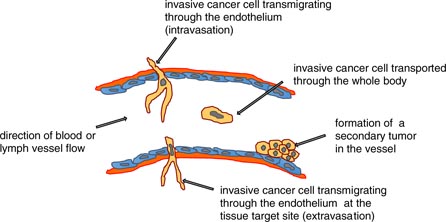
Figure 2. The transendothelial migration step of cancer cells during metastasis. The invasive and aggressive cancer cells transmigrate intercellularly through the basement membrane and the endothelial cell lining in blood or lymph vessels. The cancer cell is then transported by the vessel flow to targeted sites for secondary tumor formation. These sites can be the endothelial cell lining of blood or lymph vessels, or the invasive cancer cells can transmigrate through the endothelium and the basement membrane in order to invade the connective tissue.
Download figure:
Standard image High-resolution image1.1.1. Physical invasion strategies of cancer cells derived from solid tumors.
The invasiveness of cancer cells seems to depend on the molecular, biochemical and biophysical properties of the cells that enable them to migrate through a dense extracellular matrix network. The prerequisites for cancer cell invasion seem to depend on the dimensionality of the migration assay used. However, the ability of cancer cells of epithelial origin to migrate into three-dimensional (3D) extracellular matrices of connective tissue is not determined by a single parameter; instead, it rather depends on the balance between certain biochemical and mechanical parameters regulating the invasion velocity of cancer cells through dense 3D extracellular matrices with pore sizes of around 2 µm. Among these balanced parameters are certain properties of highly invasive cancer cells, namely (i) cell adhesion and de-adhesion dynamics (the turnover of focal adhesions and the adhesion strength), (ii) cytoskeletal remodeling dynamics, cell fluidity and cell stiffness, (iii) matrix remodeling by the secretion of extracellular matrix proteins and digestion through matrix-degrading enzymes, and (iv) the generation and transmission of protrusive (contractile) forces (figure 3) (Friedl and Brocker 2000, Webb et al 2004, Mierke et al 2008c, 2011). Each of these parameters cannot be treated as a single parameter; rather it must be related to the other parameters to reveal how strong the particular effect of a certain parameter on cell invasion and transendothelial migration is. Taken together, the balance between these invasion-regulating parameters is crucial for the efficiency of cancer cell invasion, the velocity of cell invasion and the invasion depths into 3D extracellular matrices (Zaman et al 2006). These invasiveness-determining parameters can vary depending on the cancer cell type or even be shifted towards a single parameter, but still these parameters all play a role in determining cancer cell invasiveness and invasion efficiency.
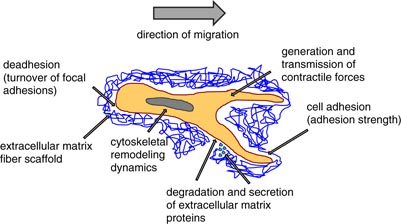
Figure 3. 3D cell motility. Cell invasion in a 3D microenvironment requires a balance of at least four mechanical properties of cells that facilitate their invasiveness through a dense extracellular matrix network. These four mechanical parameters are (i) cell adhesion and de-adhesion, (ii) cytoskeletal remodeling dynamics, cellular fluidity and cellular stiffness, (iii) matrix remodeling by secretion or degradation of extracellular matrix proteins and (iv) the generation and transmission of contractile forces.
Download figure:
Standard image High-resolution imageFor epithelial-originated cancer cells, a disruption of the balance between these parameters can switch the invasion mode from epithelial to mesenchymal migration, or even to amoeboid migration with or without traction forces. Whether this reversible transition is always fulfilled totally and holds true for all types of cancer cells is still elusive. Finally, the invasion mode of cancer cells is determined by these biomechanical and biochemical parameters. How cancer cells manage to keep the balance of these parameters is still not known. What role the tumor microenvironment such as growth factors, cytokines, chemokines, matrix-protein composition, structure and concentration, and matrix mechanical stiffness plays is not fully clear and thus is under strong investigation (Steeg 2006).
There are currently two different mechanisms under investigation. The first mechanism is the mechanical degradation of the dense 3D extracellular matrix through the secretion of matrix-metalloproteinases (MMPs) in order to facilitate cancer cell invasion (Wolf et al 2003, 2007, Friedl and Wolf 2009). Thus, the steric hindrance of the 3D extracellular matrix can be broken down and overcome by invasive cancer cells. The second mechanism is the cutting of cell–cell adhesion molecules such as NOTCH receptors, ephrins or E-cadherins from the cell surface of cancer cells by their own sheddases such as the secretases ADAM-10 and ADAM-17 (Brou et al 2000, Li et al 2008, Itoh et al 2008, Bozkulak and Weinmaster 2009, Riedle et al 2009, Singh et al 2009, van Tetering et al 2009). However, this decrease of cell–cell adhesions may facilitate signal transduction processes leading to the nuclear translocation (together with a transcription factor, LEF1/TCF4) of cell–cell-adhesion proteins such as β-catenin and the induction of gene expression supporting cell motility in order to increase cancer cell invasiveness. The enzymatic degradation of proteins building cell–cell adhesion changes the cell–cell adhesion force and subsequently also the mechanical properties such as stiffness or cytoskeletal remodeling dynamics. In addition, the sheddase ADAM-17 can cleave pro-TNF-α exposed on the cell surface of cancer cells and hence release TNF-α into the tumor microenvironment (Black et al 1997). The release of TNF-α may then activate nearby endothelial cells, which are then pre-stimulated to facilitate the transmigration of cancer cells.
Despite the shedding of membrane receptors, another mechanical parameter is involved in the regulation of the invasion speed and the invasion depth of cancer cells in dense 3D extracellular matrices, which is the physical property of highly invasive cancer cells to generate and transmit contractile forces (Mierke et al 2008a, Rösel et al 2008). Biophysical measurements of contractile forces in 3D collagen or fibrin matrices have recently been described (Bloom et al 2008, Legant et al 2010, Gjorevski and Nelson 2012, Koch et al 2012). In particular, the invasiveness of cancer cells can be analyzed by recording z-stack images using a confocal scanning microscope and a 3D cell-tracking program. Certain approaches use matrix embedded beads as displacement markers for forces exerted by the invasive cancer cells. However, other approaches use the collagen fiber structure itself to detect alterations due to force application by invasive cancer cells. Taken together, the tracking of collagen fibers is more sophisticated, but also more reliable. Moreover, the analysis of collagen fibers as markers is preferable compared to the usage of beads as markers, because the effect of marker phagocytosis or digestion is then nearly vanished, as only a minor part of collagen fibers is digested and internalized compared to the huge number of embedded bead-markers that are phagocytized in close proximity to invasive cancer cells. In addition, bead internalization also affects the mechanical properties of cancer cells such as stiffness, and, subsequently, it decreases the invasiveness of highly invasive cancer cells significantly in dense 3D extracellular matrices (Mierke 2013a).
1.1.2. Physical invasion strategies of cancer cells derived from leukemias or fibroblasts.
Despite many open questions regarding cell migration, a reliable conceptual scaffold can be developed for how a lamellipodium can support cell motility. In particular, actin filaments polymerizing below the leading edge of the cell membrane generate the pushing force (protrusion force) required for the formation of protrusions. The surface tension of the cell membrane can oppose the free anterograde expansion (outwardly) of the actin network and hence the actin filaments are pushed back into the cytoplasm of the cell, which is detectable as a retrograde (inwardly) actin flow (figure 4). In more detail, the cell–matrix adhesion receptors such as integrins couple the cytoskeleton to the external substrate through focal adhesions that ensure that these retrograde-directed forces, which are enforced by actomyosin contraction, are transformed into outward locomotion of the cell's body in the migration directions (Vicente-Manzanares et al 2009). Taken together, a basic mechanical concept of the lamellipodium is that actin polymerization facilitates the protrusion formation of the cell membrane. In addition, despite these lamellipodium-supported outward forces, protrusions from a kind of filopodia are also needed for cell motility, which are suggested to be more sensing than force generating, and invadosomes, which are needed for 3D invasion of tissue barriers (Ridley 2011). One alternative way to migrate without actin protrusion forces is the migration through membrane blebbing. These blebs are anterior cellular extensions without actin filaments. Moreover, the intracellular hydrostatic pressure generated by actomyosin contraction causes the rupture of the actin cortex (Tinevez et al 2009) and/or the focal adhesion protein-mediated linkage between actin cytoskeleton and the cell membrane (Charras et al 2006). As the membrane loses its mechanical anchorage, the intracellular pressure leads to the formation of a membrane bleb that grows until a new actin cortex is reassembled, which then may also contract, repeating the blebbing cycle (Charras and Paluch 2008). In summary, blebbing is a physiologically relevant locomotion strategy, in which the cells migrate by a directed and persistent motion (Blaser et al 2006). Moreover, the blebbing of the membrane may change the membrane tension and subsequently the mechanical properties of the whole cell. In tissue and cell cultures, several cells can switch between blebbing and polymerization-driven motility mode due to the microenvironmental conditions, in response to genetic alterations or pharmacological drugs (Lämmermann and Sixt 2009, Diz-Muñoz et al 2010, Poincloux et al 2011). At least three main points are of special interest for the analysis of the blebbing mode migration compared to the lamellipodial mode migration, such as the effect of the cell–substrate adhesion, substrate geometry and the regulation of the signaling processes between the front and the rear of a cell. Both motility modes, the blebbing and the lamellipodial migration, deal with stabilized cell–cell or cell–matrix adhesions (Renkawitz and Sixt 2010). However, the stability and physiological importance of adhesions seem to be reduced in fast-migrating amoeboid cells such as immune cells, which can migrate in the blebbing and in the lamellipodial mode (figure 5) (Lämmermann and Sixt 2009). The slow mesenchymal movement, which relies completely on focalized cell–matrix adhesions, is solely lamellipodial. The dimensionality of the microenvironment can have a broad impact on the motility of cells, and there is evidence that all migration modes can occur on 2D and in 3D microenvironments; however, a large difference is that 3D, but not 2D, microenvironments support cellular motility under minimal adhesion forces and decreased adhesion strength (Friedl and Wolf 2010). How cell polarity is involved in cell motility in 2D and 3D migration systems and whether it is connected to the protrusion type is still elusive. It has been suggested that Rho GTPases are major regulatory proteins such as Rac-1, Cdc42 and RhoA. In particular, Rac-1 is necessary for the expansion of the lamellipodium by activating the WAVE complex that then triggers the nucleation of actin by the Arp2/3 complex (Steffen et al 2006, Wu et al 2012). In addition, Cdc42 activates formins as well as the Arp2/3 complex and hence promotes actin polymerization during the assembly of filopodia and invadosomes. Moreover, RhoA can alter the actomyosin contractility directly by regulating myosin II and formins (Ridley 2006). Taken together, the Rho GTPases play an important role in protrusion-driven migration that depends on the cell type and the particular microenvironment supporting or inhibiting the cellular polarity. Finally, the coupling between actin polymerization, actomyosin contractility and cell–matrix adhesion affects the Rho GTPases that then help the migrating cells to sense their microenvironment biochemically and mechanically. In addition, Rho GTPases can help to adapt the migration mode to the geometry, chemical composition and mechanical properties of its surroundings (Millius et al 2009, Vicente-Manzanares et al 2009). Recently, it has been reported that cells migrate by forming cylindrical-shaped lobopodia with protrusions containing multiple and small blebs at their ends, which seems to be an intermediate invasion mode between the blebbing and lamellipodial modes (Petrie et al 2012). In 2D migration systems, the cells have lamellipodia, and when seeded into 3D collagen gels (of non-cross-linked bovine collagen) these cells formed invadopodia. However, when the collagen fibers can be crosslinked, the cells migrate in a lobopodia mode, in which they can sense the mechanical properties such as the elastic properties of the matrix scaffold. It has been hypothesized that the low stiffness of the 3D matrix facilitates the lobopodial migration mode, whereas the actomyosin contraction may automatically activate the deforming migration mode, where cells squeeze through the pores of the matrix (figure 5). However, it turned out that the stiffness of the microenvironment is not the major factor in the lobopodial-based migration—it is rather the shape of the stress–strain curve, as only lobopodia are formed in linearly elastic microenvironments such as the skin and the cell-derived matrix, but not in non-cross-linked 3D collagen matrices showing strain stiffening, where lamellipodia can be formed predominantly (Petrie et al 2012). This is in contrast to the work on the matrix stiffness that drives tumor invasion based on lamellipodia/invadopodia-driven cell invasion (Levental et al 2009, Alcaraz et al 2011, Pathak and Kumar 2013, Mouw et al 2014). There are still open questions regarding the fact of the stabilized functional polarity of cells that mediate persistent directional migration. How can cells detect linear elasticity and distinguish it from strain stiffening? How is the extracellular proteolysis connected or associated with lobopodial migration? Do cells migrating in a lobopodial mode secrete extracellular matrix proteins to remodel their microenvironment? Can other cells as fibroblasts use this lobopodial mode of cell migration?
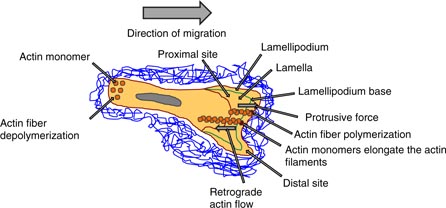
Figure 4. The physical invasion strategy. The migrating cells have a lamellipodium (2D)/invadopodium (3D) in the direction of migration and a lamella, which is underneath the lamellipodium and is separated by the lamellipodium base. The cell polymerizes actin stress fibers in the leading edge (lamellipodium) that produces a protrusive force (outwardly) towards the membrane, which pushes the cell forward in the direction of movement. Due to this anterograde expansion, the actin filaments are pushed back into the cytoplasm (inwardly) as a retrograde flow.
Download figure:
Standard image High-resolution image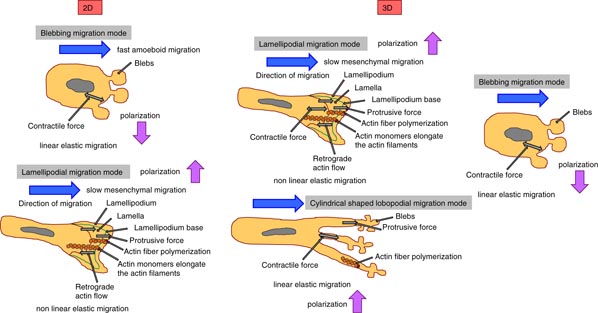
Figure 5. Three modes of cell migration in 2D and 3D microenvironments. In the 2D migration systems, the cancer cells and the immune cells can migrate in a blebbing migration and in a lamellipodial migration mode. In a 3D migration system, cancer cells and immune cells migrate in lamellipodial migration mode, blebbing migration mode and cylindrical-shaped lobopodial migration mode. The polarization and the velocity of the migratory cells depend on the migration mode.
Download figure:
Standard image High-resolution imageCompared to epithelial-originated cancer cells, leukocytes are outstanding cells, as they are scattered throughout the whole body and have the ability to migrate into any type of tissue. This behavior is definitely needed during an inflammatory response. These infiltrating leukocytes are not restricted to certain motility paths or hindered to cross the compartment boundaries. Instead, leukocytes can undergo single cell migration with high velocities that are even up to 100 times faster compared to the migration speed of mesenchymal and epithelial cell types (Friedl 2004, Pittet and Mempel 2008). How can leukocytes migrate with such a high speed through the tissue? How do leukocytes manage to undergo frequent shape changes and migrate in an amoeboid migration mode? Is the amoeboid migration always faster compared to other migration modes such as mesenchymal or lobodial migration? Indeed, it has been reported that adhesion-independent migration is driven by the protrusive flowing of the anterior actin network of the cell and supported by the squeezing induced by actomyosin contractions of the trailing edge in order to propel the rigid nucleus of the cell through narrow spaces of the matrix.
1.2. Comparison of 2D and 3D motility assays
Migration experiments have been largely performed on stiff substrates such as plastic cell-culture treated surfaces or negatively charged glass surfaces. The mode of migration has been determined in a purely 2D microenvironment, neglecting the third dimension available in tissues. In the last 30 years, the physical and molecular mechanisms regulating the motility of normal healthy cells and highly invasive cancer cells have been studied in in vitro assays using 2D surface-coated substrates (Lauffenburger and Horwitz 1996, Pollard and Borisy 2003, Ridley et al 2003). Recently, we have reported that the dimensionality of the cell-culture system used to study cell invasion cannot be ignored as it plays a key role for the mode of cellular migration and the migration efficiency (Mierke et al 2010, 2011). The 3D microenvironment of the extracellular matrix in vivo and in vitro is characterized by certain structural features, such as the pore size, connection points and fiber or bundle orientation, all of which are not found in extracellular matrix protein–coated 2D substrates, but which clearly impact the mechanical properties of the extracellular matrix and, thereby, the mechanical properties of the invading cells (Sabeh et al 2009). Certain features of the microenvironment seem to be crucial for 2D motility, such as focal adhesions, actin stress fibers, broad lamellipodia, filopodial protrusions at the leading edge and apical polarization. However, in 3D extracellular matrix–based invasion assays, these parameters are pronouncedly reduced in size or are even extremely missing in invasive cancer cells (Wozniak et al 2003, Zaman et al 2006, Doyle et al 2009, Yamazaki et al 2009, Fraley et al 2010). Instead, there are other features such as invadopodia of cells invading 3D matrices described. How these 2D structures, adhesion and migration modes can be transferred to the 3D situation has to be further revealed.
In addition, certain cellular features such as the expression of the mechano-regulatory protein vinculin, which plays an important role in facilitating 3D cell motility but plays the opposite role in 2D cell motility as it inhibits migration (Mierke et al 2010). Similarly, it was recently suggested that focal adhesions, composed of clustered integrins that physically and dynamically connect the cellular actin-myosin cytoskeleton to the extracellular matrix proteins on 2D substrates, are altered when the cells are embedded or located inside a 3D extracellular matrix (Fraley et al 2010). As a cell (20–120 µm) is much larger than the diameter of a single fibril (100 nm) of the 3D extracellular matrix, from a cellular perspective, single collagen fibrils of the extracellular matrix may appear to be one-dimensional. Moreover, this may omit the formation of large focal adhesions in 3D collagen fiber networks. However, focal adhesions of cells cultured on 2D substrates are normally of 1–10 µm in size and, hence, quite a lot larger than the diameter of a fibril of the extracellular matrix (Wehrle-Haller and Imhof 2002, Geiger et al 2009, Parsons et al 2010). In more detail, this finding may restrict the size of focal adhesions in 3D extracellular matrices and subsequently also the associated clusters of integrins and the number of focal adhesion proteins such as vinculin or focal adhesion kinase (FAK). However, it has been seen that in vitro and in vivo matrices' collagen fibers (8 µm in diameter) are composed of groups of fibrils or even larger bundles of fibrils, which have larger diameters similar in size to the focal adhesion size and hence may serve as proper focal adhesion points for invasive cancer cells. Moreover, this suggestion is supported by the findings that cancer cells can invade these 3D extracellular matrices, form focal adhesions in them, as well as transmit and generate contractile forces in this 3D microenvironment (Koch et al 2012).
Furthermore, cells in vivo can initiate the bundling of collagen fibers through the generation of contractile forces evoked by their cellular protrusions. In particular, these collagen bundles increase the surface area available for cell adhesion and may pronouncedly induce the assembly of larger focal adhesions (Smith et al 2007). In 2D motility systems, actin stress fibers play an important role by providing the amount of contractile forces needed for the regulated detachment of the cell's rear from the 2D substratum and the establishment of the actin flow at its leading edge (Sun et al 2010, Parsons et al 2012). It has been suggested that cells display fewer stress fibers inside 3D extracellular matrices compared to extracellular matrix protein–coated 2D surfaces. Indeed, in 3D motility systems, the stress fibers are either localized to the cell cortex (cortical action network) or radiate from the nucleus towards the plasma membrane to form pseudopodial protrusions (Bloom et al 2008). Additionally, this finding is supported by an inhibition experiment, where the actomyosin contractility is blocked in 2D and 3D motility systems. Indeed, the inhibition of the contractility is often substantially less effective in blocking 3D cell motility compared to 2D cell motility (Bloom et al 2008). These results suggest that the functional role of actin stress fibers depends on the dimensionality (Shih and Yamada 2010, Sun et al 2010). In contrast to these findings, we have found that the 3D cell invasion of certain highly invasive cancer cell lines can be inhibited by using inhibitors which block actomyosin-dependent contractility such as the myosin light chain kinase inhibitor (ML-7), the Rho-kinase inhibitor (Y27632) or latrunculin A (an actin-polymerizing inhibitor) (Mierke et al 2011, Mierke 2011b, 2013c). When eliminating the apical polarization of cells in 2D culture systems, the number of focal adhesions and stress fibers is reduced, and, hence, the role of focal adhesion proteins such as vinculin or FAK is fundamentally altered, and additionally, certain proteins are highly enriched in actin stress fibers, such as α-actinin or myosin II (Rehfeldt et al 2012). In addition to fewer focal adhesions and actin stress fibers in 3D extracellular matrices, cancer cells, epithelial cells or endothelial cells inside a 3D microenvironment do not form a wide lamellipodium with associated filopodial protrusions at the periphery; instead, these cells form a limited number of pseudopodial protrusions of 10–20 µm in thickness (Fraley et al 2010). This is consistent with the view that traction microscopy results lead to the suggestion that in a 2D cell-culture system, a lamellipodium actively pulls the rear of the cell through nascent focal adhesions positioned at the edge of the lamellipodium (Beningo et al 2001). Additionally, it has been reported that the primary protrusions built up from the cell body depend on FAK, talin and p130Cas, whereas secondary protrusions emanating from primary protrusions depend on Arp2/3 complex, N-WASP, WAVE1, cortactin and Cdc42 (Giri et al 2013). These protrusions play a role only in a 3D microenvironment, not in 1D or 2D migration systems, facilitating cell invasion and matrix deformation (Giri et al 2013).
Accordingly, 3D traction microscopy reveals that cells inside 3D extracellular matrices pull on the surrounding fibers of this matrix scaffold (Bloom et al 2008, Legant et al 2010). At active pseudopodial protrusions, high matrix tractions occur (Bloom et al 2008), which pull with nearly equal forces at the leading and trailing edges of the cell. As the release of pseudopodia towards the matrix collagen fibers is asymmetric, this leads to a structural defect within the collagen fiber matrix scaffold at the rear of a migrating cell. These results lead to the suggestion that pseudopodial protrusions at the trailing edge of the migrating cell are released first and pull the rear of the cell forwards through the 3D microenvironment, leading to a guided, persistent motion in a migration tunnel (Even-Ram and Yamada 2005, Lämmermann et al 2008). In contrast to a 3D microenvironment, the motion of cells in 2D is less persistent as there is no tunnel build up by the migrating cells (Doyle et al 2009), only some obstacles are secreted by the migrating cells to mark their migration path and possibly to attract chemically other following migrating cells. Finally, pseudopodia have a solely probing role in 3D extracellular matrices but are of no functional importance on 2D substrates, which are compositionally and topologically more uniform compared to 3D matrices. In addition, the pseudopodial protrusion activity in 3D extracellular matrices is modulated by focal adhesion components such as p130CAS and zyxin. For example, the migration speed of p130CAS-knock-out and zyxin-knock-out cells has been correlated with the number of protrusions generated per unit of time in 3D extracellular matrices (Fraley et al 2010). The p130CAS-knock-out cells move more slowly and the zyxin-knock-out cells move more rapidly compared to their control wild-type cells in 3D extracellular matrices, whereas the p130CAS-knock-out and zyxin knock-out cells exhibit the opposite motility phenotypes on flat 2D substrates. These results support the hypothesis that 2D substrates are not well suited for studying cellular motility, as it is not always comparable to the 3D motility situation. In addition, to further support this, vinculin knock-out cells showed a similar behavior to p130CAS-knock-out cells as their motility is the opposite in 3D extracellular matrices compared to 2D substrates (Goldberg et al 2003, Mierke et al 2008a, 2008b, 2008c, 2010, Mierke 2009). Taken together, the role of focal adhesion proteins in 2D motility systems seems not to be predictive for their migratory behavior in the more physiologically relevant 3D extracellular matrices. Moreover, there is even more different behavior depending on the dimensionality: the rate of filopodial protrusions does correlate with the 2D migration speed, whereas the rate of pseudopodial protrusions seems not to be correlated with the 3D invasion speed (Fraley et al 2010). These results lead to the suggestion that the protrusion dynamics are not required for 2D motility, but are crucial in 3D motility. Instead, other criteria of the protrusions such as their activity and their matrix-degrading properties may play a role in 3D invasion systems (Fraley et al 2010).
Recently, another type of invasion mode has been described for cancer cells with a soft cytoskeleton. This invasion mode is distinct from the mesenchymal and amoeboid migration. These soft cancer cells use a pulsating migration mode, in which slow, random migration dominates for a long time and is then interrupted by short fast, directed migration (Lee et al 2012). In particular, the soft cancer cells are surrounded by relatively stiff normal cells, and hence they migrate slowly and over only a limited, small distance (Lee et al 2012). The fast migration mode seems to be induced by myosin II–dependent deformation of the soft nucleus of cancer cells, which can be induced by the transient crowding of the surrounding non-transformed normal cells with stiff nuclei (Lee et al 2012). Moreover, the surrounding stiffer normal cells are able to move, because they have lost cell–cell adhesions due to cadherin-facilitated mismatch adhesion between normal cells and cancer cells; however, their movement is limited by the residual α-catenin-mediated cell–cell adhesions between neighboring normal cells. These findings imply that the enhanced pulsating migration of cancer cells is facilitated by the mechanical and adhesive mismatch between transformed cancer cells and non-transformed normal cells (Lee et al 2012).
1.3. Principles of cancer disease progression
The first principle of cancer disease progression is that the primary tumor reached a certain critical size, where it needs a blood supply in order to be able to grow further. Due to fewer vessels within a tumor and their vessel malfunction, tumors are usually hypoxic and nutrient-deprived. These hypoxic conditions affect gene expression and, thus, can alter the mechanical properties of these cancer cells. In addition, these conditions are supposed to facilitate cancer cell progression as these milieus may support the selection of an aggressive and highly invasive cancer cell subtype, which is able to metastasize in targeted organs. Recently, it has been reported that this particular milieu can lead to disease progression and resistance to treatment (Carmeliet and Jain 2011). Traditional anti-angiogenesis strategies to reduce the tumor vascular supply failed so far, as this treatment is restricted by insufficient efficacy and the development of resistance. Instead, initial clinical evidence revealed that the normalization of the vascular abnormalities is even better suited to treat cancer progression (Carmeliet and Jain 2011). The entry of cancer cells in the vascular system is an important step for metastasis in targeted tissue, as the cancer cells are now able to reach the targeted regions easily through the vessel flow (Al-Mehdi et al 2000). Another key point is that the tumor vessels are heterogeneous and may contain cancer cells mimicking the endothelial lining of the vessels together with endothelial cells, which are certainly altered when entering the primary tumor (Ghanekar et al 2013). The question remains whether the primary tumor must have a certain size to be able to send out highly invasive cancer cells that finally form a metastasis.
The second principle is that out of the primary tumor, highly invasive cancer cells are selected in order to migrate into the tumor microenvironment and finally build up metastases in targeted organs. These highly invasive cancer cells possess altered mechanical properties that enable them to invade the microenvironment (Mierke et al 2011). The activation of Notch-1 is associated with the development and progression of human malignancies including pancreatic cancer, which is the most lethal and is associated with a poor prognosis (Jemal et al 2010). Notch signaling has been proposed to play an important role in cell proliferation and apoptosis (Wang et al 2008). The proteins encoded by Notch genes such as Notch-1, -2, -3 and -4 are activated by interacting with their ligands such as Dll-1 (Delta-like 1), Dll-3 (Delta-like 3), Dll-4 (Delta-like 4), Jagged-1 and Jagged-2 (Miele 2006, Miele et al 2006). The Notch receptors possess small differences in their extracellular and cytoplasmic domains, but are highly similar in their structures and are connected to the cell's actomyosin cytoskeleton that determines cellular mechanics. In particular, the cytoplasmic region of Notch contains a recombination of the signal-binding protein 1 for the J-kappa (RBP-J)-association molecule (RAM) domain, ankyrin repeats, nuclear localization signals (NLS), a trans-activation domain (TAD) and a region rich in the proline, glutamine, serine and threonine residues (PEST) sequence (Okuyama et al 2008). The Notch signaling is initially activated by a receptor–ligand interaction between two neighboring cells. Upon its activation through a receptor–ligand binding, Notch is cleaved to release the intracellular domain of the Notch (ICN) through a cascade of proteolytic cleavages by the metalloprotease TNF-α-converting enzyme (TACE) and the γ-secretase. In more detail, the released ICN is then translocated into the nucleus for the transcriptional activation of Notch target genes such as the hairy and enhancer of split-1 (Hes-1), NF-κ B, cyclin D1 and c-Myc (Miele and Osborne 1999, Miele 2006, Miele et al 2006, Okuyama et al 2008, Wang et al 2008), which can either signal in multiple tissues or only in specific target tissues. These results show that the cell–cell adhesion signaling impacts gene expression and then thus supposes that the mechanical properties of cancer cells such as the cellular stiffness or cytoskeletal remodeling dynamics, and subsequently also the cellular motility, are altered. In addition, it is suggested that the acquisition of an epithelial–mesenchymal transition (EMT) phenotype and the induction of a cancer stem cell (CSC) or cancer stem-cell-like phenotype are interrelated and contribute to tumor recurrence and possibly drug resistance (Sarkar et al 2009, Grudzien et al 2010, Singh and Settleman 2010, Wang et al 2010). An EMT is associated with drastic alterations in the mechanical phenotype, as the actomyosin cytoskeleton and intermediate cytoskeleton, as well as the cell–cell adhesive junctions, are reduced to promote cell migration and invasion. In addition, the expression of the matrix-metallo-proteinase MT1-MMP is increased in cells that underwent an EMT (Yang et al 2013). This MMP has been found to increase cancer cell invasion into 3D collagen scaffolds (Woskowicz et al 2013). However, the molecular mechanism by which Notch-1 contributes to the acquisition of the EMT phenotype and the CSC self-renewal capacity is not yet fully understood. Several studies have shown that the Notch gene is abnormally activated in many human malignancies such as cervical, lung, colon, head and neck, renal carcinoma, acute myeloid lymphomas and pancreatic cancer (Shou et al 2001, Sriuranpong et al 2001, Miyamoto et al 2003, Miele 2006, Miele et al 2006, Bolos et al 2007, Aster et al 2008). In more detail, a higher expression of Notch-1 and its ligand Jagged-1 is associated with poor prognosis in breast and prostate cancer (Reedijk et al 2005). In line with this, over-expression of Notch-1 leads to the induction of the EMT phenotype by activation of mesenchymal cell markers such as ZEB1, CD44, EpCAM and Hes 1. In particular, CD44 is a transmembrane glycoprotein that can bind to hyaluronic acid and hence increase cancer cell invasion into connective tissue (Cho et al 2012). In addition, over-expression of Notch-1 leads to decreased expression of miR-200b. However, re-expression of miR-200b leads to the reduced expression of ZEB1 and vimentin, but an increased expression of E-cadherin, which switches the mesenchymal phenotype back to the epithelial phenotype (Bao et al 2011). As vimentin is an intermediate filament, the mechanical properties of these cells may be altered and hence their motility is supposed to be decreased. These findings suggest that the activation of the Notch-1 signaling leads to the acquisition of the EMT phenotype, which is in part facilitated through the regulation of miR-200b and the CSC self-renewal capacity, and these processes could be attenuated by genistein (a tyrosine kinase inhibitor). This treatment indicates an involvement of tyrosine kinases such as Rac-1 in cell invasion, as demonstrated for CD44 facilitated cancer cell invasion into 3D extracellular matrices (Bourguignon et al 2000).
However, are the fewer cell–cell adhesions mediated by Notch a prerequisite for the selection of highly invasive and aggressive cancer cells that are able to migrate out of the primary tumor? Does the Notch expression alter the mechanical phenotype of cancer cells in order to increase their aggressiveness and invasiveness?
The third principle is that metastasis results from a process similar to Darwinian evolution involving the natural selection of aggressive and highly invasive cancer cells that are capable of migration and survival at distant targeted sites. In the Darwinian evolution model, the selection of cancer cells exhibiting stable genetic changes occurs. These selected cancer cells are very rare as the selection pressure on them is extremely high and occurs at later stages in tumor progression (Hanahan and Weinberg 2000). Thus, this Darwinian evolution model is criticized, and another model has more impact on cancer metastasis and, hence, cancer progression. The recent development of novel technologies such as high-density microarray-based expression profiling, intravital imaging and the collection of invasive cancer cells from living tumors, has challenged this traditional model of metastasis (Gertler and Condeelis 2011). In particular, these novel technologies have led to new diagnostic and therapeutic markers of metastatic disease. Several studies of mammary tumors in mouse models (Giavazzi et al 1980, Mantovani et al 1981, Milas et al 1983, Wyckoff et al 2004), expression profiling of whole human breast tumors (van't Veer et al 2002, Ramaswamy et al 2003) and the collection and profiling of the invasive subpopulations of cancer cells isolated from rat and mouse mammary tumors (Wang et al 2005a) have suggested that the metastatic ability seems to be acquired in much earlier stages of tumor progression than predicted by the Darwinian evolution model. Thus, the information of the metastatic ability seems to be encoded throughout the bulk of the primary tumor, involving certain transient changes in gene expression. However, these two models can be combined, as after the genetic alterations the microenvironment may also have an impact on the invasive and metastatic phenotype that may even occur early in cancer disease progression (Gertler and Condeelis 2011). The merged 'novel' model is then called the tumor microenvironment invasion model, which states that the tumor microenvironment initiates the expression of genes that induce cell motility, invasion and metastasis (Wang et al 2005a, 2005b, 2007a, 2007b). In particular, the tumor microenvironment invasion model proposes that the oncogenic mutations in cancer cells within the primary tumor change the microenvironment, which then is able to induce cellular motility in cancer cells and stromal cells. The alterations of the tumor microenvironment are in particular an increased microvascular density (Leek and Harris 2002), inflammation (Condeelis and Pollard 2006) and hypoxia (Giraudo et al 2004). These microenvironmental alterations lead to transient and epigenetic changes in the gene expression of cancer cells and stromal cells, similar to the alterations that drive morphogenetic cell movements in a developing embryonic organ such as the EMT. Furthermore, the tumor microenvironment invasion model states that microenvironments causing invasion and metastasis can appear randomly in time and location in the primary tumor, leading to repeated episodes of invasion and systemic tumor cell dissemination during tumor progression (Wang et al 2007a). In line with this model, using intravital imaging of experimental mammary tumors, just a small proportion of cancer cells can migrate that are randomly distributed within the primary tumor and are frequently localized in close proximity to perivascular macrophages (Condeelis and Segall 2003, Wyckoff et al 2007, Kedrin et al 2008). In particular, this model can be supported by the result that micrometastases are genetically heterogeneous, and hence it is suggested that the invasive behavior is not characterized by a stable genetic phenotype (Klein et al 2002). In addition, expression profiling of invasive cancer cells collected from primary mammary tumors revealed an invasion signature (an expression pattern) that involves motility pathways involved in the migratory and chemotactic activity in vivo (Wang et al 2004, 2007a, 2007b, Condeelis et al 2005). Among the highly up-regulated molecules in the invasive mammary cancer cells collected in vivo was Mena (Wang et al 2007b), which was consistent with results stating that high Mena expression levels are associated with poor clinical prognosis for breast cancer patients (Di Modugno et al 2004, 2006). Mena is an actin regulatory protein and influences several of the invasion pathways by controlling the polymerization of actin that is initiated in common by these invasion pathways (Wang et al 2007b, Philippar et al 2008). Finally, Mena has been proposed to be a prognostic biomarker for metastasis (Robinson et al 2009). In more detail, in invasive migratory cancer cells, Mena isoforms that show a heightened sensitivity to epithelial growth factor (EGF) and increased protrusive as well as migratory properties are up-regulated, since other isoforms of Mena are pronouncedly down-regulated (Gertler and Condeelis 2011). In addition, microRNAs have recently been shown to regulate the properties of the microenvironment by driving gene expression in stromal cells that in turn may impact the mechanical properties of the microenvironment (Chou et al 2013). Anomalies of the ECM impact the behavior of stromal cells and may drive the tumor-associated angiogenesis and inflammation that finally alters the normal microenvironment to a tumorigenic microenvironment (Lu et al 2012).
The fourth principle is that several parameters of the cancer cells act together, such as enzymes, molecules of the contractility apparatus and the extracellular matrix structure and composition, to develop a feedback loop in order to regulate their motility in 3D systems. In addition to cell–cell adhesion molecules, the interplay between the growth of pseudopodia along the 1D tracks provided by the collagen fibers, the amount of contractile forces and local enzymatic digestion of the extracellular matrix by MMPs such as MT1-MMP, MMP2, MMP3 or MMP9 has been suggested to be different from 2D substrates due to different shapes of membrane protrusions and the crucial importance of MMPs in 3D invasiveness. 3D culture models are needed to unravel the functional consequences of specific mutations and copy number changes on signal transduction pathways during cancer progression in order to develop potential therapeutic drugs (Weigelt and Bissell 2014). As cellular tractions on collagen fibers may activate MMPs (Ellsmere et al 1999), the interplay between pulling by cell protrusions, the MMP activity and the overall cell migration may be regulated via a feedback loop. However, when the cancer cells migrate without the degradation of the extracellular tissue, and still transmit and generate forces towards collagen fibers, how is cellular motility then regulated? Does the possible feedback loop affect also sheddases that can cut the cell–matrix adhesion receptors from the cancer cells' membrane surface?
1.3.1. Basement membrane barrier crossing.
The basement membrane barrier crossing of cancer cells has not yet been clearly defined and investigated, but is still the focus of mechanical-driven cancer research. How certain cancer cells of a primary tumor are able to migrate out of the primary tumor by crossing the basement membrane of the tumor and thus overcoming the tumor boundaries is still not precisely known. In particular, it is not even known whether these cells come from the inner or outer mass of the tumor. It has only been suggested that the cells in the outermost regions of the tumor have less surface tension (Foty et al 1996). Moreover, it has been suggested that a subgroup of highly invasive cancer cells can manage to weaken their cell–cell adhesions, migrate through the crowd of basal cancer cells, which do not possess the potential to become highly invasive or to form a metastasis, and finally cross the basement membrane of the tumor. How these cancer cells get the ability to down-regulate their cell–cell adhesions and possibly up-regulate their cell–matrix adhesions is still under investigation. This view is supported by the differential adhesion hypothesis formulated by Foty and Steinberg (2005), stating that the tissue-spreading and cellular segregation phenomena during tissue development arise from surface tensions of the tissue evoked by differences in cell–cell adhesiveness. Whether surface tension or other alterations in the mechanical properties of highly invasive cancer cells lead to their ability to migrate out of the primary tumor has to be investigated further.
In particular, cancer cells need to migrate out of the primary tumor in order to transmigrate into blood vessels. First, cancer cells break through the basement membrane, a thin, continuous and dense sheet-like structure composed of a network of collagen IV, proteoglycans and laminin that separates normally epithelial tissues from adjacent connective tissues (Yurchenco 2011). They can then migrate through the stroma composed of fibrillar collagens, proteoglycans and various glycoproteins (Boot-Handford and Tuckwell 2003, Egeblad et al 2010, Naba et al 2012). Furthermore, the crossing of basement membranes by cancer cells is crucial for the onset of metastasis, as it must occur before cancer cells are able to transmigrate into lymphatic or blood vessels (intravasation) and also when they penetrate into the target organ tissue (extravasation), where they will possibly build up secondary tumors. The detection of circulating cancer cells can be correlated with the aggressiveness of the disease, increased metastasis and decreased time to relapse. There is an ongoing discussion whether cancer cells undergo an EMT that leads to more mesenchymal and more stem cell–like cancer cells (Plaks et al 2013).
While some studies describe distinct adhesion structures containing 'adhesome' proteins such as vinculin, paxillin and zyxin in cells migrating in different 3D matrices, others report that adhesome proteins do not form distinct aggregates and can be detected diffusely in the cytoplasm (Cukierman et al 2001, Tamariz and Grinnell 2002, Li et al 2003, Petroll and Ma 2003, Wozniak et al 2003, Martins and Kolega 2006, Provenzano et al 2009, Fraley et al 2010, Deakin and Turner 2011, Hakkinen et al 2011, Kubow and Horwitz 2011). However, these studies used different matrices (cell-derived matrices or collagen I of different concentrations and methods of extraction) and cells that were either fully embedded in 3D matrices or just seeded on top of the matrices (Harunaga and Yamada 2011). Possible explanations for those diverse results may be that first the elasticity of 3D matrices increases by increasing the distance to the rigid glass surface, which may affect the assembly of distinct adhesions (Fraley et al 2011, Harunaga and Yamada 2011). Second, the organization of the 3D matrices can influence the assembly of adhesion structures, as the 3D collagen fibers are too thin for cell adhesions to assemble. In line with this, extensive collagen bundling can increase the width, allowing the assembly of adhesions. In addition, the usage of non-pepsinized or pepsinized collagen may also affect the formation of focal adhesions in 3D culture systems. Third, over-expression of fluorescently labeled adhesome proteins may interfere with the visualization of distinct aggregates, which would be seen when fluorescently labeled adhesome proteins are expressed at low levels (Kubow and Horwitz 2011, Deakin and Turner 2011). However, the mechanical properties and their impact on basement membrane crossing are still elusive. In particular, the impact of mechanical properties on the focal adhesion formation in a 3D microenvironment is under much discussion and looks promising, but has still not been shown.
1.3.2. The integrin-dependent and blebbing mode of cell invasion into connective tissue.
The actual model of cell migration is described as a multistep process. In particular, the F-actin polymerization at the leading edge of a migrating cell pushes out a membrane protrusion that is anchored to an extracellular substrate by the transmembrane receptors of the integrin family and mechanically coupled via focal adhesion proteins to the actomyosin cytoskeleton. The integrins transduce the internal contractile force that is generated when myosin II acts on the actin network by contracting it. The contraction imposes retrograde pulling forces on the integrins, which then facilitate the forward locomotion of the whole cell body (Lauffenburger and Horwitz 1996, Mitchison and Cramer 1996, Giannnone et al 2007). In particular, the integrin composition of each cell type defines the kind of substrate a cell can use for its adhesion-driven mode of cell migration. This linkage between substrate-specific adhesion and cell migration limits the migrating cells to possibly preformed pathways, which seems to be essential for many of the precise cell trafficking and positioning processes underlying the compartmentalization and patterning processes during development and regeneration, and also in cancer disease and inflammatory processes (Hynes and Zhao 2000, Hynes 2002).
Another mode of cell migration is the blebbing mode. In particular, the leading front of a migrating cell can either protrude as an actin-free membrane bleb that is formed by outwardly-directed actomyosin-driven contractile forces, or as an actin-rich pseudopodium, a site where polymerizing actin filaments push the membrane outwardly (Mitchison and Cramer 1996, Mogilner 2006, Charras and Paluch 2008, Lammermann and Sixt 2009, Renkawitz et al 2009). The outward pushing of filaments can cause the membrane to protrude if the expanding actin network experiences a retrograde counter-force, which is usually provided by integrins (Renkawitz et al 2009). In more detail, chemotactic dendritic cells mechanically adapt to the substrates' adhesive properties by switching between integrin-dependent and integrin-independent migration. When engaging the integrin–actin clutch mechanically, the actin polymerization was entirely turned into protrusions; however, on disengagement of integrin–actin connections, actin is able to slip and mediate a retrograde flow (Renkawitz et al 2009). Moreover, accelerated retrograde flow can be balanced by an increased actin polymerization to keep the cell's shape and protrusion velocity constant due to alternating environmental conditions.
Finally, this behavior predicts that in this adaptive response in polymerization dynamics, the migrating cells must not use the tracks of adhesive substrates that may force them to go a certain migration route. However, directional guidance was exclusively provided by a soluble chemoattractive gradient, which equips amoeboid migrating cells with an extraordinary flexibility to migrate in almost every type of tissue.
1.3.3. Transendothelial migration.
The migration of cells through microspaces plays a role in many diseases, such as the immune response upon wound healing or metastasis. Leukocytes cross the endothelial lining of blood or lymph vessels in order to enter tissues during inflammatory and immune responses (Butcher 1991, Muller 2003). A main restriction may be the relatively large and less deformable nucleus of interphase cells that strongly hinders the cells deforming themselves to squeeze through the restriction. In this process of overcoming narrow spaces, cellular mechanical processes seem to be involved, such as the stiffness of the nucleus, the overall cellular stiffness, the cytoskeletal remodeling dynamics and the generation and transmission of contractile forces (figure 6). The alteration of the mechanical properties of transmigrating cells involves the restructuring of microfilaments, intermediate filaments and possibly microtubuli. How much these three main components of the cytoskeleton contribute to the mechanical properties of cancer cells and 'barrier' building endothelial cells is still not clear. However, intermediate filaments have come to be in the focus of biophysical research, as they are suggested to interact with the actomyosin cytoskeleton providing cellular mechanical properties regulating motility.
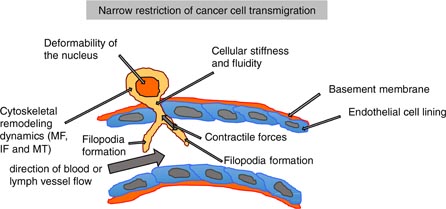
Figure 6. Narrow restrictions for the transendothelial migration of cells. The mechanical properties of the nucleus such as deformation play a role in the transmigration process. Other mechanical parameters of the cell, such as the cellular stiffness or fluidity, cytoskeletal remodeling dynamics of microfilaments (MF), intermediate filaments (IF) and microtubuli (MT), also play key roles. Another important mechanical property is the ability of cells to generate and transmit contractile forces. During transendothelial migration, the cells form filopodia that sense the mechanical microenvironment and help to determine the site for transendothelial migration within the endothelial cell lining.
Download figure:
Standard image High-resolution image1.3.3.1. Epithelial-originated cancer cell transmigration.
The transmigration of cancer cells through the endothelial lining of blood or lymph vessels seems to be a crucial step in the process of metastasis. The impact of the endothelial cell lining of vessels on the regulation of cancer cell invasiveness into 3D extracellular matrices is still elusive. The precise regulation of cancer cell transendothelial migration seems to be a complex scenario, which is not fully characterized yet. In numerous previous studies, the endothelium has been reported to act as a barrier against the invasion of cancer cells (Al-Mehdi et al 2000, Zijlstra et al 2008). During the intravasation and extravasation, cancer cells undergo enormous elastic deformations to transmigrate through the endothelial cell–cell adhesions (Wirtz et al 2011). In addition, the endothelium even reduces pronouncedly the invasiveness of cancer cells and hence their ability to form metastases (Van Sluis et al 2009).
In contrast, several recent reports have proposed a novel paradigm in which endothelial cells actively regulate the invasiveness of certain cancer cells by increasing their dissemination through vessels (Kedrin et al 2008) or by enhancing the invasiveness of certain cancer cells (Mierke et al 2008a). Although several adhesion molecules have been identified and characterized to function in tumor–endothelial cell interactions and, hence, facilitate cancer metastasis, the role of endothelial mechanical properties during the transmigration and invasion of cancer cells is still more or less elusive. However, it has been suggested that alterations in the mechanical properties of endothelial cells may favor one of the two main functions of the endothelium in cancer metastasis: first, they act either as a barrier and, secondly, they serve as an enhancer for cancer cell invasion. Alterations of the endothelial mechanical properties are induced by interacting cancer cells, when these cancer cells belong to an aggressive and highly invasive cancer cell type; however, when the cancer cells are non-invasive, the mechanical properties of the endothelium remained unchanged (Mierke 2011).
The main biochemical pathway of the tumor–endothelial interaction has been reported to involve cell adhesion receptors and integrins such as platelet endothelial cell adhesion molecule-1 (PECAM-1) and αvβ3 integrins, respectively (Voura et al 2000). As integrins provide a link between the extracellular matrix and the actomyosin cytoskeleton (Neff et al 1982, Damsky et al 1985, Riveline et al 2001), the connection between integrins and the actomyosin cytoskeleton is facilitated through the mechano-coupling focal adhesion protein vinculin (Mierke et al 2008b). Moreover, the mechano-regulatory function of vinculin determines the amount of cellular counter-forces that maintain cellular shape, morphology and cellular stiffness (Rape et al 2011, Mierke et al 2008b, 2010). However, a biomechanical approach investigating the endothelial barrier breakdown precisely and in depth in the presence of co-cultured invasive cancer cells is still elusive. As microrheological measurements such as magnetic tweezer microrheology turned out to be suitable for the analysis of the endothelial cell's mechanical properties such as cellular stiffness during co-culture with invasive or non-invasive cancer cells compared to monocultured endothelial cells, endothelial stiffness is found to be influenced by the type of co-cultured cancer cells. In particular, highly invasive breast cancer cells can influence the cellular mechanical properties of co-cultured microvascular endothelial cells by lowering the stiffness of endothelial cells, whereas non-invasive cancer cells have no effect on endothelial cell stiffness (Mierke 2011). However, the effect of neighboring macrophages or fibroblasts is still elusive, but is supposed to be critical for the regulation of endothelial cell lining permeability during the transendothelial migration of cancer cells. Additionally, using the nanoscale particle tracking method, diffusion measurements of actomyosin cytoskeletal-bound beads, which serve as markers for structural changes of the intercellular cytoskeletal scaffold, are useful to determine the acto-myosin-driven cytoskeletal remodeling dynamics. Hence, the cytoskeletal remodeling dynamics of endothelial cells are shown to be increased in co-culture with highly invasive cancer cells, whereas they are not altered by non-invasive cancer cells (Mierke 2011). These findings demonstrate that highly invasive breast cancer cells can actively alter the biomechanical properties of co-cultured endothelial cells. Finally, these results may provide an explanation for the breakdown of the endothelial barrier function provided by the endothelial monolayers lining vessel walls.
However, it has been suggested that the endothelial cell's actin cytoskeleton serves as a migration scaffold for transmigrating cancer cells. The endothelial cell lining of vessels represents a barrier and, thus, is a key rate-limiting step against the transmigration, invasion and metastasis of invasive cancer cells (Zijlstra et al 2008). In particular, the endothelial vessel wall has been commonly considered as a strong tissue barrier towards the dissemination of cancer cells by pronouncedly reducing the invasiveness and, subsequently, the metastatic potential of cancer cells (Wittchen et al 2005). Recent results lead to the establishment of a novel role for the endothelial cell lining of vessels. In this novel role, endothelial cells enhance the invasiveness of certain cancer cells. First, breast cancer cells show increased dispersion and clearance through hematogeneous dissemination adjacent to blood vessels (Kedrin et al 2008). Secondly, the invasiveness of certain cancer cell lines is endothelial-cell-dependent and, thus, enhanced in highly invasive cancer cells, whereas in weakly invasive cancer cells the endothelium acts as a classical barrier for cancer cell invasion (Mierke et al 2008a). Although the process of cancer cell invasion and metastasis has been the subject of numerous research articles, the molecular and mechanical mechanisms of cancer cell transendothelial migration are still not yet fully understood.
The physical and biochemical aspects of the cancer cell intravasation process involves the interaction of at least three cell types, such as an invasive cancer cell, a macrophage and an opposing endothelial cell. All three cell types will engage the mechano- and biochemical-transduction properties of the cytoskeleton of all three neighboring cells. In order to reveal the cancer cell–induced signals in endothelial cells, a 3D assay can be used in which the real-time intra-endothelial signaling events evoked by invasive cancer cells or macrophages are analyzed and compared to monocultured endothelial cells (Khuon et al 2010, Dovas et al 2013, Roh-Johnson et al 2013). In more detail, this assay involves the assembly of a vasculature network in a 3D collagen matrix using endothelial cells that express a fluorescent resonant energy transfer-based biosensor reporting the activity of myosin light chain kinase (MLCK) in endothelial cells in real time (Chew et al 2002). As expected, endothelial cells react to mechano-sensing events in the 3D collagen matrix. For example, the 3D microenvironment induces lumen formation, and endothelial cells show basal-apical polarity in the proper orientation indicated by α 4 laminin deposition. As hypothesized before, it could be confirmed that invasive cancer cells affect the MLCK-mediated actomyosin function within the underlying endothelium. In addition, cancer cells are capable of transmigrating in at least two different cellular ways: first, via transcellular routes (through individual endothelial cells) and, secondly, via paracellular routes (though the endothelial cell–cell junctions) (Khuon et al 2010). In particular, when cancer cells use a transcellular invasion path, they trigger MLCK activation in this endothelial cell, which correlates with increased, located and spatial restricted phosphorylation of the myosin-II regulatory light chain (RLC) and localized endothelial myosin contraction. Indeed, this has been functionally analyzed by using endothelial cells expressing a RLC mutant that cannot be phosphorylated, hence, the intravasation events of cancer cells migrating intracellularly through the endothelial cell body are reduced. In summary, (i) invasive cancer cells are capable of undergoing transcellular migration; (ii) cancer cells induce transient and local MLCK activation as well as myosin contraction in adjacent endothelial cells at the site of transmigration and tissue invasion and (iii) the transcellular invasion path through endothelial cells depends on the phosphorylation of myosin-II RLC. Finally, all these findings demonstrate that the endothelium fulfills an active role in cancer cells' intravasation and also possibly in extravasation.
1.3.3.2. Leukemia cell transmigration.
Before leucocytes transmigrate (extravasate), they adhere to the endothelium and roll on it in order to transmigrate at a certain site. In particular, there are contact and lubrication forces involved, and the cell–cell interactions are altered by the blood flow (Subramanium et al 2013). In addition, the interfacial contact will increase due to the initial contact that enhances leukocytes rolling on the endothelium. The velocity of the rolling process depends on the cell compliance (Subramanium et al 2013). In general, neutrophil granulocytes need to traverse rapidly through narrow constrictions that are even smaller than their own cellular diameter of approximately 7–8 µm. For example, they can pass through capillaries with diameters of 2 µm, and during the transmigration through transendothelial cell–cell adhesion spaces as well as interstitial spaces they migrate through pores ranging from 0.1 to 10 µm (Doerschuk et al 1993). In healthy processes, the ability of neutrophils to migrate through narrow constrictions is necessary to keep arteries and capillaries still under flow without an accumulation of white blood cells. In particular, the increased cellular stiffness of neutrophils leads to neutrophils sticking in arteries and capillaries (Worthen et al 1989), and finally to their accumulation in post-capillary venules which supports inflammation within the vascular neighborhood (Downey et al 1993).
Besides the regulation of the neutophils' mechanical properties through alterations of the actin filaments and microtubuli cytoskeletal filaments (Tsai et al 1994, Ting-Beall et al 1995, Tsai et al 1998), the main focus is on the alteration of the multilobed nuclear morphology, which is suggested to mediate the passage through narrow spaces by the alteration of mechanical properties such as the deformation of the neutrophil's nucleus (Hirsch 1959, Lautenschläger et al 2009). In more detail, a round-shaped nucleus may sterically hinder the cells to deform in order to migrate through a narrow hole such as a pore. In contrast, a multilobed neutrophil's nucleus may support the transmigration through narrow spaces as individual lobes may pass more easily through these narrow constrictions compared to a bulky nucleus (Rowat et al 2013). This hypothesis is supported by the finding that cells with a lobulated nuclear shape are less hindered by the migration through 8 µm-pore-sized membranes compared with cells possessing round nuclei (Downey et al 1990, Erzurum et al 1991). However, the hyperlobulated shape of the nucleus is not the only parameter determining the deformability of the nucleus. The intermediate filament composition and individual filament concentration seem to play prominent roles in regulating the mechanical properties of the nucleus. During the process of granulopoiesis, which can be modeled by using the human promyelocytic leukemia HL-60 cells, major alterations are detected in the expression levels of two key nuclear envelope proteins, namely the integral nuclear membrane protein, lamin B receptor (LBR, strongly up-regulated) and lamin A (strongly down-regulated). Lamin A is a key structural protein forming a network underneath the inner nuclear membrane and thus affecting the mechanical stiffness of the nucleus (Olins et al 2000, Lammerding et al 2004, 2006). Beside the morphological shape of the nucleus, it has been hypothesized that these reduced levels of lamin A can increase the nuclear deformability and thereby support cell migration through micrometer-scale constrictions (Rowat et al 2013). In particular, they showed that levels of lamin A have a predominant effect on the ability of the neutrophils to pass through narrow constrictions, whereas the altered morphological shape of their nucleus is not necessary for the rapid migration through micrometer-scale pores (Rowat et al 2013).
Besides these mechanical properties, the particular interaction between leukocytes and the endothelial lining of the vessels is also essential for their ability to migrate through the endothelium into the connective tissue. The leukocyte transendothelial migration (extravasation or diapedesis) is restricted to specific regions of the vasculature such as the capillaries surrounding an inflammation or high endothelial venules in secondary lymphoid organs through the highly localized regulation of the cell–cell adhesion receptor expressions. The current concept of leukocyte transendothelial migration during inflammation is that the secretion of cytokines during the initial inflammatory response leads to an induction of endothelial cell surface receptors such as E- and P-selectins, which is followed by VCAM-1 (vascular cell adhesion molecule-1) expression in order to reduce the flow speed of leukocytes and to facilitate interactions between the two cell types by establishing cell–cell receptor interactions during tethering and rolling steps of the leukocyte transmigration (figure 7). In particular, this preliminary and initial interaction enables the leukocyte to recognize chemokines presented on the endothelial cell surface that in turn stimulate the leukocyte's β1 and β2 integrin activation, resulting the firm adhesion of leukocytes to endothelial cell adhesion receptors of the immunoglobulin superfamily such as ICAM-1 (intercellular adhesion molecule-1) and VCAM-1. Then, the arrested leukocyte crawls on the endothelium in order to decide where to migrate through the endothelium (Butcher 1991). However, the precise mechanisms for the recognition of the transmigration space are still elusive. What roles do mechanical sensing processes play in finding a transmigration site? Is this mechanical sensing process used by each of the two interacting cell types, the leukocyte and the endothelial cell? During transendothelial migration, the leukocyte engages other transmembrane receptors such as PECAM-1 (platelet/endothelial cell adhesion molecule-1), the JAM (junction adhesion molecule) family and CD99, which can also signal and contribute to the transendothelial migration (Ostermann et al 2002, Schenkel et al 2002, Muller 2003). Leukocytes have been described to migrate either through junctions between ECs (paracellular migration), or directly through the entire endothelial cell (transcellular migration), as shown in figure 7 (Marchesi and Florey 1960, Feng et al 1998). These two pathways may involve different biochemical signaling mechanisms and morphological changes in the endothelial cells.

Figure 7. Leukocyte transendothelial migration during the wound healing process. The endothelium is stimulated by cytokines at the site of the inflammation due to tissue injury. The E-selectin and P-selectin receptors are up-regulated on endothelial cells that lead to a reduction of the leukocytes' flow speed. Then, ICAM-1 and VCAM-1 on endothelial cells are up-regulated, mediating the tethering and rolling of leukocytes on the endothelial vessel lining. This is allowed by the post-adhesion strengthening and crawling of leukocytes. After this, there are two possibilities of leukocyte transendothelial migration: the paracellular migration through the endothelial cell–cell adhesions involving the down-regulation of cell–cell adhesion molecules such as PECAM-1, JAM-1 and CD99 (1) and the transcellular migration through a living endothelial cell (2).
Download figure:
Standard image High-resolution imageDuring firm adhesion, the turnover of focal adhesions can altered and ICAM-1- as well as VCAM-1-faciliated signaling may enhance the actomyosin contractility of the endothelial cell and possible also the overall endothelial cell stiffness. However, these mechanical alterations in endothelial cells have not been investigated in detail. In addition, VCAM-1 expression may cause the disruption of cell–cell adherens junctions.
1.4. Principles of inflammation
These principles are so far not described as hallmarks like in cancer disease. Here, we will define the important steps also as hallmarks. The precise targeting of leukocytes to their targeted grounds is a key strategy for the victory of the immune defense system (Petri et al 2008). Although this complex scenario has been the subject of intensive research for several decades (Springer et al 1985), many of the involved signaling pathways are still not yet understood (Lefort and Ley 2012). However, different sequential leukocyte recruitment steps have been identified that each involve the distinct functioning of particular members of the integrin family of adhesion molecules (Petri et al 2008). These cell membrane proteins have a dual role of sensing and interacting with the surrounding microenvironment (Abram and Lowell 2009). Even though many stimuli are known to activate integrins, the molecular details of the involved signaling pathways are still elusive and, hence, the subject of current research. A novel aspect is the impact of the microenvironment in the integrin regulation at the sites of inflammation. However, recent studies identified physiological interactions on all levels of integrin activation, in particular the modulation of signaling receptor and ligand expressions, the modifications of signaling pathways, and the antagonizing ligand binding of integrins (Herter and Zarbock 2013).
Although several details of the molecular mechanisms have been revealed, the current knowledge of the involved pathways is still fragmentarily investigated. In particular, the mechanical aspects of these inflammatory processes have been so far ignored. Some steps, such as selectin-mediated slow leukocyte rolling and transmigration, are better understood than the G protein–coupled receptor (GPCR) signaling that finally leads to the integrin activation. In particular, several in vitro results, such as the rolling of monocytes at the site of the inflammation, have yet to be confirmed under physiological conditions. Moreover, the interaction among different integrins and their activation during crawling and transmigration are still not yet clearly understood. Taken together, integrins are adhesion proteins affecting a wide variety of cellular functions (Hynes 2002). In particular, beyond their key role in immune surveillance and leukocyte trafficking, integrins are important in cell development, hemostasis and cancer, and influence key cell cycle events (Hynes 2002). During the inflammatory processes, integrins are linked to several crucial steps in leukocyte recruitment that include the anchoring of leukocytes to the extracellular matrix (inside-out signaling) and the facilitating of signals in response to the surrounding microenvironment either by the binding of extracellular matrix proteins or ligands expressed on the cell surface of other cells (outside-in signaling) (Abram and Lowell 2009).
In order to fulfill these different functions, integrin assembly constitutes obligate noncovalently-bound heterodimers (Campbell and Humphries 2011). At least 18 α- and 8 β-subunits have been identified, which generate 24 specific integrins. However, several different splice variants are known that imply a much larger biological diversity of the integrin family than currently suggested (Hynes 2002). The different leukocyte subsets in the circulation express different combinations of integrins on their cell surface. In particular, neutrophils express mostly β2-integrins, but also low amounts of β1- and β3-integrins. However, monocytes express β1- and β2-integins, and lymphocytes possess an even broader pattern of β1-, β2- and β7-integrins that differs from subtype to subtype and depends on the activation state (Harris et al 2000).
The integrins possess a large extracellular domain, a transmembrane domain, and a short cytoplasmic tail (Luo et al 2007). In most cases, the intracellular tail of the β-subunit links the molecule to the actin or intermediate cytoskeleton. The integrin is kept in an inactive and closed state by close association of the α- and β-chains. In this configuration, the extracellular chains are in close proximity and form a ligand-binding site at the N-terminal ends featuring an I domain (or A domain) that may lie on either the α-chain or on the β-chain. Furthermore, the adhesion binding site of the integrins is metal ion–dependent and, thus, the integrin requires the presence of calcium or magnesium as a coenzyme for its activation.
In particular, the ligand affinity (strength of monovalent protein/ligand interaction) and avidity (ability to form multiple interactions such as the combined, synergistic formation of bonds) are actively altered by the activation of characteristic signaling pathways in an inside-out fashion (Lefort and Ley 2012). The external binding of ligands to the integrin evokes an outside-in signaling that provides profound alterations to cellular physiologies (Abram and Lowell 2009).
On resting cells, integrins are displayed in their closed or 'bent' conformation on the cell surface and present a low binding affinity for ligands (inactive form) (Luo et al 2007). Upon activation, the conformation of the integrin molecule stretches and elongates to an extended shape and becomes a high-affinity conformation for its ligands (Luo et al 2007). In this activated conformation, the α- and β-subunits are farther apart which can reflect the active rearrangement by the binding of the cytoskeletal proteins talin-1 and kindlin-3 to the cytoplasmic β-chain of the integrin (Rose et al 2007). In particular, shear stress can passively move the two integrin subunits apart and hence stabilize ligand binding. In more detail, shear stress may constitute a prerequisite for physiological β2- and α4β1-integrin functioning at the vessel wall (Alon and Dustin 2007). However, the precise mechanism is still not clear. In addition, crystallographic analysis has revealed a third, intermediate conformation for LFA-1, supporting the current model of at least three distinct activation states (Luo et al 2007). Moreover, it has been proposed that each of these three conformations only describes a favored conformation for a defined activation level, whereas the other two conformations may exist at the same time (Luo et al 2007).
Furthermore, the regulation of the avidity is an important feature of integrin adhesiveness (Carman and Springer 2003). However, the contribution of avidity to adhesiveness, its investigation in vivo and the cell's active contribution to its regulation is still elusive, as changes of avidity and affinity physiologically often appear at the same time and cannot be distinguished so far (Carman and Springer 2003). In particular, the crosslinking of integrins is used to form artificial integrin clusters to study outside-in signaling that facilitates a strong stimulus into the cell (Chan et al 2000). For example, high-resolution mapping of the LFA-surface distribution has revealed at least three different avidity patterns: randomly distributed molecules, ligand-independent nanoclusters and ligand-facilitated macroclusters (Cambi et al 2006). The preformed nanoclusters can solely be dynamically recruited to the cell–cell interface to form macroclusters (Cambi et al 2006).
Leukocyte recruitment into tissues underlies several consecutive steps: in particular, leukocyte recruitment into inflamed tissue involves a defined cascade of events, starting with the capturing of free-flowing leukocytes to the endothelial vessel wall, which is followed by rolling, the leucocyte adhesion to endothelial cells, the post-adhesion strengthening on the endothelium, the further crawling of leukocytes over the endothelium, and finally the transmigration of the leukocytes (figure 7). During all of these consecutive steps, different integrin receptors have to be activated via inside-out signaling. Each step of leukocyte transmigration requires a different set of integrins that differs among leukocyte subsets. Similarly, different activating stimuli trigger distinct signaling pathways on the endothelium and on the leukocytes during every step.
1.5. Physical perspectives in cancer progression
Up to now, the potential of the physical understanding of cancer has not yet been fully revealed. It may serve to provide the missing understanding of cancer progression and reveals further insights into cancer disease and especially metastasis. Many of the following questions are still unanswered.
What roles do contractile forces play in the regulation of cancer cell motility through the extracellular matrix and through the endothelial lining?
What role does the mechanical stiffness of highly invasive cancer cells play in facilitating their migration speed and transendothelial migration efficiency?
What role do the mechanical properties of interacting endothelial cells play for cancer cell transendothelial migration and tissue invasion?
How do interacting macrophages and the release of cytokines or chemokines that possibly influence the mechanical properties of cancer cells and endothelial cells affect cancer cell invasiveness?
What impact do the mechanical properties of the microenvironment of tumors have on the ability of cancer cells to sort out a subtype of cells that is able to migrate through tissue and to metastasize?
From a physicist's point of view, the process of cancer progression is clearly a mechanically driven process that in turn affects the biochemical signaling pathways. For example, the signal transduction pathways evoked by alterations in cellular mechanics are still not yet fully revealed and hence need to be studied in more detail.
In order to investigate the mechanical aspects of cancer progression and its subsequent metastasis, the focus of our physical-driven cancer research is on the selection of aggressive cancer subtypes showing increased invasiveness and metastasis. The progression of the metastatic process evolves step-by-step, but occurs rarely among the huge number of cancer cells at the primary tumor site. In particular, cancer cells spread from the primary tumor, cross tumor boundaries and migrate or flow through widely different microenvironments, including the tumor stroma, the blood vessel endothelium, the vascular liquid system (blood flow) and the targeted tissue at a secondary tumor site (Chambers et al 2002, Steeg 2006), where the probability of metastasis is more likely to occur compared to other non-targeted sites. These are all processes where the mechanical properties of cancer cells play a key role. Together with enhanced cellular invasiveness, cellular morphology, cytoskeletal architecture and biomechanics, cancer cells are able to remodel and adapt their microenvironment, including its mechanical properties such as stiffness and steric hindrance, to facilitate tumor progression and finally to metastasize in targeted organs (Brábek et al 2010, Mierke 2011b, Wolf et al 2003).
In addition, it has been reported that the tumor microenvironment seems to be no passive compartment in the process of the cancer disease progression. The tumor microenvironment is, hence, rather an active compartment, which can be critical for providing the physical properties for malignant cancer progression (Engler et al 2006, Mierke et al 2011) Moreover, the tumor microenvironment seems to be highly critical for all steps of the cancer metastasis process. In addition, the endothelial microenvironment, such as the blood or lymph vessels of a tumor, is an active element for providing cancer cell invasion into dense 3D extracellular matrices, where the pore size is even smaller than the cancer cell's diameter (Mierke 2011).
The external physical properties of the tumor microenvironment can be of a geometric or mechanical nature, and can have differences in the dimensions of the space that is available for invasive cancer cells (Paszek et al 2005, Kumar and Weaver 2009, Provenzano et al 2009). In particular, the structural and mechanical properties of the tumor microenvironment (tumor stroma) can be described mechanically by the matrix stiffness, pore size, fiber crosslinking points, crosslinking proteins, extracellular matrix fiber network composition, and their thickness, bending properties and spatial orientation (Lee et al 2012, Parekh and Weaver 2009, Storm et al 2005). All of these parameters of the tumor microenvironment are altered by the primary tumor (Ng and Brugge 2009), by aggressive and highly invasive cancer cells migrating into the external matrix (Branch et al 2012) or tumor-associated cells such as endothelial cells, macrophages or fibroblasts (Harris et al 1981, Geiger et al 2009) to adapt the external microenvironment as an optimal substrate for the single or collective invasion of cancer cells..
When cancer cells are too stiff or too soft, however, they are probably no longer able to deform in order to squeeze through highly crosslinked collagen fibers or bundles of the extracellular matrix scaffold and, subsequently, to regulate their migration efficiently (Guck et al 2005, Levental et al 2009). Finally, the mechanical properties of a primary tumor's microenvironment seem to play an important role in governing the migratory and metastatic behaviors of cancer cells. In particular, the matrix stiffness of the local tumor microenvironment may be sufficient to facilitate the selection of an aggressive (highly invasive) cancer cell subtype, possibly in a similar way as demonstrated for the differentiation of mesenchymal stem cells into distinct lineages, which is consistent with differences in tissue compliance (Engler et al 2006).
The physical interactions between a cancer cell and the extracellular matrix, a collagen-rich scaffold on which the tumor grows, play a key role in allowing the cancer cells to migrate from a primary tumor into the nearby tissues by crossing the tumor boundary and compartment boundaries, and, if the tumor is strongly malign, the cancer cells can also physically interact with invading blood or lymph vessels. During the steps of intravasation and extravasation, cancer cells seem to undergo large extreme elastic deformations to penetrate endothelial cell–cell contacts and even migrate through the living endothelial cell lining of vessel walls, which is still intact and reassembles to a closed monolayer even after the transmigration event of a cancer cell (Mierke et al 2011). After entering the vascular system, a cancer cell must deal with the migration velocity evoked by the vessel flow and be able to adhere to the endothelial vessel walls, which influence the cancer cell's binding efficiency to the endothelial vessel lining and hence determine the translocation sites where, in close proximity, a secondary tumor will initiate and grow.
Finally, a clear understanding of the role of physical interactions and mechanical forces, and their interplay with biochemical alterations, will provide novel and important insights into the progression of cancer, will lead to novel strategies to fight against cancer and may provide the basis for new therapeutic approaches.
1.5.1. Physical aspects affect hallmarks of cancer.
All of the current postulated eight hallmarks of cancer still miss the mechanical properties of cancer cells and their microenvironment, indicating that the established view on cancer is not from a physical position. Here, we address and focus on the physical view on cancer disease.
As current genetic and molecular biological classical approaches have not captured the full complexity of metastasis, to get even more insights into the malignant cancer disease progression, classical physical approaches need to be adapted and new biophysical methods have to be developed in order to use them in the field of cancer research. In particular, these novel directions and approaches have pronouncedly altered the classical field of current cancer research and have broken down the classical view on cancer disease. In particular, there is agreement that physical aspects in cancer research can no longer be ignored and must be included into the current cancer research. This means that at least a ninth hallmark, which includes the aspect of physics into classical cancer research, is necessary to understand the complex regulatory scenario of cancer metastasis. In particular, this ninth hallmark describes that the primary tumor and the tumor microenvironment alter the survival conditions and cellular properties of a certain set of cancer cells, which subsequently favors the selection of an aggressive (highly invasive) subtype of cancer cells. Moreover, this aggressive subtype of cancer cells may be able to reduce cell–cell adhesions to neighboring cells affecting the mechanical properties of neighboring and interacting cells, cross the tumor boundary of the primary tumor, including the tumor-surrounding basement membrane, and migrate into the tumor stroma consisting of an extracellular matrix scaffold and embedded cells. Taken together, each of these steps alters the mechanical properties either of the invading cancer cells, their microenvironment, the neighboring cells representing a stable barrier and the interacting endothelial cells. In principle, this novel ninth hallmark can be included after the hallmark of avoiding immune destruction and before the hallmark of activating invasion and metastasis (Mierke 2013b). In particular, the novel ninth hallmark, the selection of an aggressive subtype of cancer cells, can have the capability to down-regulate cell–cell adhesions, possibly up-regulate cell–matrix adhesions and regulate the mechanical properties of cancer cells that facilitate their transmigration through the basement membrane and their migration into the connective tissue. In addition, these nine hallmarks can be grouped into three major groups: neoplasm formation (hallmarks 1–3), transformation of cancer cells into aggressive and invasive cells (hallmarks 4–5) and tumor growth (hallmarks 6–9) (Mierke 2013b). By investigating the eight classical hallmarks, the physical aspect can also be included in each of them to refine this hallmark.
1.5.2. Physical aspects affect hallmarks of inflammation.
As mentioned above for cancer disease, the physical view on inflammatory disease has yet to be formulated. Here, the focus is now on the physical view on inflammation. The whole process of inflammation can be divided into the following hallmarks: hallmark 1: external stimulation via cytokines and chemokines after an inflammatory event; hallmark 2: leukocytes' initial recruitment of leukocytes to the endothelium via capturing, tethering and rolling; hallmark 3: leukocytes' adhesion to the endothelium and post-adhesion mechanical strengthening; hallmark 4: leukocytes' intravascular crawling on the endothelium; hallmark 5: leukocytes' transendothelial migration; hallmark 6: leukocytes' detachment from the endothelium; hallmark 7: leukocytes' migration through the connective tissue to the inflammation site; hallmark 8: possibly the differentiation of monocytes in macrophages via integrin outside-in signaling; and hallmark 9: integrin regulation at the site of inflammation (figure 8). All of these nine hallmarks of inflammation involve mechanical properties together with biochemical ones. As the strengthening of the adhesion may be in response to the mechanical stimulation upon the initial adhesion between the leukocytes and the endothelial cell lining, crawling may also be induced via mechanical strengthening, and then the transendothelial migration of the leukocytes is suggested to involve alterations of the mechanical properties of endothelial cells and leukocytes.
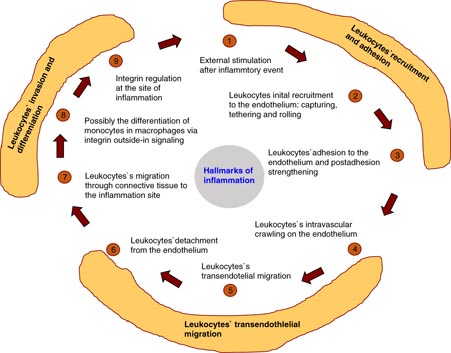
Figure 8. Nine consecutive hallmarks of inflammation. The nine hallmarks can be grouped into three main steps such as the recruitment and adhesion of leukocytes to the endothelium, their transmigration and their invasion through the connective tissue.
Download figure:
Standard image High-resolution image1.5.3. Biophysical measurements to identify the malignant potential of cancer cells and to detect early tissue injury.
The optical cell stretcher method has been used to analyze the deformability of cancer cell lines and primary cancer cells isolated from primary breast tumors (Guck et al 2005, Fritsch et al 2010). Recently, this method has been used to characterize the role of intermediate filaments by using keratin knock-out keratinocytes and wild-type keratinocytes isolated from mice. In particular, it has been reported that cells lacking keratins are more deformable compared to wild-type cells and, in particular, their 2D motility is increased compared to wild-type cells (Seltmann et al 2013b). Moreover, a connection between the intermediate filament (keratin) cytoskeleton and the actomyosin cytoskeleton has even been reported (Bourdeleau et al 2012).
Recently, 3D force assays have been suggested to play an important role in determining the invasive and metastastic potential of cancer cells. In order to qualitatively determine the forces that cells exert in a 3D microenvironment, gel contraction assays have been developed and used in numerous studies (Bell et al 1979, Cooke et al 2000, Berendsen et al 2006, Smith et al 2006). In particular, the cells are mixed with collagen prior to the gelation into a disk. The floating gel disk has free boundaries or is even only loosely attached to the wall of the culture dish and contracts when the cells exert contractile forces. Directly from the size reduction of the gel (mirrored in the reduced gel diameter), a qualitative description of the contractile forces can be obtained. A quantitative value of the average forces generated by the cells inside the gels can also be obtained when the cell number and the viscoelastic properties of the gel are known and when the spatial cell density distribution and overall cell orientation is homogenous and isotropic within the gel. However, some of these prerequisites cannot always be satisfied. For example, cells can remodel their extracellular matrix by compacting the matrix, by secretion of matrix-degrading enzymes or membrane-bound sheddases (cut cell–matrix or cell–cell adhesion receptors or membrane-bound metallo-proteinases from the cell surface) or by the secretion of new matrix proteins such as fibronectin and, thus, the local and global viscoelastic properties of these gels may be dramatically altered over time (Bell et al 1979, Leung et al 2007). However, the matrix remodeling and restructuring can be monitored over time by twisting micrometer-scale ferrimagnetic beads embedded within the gel and measuring the angular bead rotation, but these measurements do not have the sensitivity and spatial resolution needed for a quantitative analysis of cell forces (Leung et al 2007). In addition, cells can alter their biomechanical properties in response to the rheological properties such as the rigidity, the structural properties such as mesh size, fiber orientation and cross-links and the biochemical properties such as adhesive ligands' concentrations of the extracellular matrix (Pelham and Wang 1997, Discher et al 2005, Paszek et al 2005). Moreover, actual models of cell migration in 3D matrices are limited due to the ignorance of active cell responses to the matrix (Zaman et al 2005, 2006).
For a more-detailed quantitative estimate of cell tractions and contractile forces of single cells in a 3D extracellular matrix, it seems to be necessary, at least in principle, to extend the 2D traction microscopy method (Mierke et al 2008a) to the third dimension (Kumar and Weaver 2009, Yu et al 2011, Koch et al 2012, Mouw et al 2014, Rubashkin et al 2014). First, instead of a polyacrylamide hydrogel, a reconstituted connective tissue matrix such as a collagen matrix should be used. In particular, invasive cancer cells are able to spontaneously invade deep into collagen matrices. Second, instead of a single layer of fluorescent bead-markers at the gel surface, the bead-markers need to be randomly dispersed throughout the 3D collagen matrix. Third, from alterations in the bead-marker position (either measured over time before the initiation of cell adhesion, or after cell treatment with drugs relaxing the contractile forces of adherent cells and finally inducing cell detachment), certain measures of contractile force generation such as the elastic strain energy can be analyzed. Fourth, instead of bead-markers, which may be subject to phagocytosis by cancer cells, the displacement of collagen fibers can be tracked directly and can serve as measure of the contractile forces of invasive cancer cells within the matrix. However, the measurement of contractile forces in 3D matrices has to deal with obstacles that the boundary conditions, such as the free upper gel surface (usually overgrown with cells that have not yet invaded) and the fixed lower surface, as well as the non-linear rheological properties of the gel, need to be considered, and the Green's function under such conditions is unknown. In addition, the invasive cancer cell may have generated a migration path through the gel by secreting matrix-degrading enzymes and extracellular matrix proteins, as well as releasing cellular particles such as membrane and cytoskeletal components.
Taken together, a way to overcome these problems is to use the elastic strain energy stored in the matrix as a robust estimate of contractile cell forces (Butler et al 2002). Thus, the elastic strain energy can be calculated from the local matrix strain between adjacent fluorescent beads or between adjacent collagen fibers, and only the matrix rheological properties, but not the boundary conditions, need to be known. The calibration measurements of the deformation field in collagen matrices are performed by the application of point forces generated with magnetic tweezers towards the collagen matrix (Kollmannsberger and Fabry 2007). However, it is still suggested that aggressive cancer cells display increased invasiveness by becoming more contractile compared to non-invasive and less-contractile cancer cells (Mierke et al 2008a, 2008b, 2008c, Mierke et al 2011, Guck et al 2005). Moreover, a substance that can alter the contractile properties of cancer cells may provide novel therapeutic strategies to inhibit or decrease the invasiveness of cancer cells. In addition, the contractile force transmission and generation of cancer cells depends also on their cytoskeletal stiffness or on the inverse, their deformability, which may possibly interfere with the ability to transmit and generate contractile forces.
2. Cellular responses to mechanical stress
Cells can feel if the rigidity of their microenvironment changes and can adapt to the altered mechanical needs, if the cell's keratin network is intact. However, if the keratin network is altered regarding keratin 8 and 18, then the connection between the keratin network and the actin cytoskeleton is no longer functional or able to provide assistance in reacting to mechanical alterations of the substrate such as the rigidity (Bordeleau et al 2012).
Actin filaments, microtubules (MTs) and intermediate filaments (IFs) build up the cytoskeleton that is responsible for the cellular shape and the tissue integrity and hence provides resistance to external forces, coordinates cell motility, regulates intracellular movements and facilitates signal transduction processes (Fuchs and Cleveland 1998, Herrmann et al 2003). Despite the balanced forces within cells, processes such as cell invasion into dense extracellular matrices require single cells or a collection of cells that need to be highly deformable. The functions of actin filaments and MTs have been widely investigated, but the role of the IFs in providing the mechanical properties of cells and hence supporting their invasiveness and transendothelial migration ability is still not yet understood. IFs are encoded by a gene family of approximately 70 members in humans and build up the IF cytoskeleton by assembling long and apolar keratin heterodimers that consist of at least one type I keratin and one type II keratin. The cell type–and differentiation state–specific expression of keratin isotypes may contribute pronouncedly to the biomechanical properties of cells and whole tissues (Herrmann et al 2003). Basal epidermal keratinocytes express the keratin pair K5/K14 that is down-regulated upon the differentiation of skin tissue, whereas K1/K10 is up-regulated, and, subsequently upon tissue regeneration, the expression of K1/K10 is decreased and in turn the expression of K6/K16 is transiently increased (Fuchs and Green 1980). As cell fragility diseases are caused by keratin mutations that lead to a destroyed keratin cytoskeleton, major contributions of keratins to cell and tissue integrity have been suggested, but not yet revealed (Omary 2009). In more detail, when the keratin expressions are down-regulated, the cell–cell and cell–matrix adhesions are also pronouncedly reduced, which subsequently affects possibly the EMT and the invasive and metastatic behavior of cancer cells. In particular, cell shape changes, and the cellular stiffness of cancer cells (figure 9), which is necessary to migrate through the dense surrounding tissue of pore sizes below the cell's nucleus (Wolf et al 2007, Kumar and Weaver 2009, Fritsch et al 2010), may also be affected. However, small deformations of cancer cells may solely impact the cortical actin, which has been shown to act as the dominant contributor to cell stiffness (Rotsch and Radmacher 2000, Guck et al 2005, Nawaz et al 2012). MTs have been shown to have no impact on cell stiffness in this regime of small deformations (Bausch and Kroy 2006, Lulevich et al 2010). For large deformations of cells, it has been proposed that IFs are involved to maintain cellular integrity (Janmey et al 1991). Moreover, this is supported by studies of vimentin-deficient cells that have been shown to be less stiff compared to vimentin wild-type cells at high external stresses, whereas for low external stresses no impact on vimentin expression could be detected (Wang and Stamenovic 2000). However, there remain open questions. What impact do IFs have on the intermediate deformations of cells? How is the IF cytoskeleton connected to the actin cytoskeleton? The last question can be answered partly by a recent study that describes the formation of a complex between 14-3-3σ (stratifin), actin and intermediate filaments regulating cellular movement (Boudreau et al 2013).
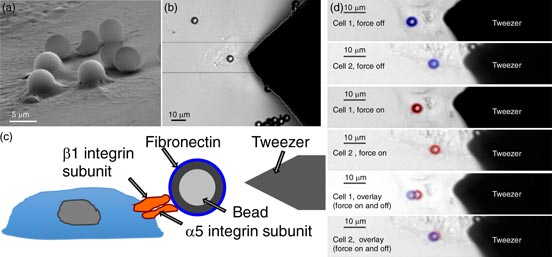
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9. Cellular stiffness can be measured by using magnetic tweezer microrheology. (a) A scanning electron microscopic image of superparamagnetic beads that are bound to cancer cells. (b) A bright-field image of a cancer cell with a bound bead and the magnetic tweezer needle. (c) A schematic image of a cell that has bound a fibronectin-coated bead to its α5β1 integrin receptor on the cell surface. The tweezer needle can apply forces to the superparamagnetic beads through generating a magnetic field radial from the needle tip by adjusting the current through the needle coil. In more detail, the magnetic field strength can be regulated. (d) Two cancer cells with a bound bead are shown in a force-off condition (the two upper images) and in a force-on condition (the two middle images). The lower images are an overlay of cell 1 or cell 2, respectively, with or without external force application.
Download figure:
Standard image High-resolution image{kind=link}
The effect of the keratin IF cytoskeleton on cellular stiffness and hence invasiveness has been analyzed using keratinocyte cell lines devoid of their keratin cytoskeleton, wild-type cell lines and a rescue cell line re-expressing the single keratin pair K5/K14 (Fuchs and Green 1980, Seltmann et al 2013a, 2013b). The keratin knock-out cells showed pronouncedly reduced cellular stiffness compared to wild-type cells and rescue cells (Seltmann et al 2013a). In addition, these keratin knock-out cells showed less motility in transwell-membrane assays, and even in 3D spheroid culture assays, these cells displayed less invasiveness. Taken together, these experiments are of major impact as they demonstrate clearly the pronounced effect of the IF cytoskeleton on cellular mechanics upon external mechanical stimulation and on the functional properties such as the migration and invasion of cells.
2.1. The role of external forces in cancer invasion and transmigration
Upon the stimulation of cells with external applied forces, the cells can react drastically, by just moving away in the opposite direction of the force. This phenomenon has been frequently observed in certain cancer cells that have been touched with the needle tip of the magnetic tweezer, moving it softly over the cell surface. However, it is still elusive whether macrophages or cancer-associated fibroblasts support cancer cell transendothelial migration by applying external forces on cancer cells at the onset of the transmigration process.
Besides other cell types, integrin-mediated cellular adhesion and migration across tissue boundaries and the sensing of mechanical properties of the extracellular microenvironment are fundamental in normal cellular processes and in cancer disease progression. Integrins have large extracellular domains, single-pass transmembrane domains (TMDs) and short cytoplasmic tails (Humphries 2000, Hynes 2002). In their ground state, integrins cannot adhere, but after receiving cellular signals, they can bind to extracellular matrix ligands (Tadokoro et al 2003) and facilitate upon their receptor ligation intracellular signaling cascades (Harburger and Calderwood 2009). This mechanical strengthening of the receptors upon cell adhesion facilitates cell migration, protects from apoptosis and regulates the differentiation of cells. Thus, integrins signal in both outside-in and inside-out directions through the cell membranes without the direct interaction of their TMDs or any kind of ligands. The TMDs seem to solely facilitate the signals across cell membranes and regulate the integrin affinity towards ligand binding. The resting structure of TMD has dimerized TMD helices (Gottschalk et al 2002, Hoefling et al 2009, Lau et al 2009), has a low affinity to extracellular matrix ligands and does not trigger signaling cascades, leading to only weak cell adhesion. The TMDs are separated when fully activated (Hantgan et al 1999, Xiao et al 2004) and have a high affinity to soluble ligands facilitating intracellular signaling cascades in order to provide strong cell adhesion and to mediate cell migration. The integrin αvβ3 (ligand vitronectin) is crucially involved in angiogenesis and tumor metastasis. Different TMD constructs have been functionally analyzed. TMD structures such as the glyophorin A (GpA) TMD (intermediate confirmation) have been generated that regulate integrin-controlled cellular functions through their design to investigate their impact on cell adhesion, migration or signal transduction (Gottschalk et al 2002, Müller et al 2013). It has been suggested that a mutant, which has a low basal affinity in the absence of forces due to the strong TMD interactions, can be activated by external forces leading to strong force-initiated adhesion. This is physically based on the distinction between the equilibrium property 'affinity' from the non-equilibrium property 'adhesiveness under force'. However, as the applied force may lead to a conformational change by pulling the protein over prohibitive energy barriers, adhesiveness under force can be high even if the integrin affinity in a force-free environment is low. The abrogation of the TMD dimerization should lead to constitutive integrin activation (Lemmon et al 1992, Zhu et al 2009). The expression of a TMD-GpA chimera leads to cell surface expression of the functional αvβ3 dimer, but with decreased affinity to a soluble ligand. However, the TMD-GpA chimera strengthened cell adhesion compared to wild-type cells under external shear forces that are applied either by atomic force microscopy or by a spinning disk device. In addition, the TMD-GpA chimera blocked the cells' capacity to promote αvβ3-facilitated migration or intracellular signaling and co-localization of the integrin, with talin being crucial for total integrin activation and signaling. Taken together, the integrin state induced by incorporating the GpA-TMD sequence shows the signature of a force-activatable integrin with properties in-between the resting state and the fully activated integrin state that may represent a physiologically relevant intermediate integrin state.
2.2. The role of internal forces produced by the cancer cell
Although the nature of the cell invasion is foremost a mechanical process, previous cancer research has focused largely on the gene regulation and signaling that underlie uncontrolled cell growth. In particular, the proteins encoded by the genes and signals involved in the mechanical regulation of the invasion and transendothelial migration of cancer cells, such as the role of adhesion molecules and matrix-degrading enzymes, are the novel focus of cancer research (Rolli et al 2003, Wolf et al 2003, Paszek et al 2005). However, the mechanical processes and the underlying physical models themselves that control cancer cell invasion, such as cell adhesion, changes of cell shape, cell movements and motility, and the generation of forces, are currently not well understood (Friedl and Brocker 2000, Ridley et al 2003, Zaman et al 2006). Moreover, many of the most elementary questions regarding the forces during cancer cell invasion have not yet been successfully answered. Do cancer cells push against the tissue to propel themselves forward, or instead do cancer cells grab the tissue matrix in front of the leading edge and then pull on the extracellular matrix? How strongly do they push or pull on the extracellular matrix? How strong is the adhesive interaction of cancer cells with their matrix? What is the size of the holes through which cancer cells are able to squeeze? What are the forces during amoeboid versus mesenchymal invasion strategies? What impact has a confinement of the invasion and the transendothelial migration of cancer cells?
The major part of our current knowledge on cellular motility, mechanical cell tensions and cellular forces is still derived from 2D migration studies of cells cultured on planar substrates (e.g. tissue culture plastic or glass, or polyacrylamide hydrogels) as described above. In particular, the methods to analyze traction forces during cell migration in 2D culture systems have been used for several decades (Pelham and Wang 1997, Harris et al 2000) and were more recently developed into quantitative tools (Dembo and Wang 1999, Butler et al 2002, Raupach et al 2007, Sabass et al 2008). The principal idea behind all of these methods is the measurement of the deformations in an elastic substrate such as polyacrylamide with a known elastic modulus on which adherent cancer cells are seeded. When cancer cells adhere and spread, they generate and transmit tractions and thereby deform the underlying substrate. In more detail, the tractions can then be determined from the substrate displacements using a continuum mechanics theory. During cell adhesion, force measurements have been performed by tracking the displacement field of small fluorescent beads that are embedded near the gel substrate surface. An advantage of this method is that the elastic modulus of the polyacrylamide substrate can be adjusted over a wide range by changing the acrylamide and bis-acrylamide cross-linker concentration (Pelham and Wang 1997, Yeung et al 2005). The spatial resolution of the traction map obtained approaches 1 µm under ideal standard conditions, which is even sufficient to detect and measure the forces from individual focal adhesions (Sabass et al 2008).
Thus, 2D traction microscopy has brought new insights into the mechanobiology of cancer cells, helping us to understand cancer cell migration (Mierke et al 2008a, 2008b, Raupach et al 2007, Runz et al 2008). In particular, cancer cells sense their microenvironment and respond to its mechanical properties such as the stiffness of their extracellular matrix by a dynamic regulation of adhesion receptor (integrin) clustering, the assembly of focal adhesion complexes and the remodeling of their cytoskeletal architecture (Discher et al 2005). In turn, the generation and transmission of contractile forces and subsequent cell migration and transendothelial migration are pronouncedly regulated by the mechanical properties of the extracellular matrix (Pelham and Wang 1997).
What role do forces play during cell invasion through 3D connective tissue and during the process of transendothelial migration? What about the existence of a force-feedback mechanism from the microenvironment towards the cancer cells? How does such a force-feedback mechanism play out in a 3D microenvironment? Force generation and transmission, migratory behavior, cell adhesion, focal adhesion formation, cytoskeletal organization and the dynamics of cancer cells in a 2D culture have been demonstrated to substantially differ from the situation in a 3D environment where cancer cells are embedded in a flexible, degradable 3D extracellular matrix (Cukierman et al 2001, Zaman et al 2006).
Hence, the migration speed in a 3D microenvironment, regardless of the cell type, is regulated by the balance of four biophysical processes (Zaman et al 2006). (i) Contractile forces need to be generated that help the cell to pull itself through a dense matrix scaffold. (ii) In more detail, contractile forces need to be transmitted to the surrounding extracellular matrix via integrin cell adhesion receptors. Moreover, the cellular adhesive bonds need to be sufficiently strong under the load imposed by the contractile forces, but they also need to be reversible in order to de-adhere in time to provide cellular motility. (iii) As the cancer cell squeezes through the pores of the matrix network, the cancer cells' resistance of elastic and frictional forces against these cell shape alterations are supposed to be sufficiently low, whereas otherwise the cancer cell's cytoskeleton cannot dynamically remodel itself to accommodate the necessary shape alterations. (iv) The resisting and mainly elastic forces imposed by the extracellular matrix when it is deformed as the cell squeezes itself through the matrix scaffold are suggested to be sufficiently small; if not, the invading cancer cell needs to degrade the matrix network enzymatically to decrease matrix resistance and steric hindrance. However, matrix resistance plays no role in 2D migration where cell adhesion, de-adhesion and the ability to remodel cytoskeletal structures are the only important mechanical parameters that influence the migration speed, whereas the forces needed to overcome the viscous drag imposed by the liquid microenvironment are negligible. The only forces needed in 2D migration are protrusive forces in the direction of cell movement through the de-novo polymerization of the actin filaments.
2.3. Mechanical properties of the microenvironment sensed by cancer cells
Cells are able to sense the mechanical properties of their microenvironment and to respond towards mechanical alterations by adapting their mechanical properties or by expressing a certain set of genes. This review focuses on two structurally and functionally similar protein families, the talins and the kindlins. These proteins bind the cytoplasmic tails of β-integrin subunits and, thereby, are essential for the activation and signaling of integrins (Kloeker et al 2004).
An unexpected result has been that a lack of kindlin 3 leads to severe integrin activation defects in platelets upon talin stimulation (Moser et al 2008). A role for kindlin in integrin function first found in C. elegans kindlin (UNC-112), which was essential for the organization of integrins at muscle attachment sites (Rogalski et al 2000). Ten years ago, mutations in a human kindlin (UNC-112 homologue) were shown to cause Kindler syndrome (Siegel et al 2003, Jobard et al 2003), which is a rare autosomal recessive disease that is characterized by skin blisters, photosensitivity, mucosal erosion and gastrointestinal ulcers. The expression of kindlin 1 (= FERMT1) is largely restricted to epithelial cells, and kindlin 1–deficient cells exhibit phenotypes displaying defects in spreading, polarity, migration, survival and extracellular matrix organization (Karakose et al 2010). There are three closely related kindlins—kindlin 1, kindlin 2 and kindlin 3—in humans, and all three have been shown to regulate integrin activation. For example, mutations of kindlin 3 have been reported to facilitate leukocyte adhesion deficiency type III (LADIII) (Karakose et al 2010, Plow et al 2009). Many points regarding the kindling regulation of integrin are unclear; however, it is known that kindlins cooperate with talin during the activation of integrins and that a direct interaction of kindlin with the β-integrin tail is essential, but not solely required for integrin activation. In particular, the focus of a recent study was on the structure of talin, the mechanisms by which the RAP1A GTPase effector RIAM binds and then recruits talin to dynamically growing adhesions at the leading edge of cells (Shattil et al 2010, Anthis and Campbell 2011, Ye et al 2011), and on the way that force-induced conformational alterations in the talin rod lead to a switch from talin–RIAM to talin–vinculin (Ziegler et al 2006, Peng et al 2011) complexes that stabilize focal adhesions (Calderwood et al 2013). In addition to talin F3 and β-integrin tail interaction, the talin head domain interacts with the cell membrane and, thus, plays an important role in integrin activation. In line with this, the talin head has many basic residues along its membrane-proximal surface that can bind acidic phospholipids (Elliot et al 2012). In addition, mutations in basic residues in F2 (Anthis et al 2009) and in an inserted loop in F1 (Goult et al 2010) impair membrane binding dramatically and subsequently inhibit integrin activation. In more detail, the acidic phospholipids have been shown to greatly increase the affinity of β3 tails for the talin head domain (Moore et al 2012), suggesting that the acidic plasma membrane phospholipids such as phosphatidylinositol-4, 5-bisphosphate (PtdIns(4,5)P2) play a key role in helping to orient the talin head domain so that it now can bind and activate integrins (Elliot et al 2010). However, is this model of integrin activation via kindlins and talin a general model for all integrins regardless of the cell type and on which membrane they are located? It has been shown that the activation of integrins is assessed by measuring the binding of soluble ligands or reporter antibodies that selectively or preferentially bind the active integrin (Bouaouina et al 2012). Whereas overexpressing the talin head domain is sufficient for integrin activation, full-length talin also activates integrins, but to a lesser degree due to its autoinhibition (Goksoy et al 2008, Goult et al 2009, 2013, Song et al 2012). Mutagenesis confirms that the activation relies on interactions between the talin head and the β-integrin tail (Han et al 2006, Goksoy et al 2008, Bouaouina et al 2012). The interaction of full-length talins with integrins is implicated in the clustering of integrins (Brown et al 2002, Helsten et al 2008, Saltel et al 2009), and as many integrin ligands are multivalent, this may increase the adhesion strength and, subsequently, increase the integrin's avidity (Bunch 2010).
Recently, it has been reported that kindlins have key roles in integrin activation and signaling (Plow et al 2009, Karakose et al 2010, Bouaouina and Caderwood 2011, Ye and Petrich 2011). In particular, kindlins directly bind β-integrin tails, whereas kindlin or integrin mutations that inhibit this binding can impair talin-mediated integrin activation suggesting a direct kindlin–integrin interaction necessary for maximal integrin activation. However, kindlin over-expression does not normally activate integrins (in certain cases it even suppresses activation) (Harburger et al 2009), but when co-expressed with the talin head, kindlins can strongly potentiate αIIbβ3 activation (Ma et al 2008, Montanez et al 2008, Harburger et al 2009). However, even when the kindlins are co-expressed with the talin head domains, over-expression of kindlins cannot activate α5β1 integrins (Harburger et al 2009). In particular, kindlin co-expression can suppress talin head-mediated β1 integrin activation, but the molecular mechanisms are not yet understood. However, β1 integrin activation is kindlin-dependent, as kindlin knock-out or knock-down inhibits β1 integrin activation (Montanez et al 2008, Malinin et al 2009, Qu et al 2011). Furthermore, mutations that inhibit talin binding to β-integrin tails block both talin-driven and kindlin-driven integrin activation, whereas mutations that inhibit kindlin binding still permit talin-mediated activation, but abolish the kindlin enhancement effect (Ye et al 2010). Taken together, these findings suggest that kindlins possess a role in talin-facilitated integrin activation.
2.4. Physical aspects of cell invasion and transmigration
A main physical aspect of cell invasion and transendothelial migration is the measurement of the mechanical properties of the microenvironment through mechanical sensing receptors such as the integrins. The composition and structure of extracellular matrices may vary widely and may depend on the tissue type and the state of health or disease of the tissue (Delmas 2004, Jaalouk and Lammerding 2009). In health and disease, the extracellular matrix in which cells are living provides microenvironments with various mechanical properties that vary on a length scale of a few micrometers to several millimeters (Kumar and Weaver 2009, Carey et al 2012). Biophysical measurements can be used to determine the mechanical properties of the matrix such as substrate elasticity, topography and roughness and how this influences cellular processes such as spreading, migration, phagocytosis and differentiation (Beningo et al 2002, Discher et al 2005, Engler et al 2006, Friedl et al 2012). The ability of cells to sense and respond to the mechanical properties of the extracellular matrix depends partly on the application of actomyosin-dependent contractile forces (Nishizaka et al 2000). Indeed, the cellular response after mechanical properties of the extracellular matrix and force-induced matrix deformation can be measured by characterizing the deformation fields created in the extracellular matrix (Mohammadi and McCulloch 2014). However, unequal to synthetic hydrogels such as polyacrylamide gels that exhibit nearly linear elastic deformation, natural matrix biopolymers such as collagen demonstrate complex mechanical behavior after the application of forces. For example, collagen matrices have been reported to exhibit non-linear viscoelastic behavior when subjected to cellular forces and can even undergo matrix or collagen fiber strain-stiffening (Strom et al 2005, Motte and Kaufmann et al 2013), fiber alignment (Vader et al 2009) or irreversible network compaction (Achilli et al 2012), depending on the magnitude and duration of the deformation.
Elastic or inelastic matrix deformations of the extracellular matrix can be observed by adherent cells and hence detected as inhomogeneous physical properties of the extracellular matrix, such as the presence of a rigid foundation (Sen et al 2009, Leong et al 2010) subjacent to the matrix or of adjacent cells that are located at several hundred microns' distance (Reinhardt-King et al 2008, Winer et al 2009). Notably, it has been found that adherent cells can mechanosense relatively far on fibrillar matrices compared to elastic hydrogels (Rudnicki et al 2013), which can be explained by the non-linear strain-stiffening behavior of fibrillar matrices (Winer et al 2009) and/or by fiber alignment in collagen gel networks (Ma et al 2013). Through the formation of filopodia, cells can sense the mechanical properties of their immediate microenvironment (Mattila and Lappalainen 2008). All three main functions of filopodia, such as sensory, exploratory and matrix remodeling functions, are dependent on the generation of tensile forces and by the resistance of the matrix to deformation (Mogilner and Rubinstein 2005).
Recently, the impact of physical boundaries on cell behavior and cytoskeletal organization have been investigated, and thus a new model system has been introduced in which nylon meshes with defined openings (200 mm wide and larger) provide defined physical boundaries in the plane of the supported matrix (Mohammadi et al 2014). In particular, with the use of micropatterning techniques, cell spreading is controlled by the area and shape of a patterned, highly adhesive surface so that cells conform to a configured geometry to which they are restricted. With this approach it seems to be that the magnitude and spatial distribution of the cell-generated forces are not primarily dependent on the cell spreading area (Wang et al 2002). However, a more important determinant of traction forces is the distance from the cell centroid to the perimeter of the adhesive area: when comparing cells of equal areas, more elongated cells generate stronger traction forces (Rape et al 2011).
A similar model was used to examine pre-configured patterns of traction forces in 3D gels, which revealed that the strongest inwardly directed traction forces are generated from the tips of long, matrix-probing pseudopodia (Legant et al 2010). Additionally, with the use of matrices of defined geometry, the direction and magnitude of local forces can be correlated with the area and elongation of mature focal adhesions (Balaban et al 2001). Thus, the results of such studies have suggested a linear relationship between the net traction force and the size of focal adhesions in unconstrained spread cells, if a thin matrix protein coating such as collagen on a rigid foundation (i.e. out-of-plane physical boundaries) such as glass (Chen et al 1997) or polydimethylsiloxane gel surfaces has been used (Balaban et al 2001). Are the same focal adhesions formed on 2D substrates responsible for the generation and transmission of contractile forces in 3D matrices? Recently, it has been reported that while focal adhesion proteins such as vinculin and FAK contribute to cell-induced matrix deformation in 2D models, these adhesion molecules are not significantly involved in the generation of cellular forces in 3D microenvironments (Mohammadi et al 2014). Taken together, these findings highlight that rigid physical boundaries are important in assessing cellular function and contractility.
Cell migration speed alternates between fast and high persistent and slow and low persistent movements. Recently, it has been reported that the movement of the interphase nucleus during cell migration switches between two modes: a rotation mode and a translocation mode (Kim et al 2014). In the rotation mode, the cell is rounded, and in the translocation mode, the cell is elongated. The switch between the nuclear modes and the cell morphology has been shown to be regulated by the actin cap that couples through the LINC complex between the nuclear envelope as well as the nuclear lamina and the actin cytoskeleton (Kim et al 2014).
3. Theoretical aspects of basement membrane crossing, cancer cell invasion and transendothelial migration
Many aspects of the basement membrane crossing, cancer cell invasion and transendothelial migration, such as the effect of crowding, the role of confinement, the effect of the polymerization of cytoskeletal components, the impact of cellular deformability and the effect of mechanical stress applied on interacting cells such as the endothelial cell lining, have not been investigated.
The development and maintenance of epithelial tissue requires finely balanced rates of cellular growth and cell death. There seems to be only a small difference between the mechanical and biochemical mechanisms that facilitate the proper feedback control of tissue growth and the same mechanism that leads to deregulated growth supporting tumorigenesis. How this is precisely regulated is still elusive. In particular, in crowded regions of the tissue, a proportion of cells can lose their cell–cell adherence junctions and subsequently their apical area, before they are squeezed out by their neighbors that still display cell–cell adherence junctions (called the path of delamination). Whether the selection of an aggressive cancer cell type starts in the same way is still under investigation. However, it is then even possible that these loose contactless cells sense their microenvironment as a migratory path on which they are able to migrate in order to invade connective tissue. Taken together, the system can omit overcrowding by squeezing some cells out that are able to flow or migrate away from the crowded region or can be removed by phagocytosis after the apoptosis process has been induced. Indeed, this process of delamination has been shown to be mechanistically distinct from apoptosis-mediated cell extrusion and precedes the first signs of cell death (Marinari et al 2012). Finally, this model provides a simple mechanism that buffers epithelial tissue against variations in cellular growth due to increase of growth factors. Moreover, this live-cell delamination connects mechanistically epithelial hyperplasia and cell invasion, which may help to understand the early stages of the onset of cancer disease.
A confinement may alter the dimensionality of cell motility on planar substrates by forcing them to migrate in a caged 3D system. A long time ago, it was reported that fibroblasts alter their direction of migration in vitro due to the interaction with other cells (Abercrombie and Heaysman 1953). This phenomenon has been named contact inhibition of locomotion (CIL), and it has been suggested as the main force that mediates the wound healing of epithelial tissue (Abercrombie and Ambrose 1962, Abercrombie 1979). In more detail, CIL is defined as the ability of a cell to change the direction of migration after contact with another cell that represents a confinement. The cell–cell adhesion is built by four consecutive steps: (i) cell–cell adhesion, (ii) inhibition of membrane protrusions such as filopodia at the site of contact, (iii) repolarization of the cells through generation of a new protrusion away from the initial site of cell contact and (iv) migration in the direction of the new protrusion (Mayor and Carmona-Fontaine 2010). However, in cancer disease, malignant mesenchymal cancer cells showed a reduced CIL response that enables them to invade other monolayers or polylayers of cells supporting cancer metastasis (Abercrombie and Heaysman 1954, Abercrombie and Ambrose 1962, Abercrombie 1979). In particular, Eph-ephrin signaling has been reported to play a role in the regulation of the invasiveness of prostate cancer cells, as these cancer cells showed a reduced response to CIL towards stromal fibroblasts (Astin et al 2010). However, several of the following questions remain unanswered. How can this finding be transferred to the interaction of malignant cancer cells and the endothelial cell lining of blood and lymph vessels? Why can solely invasive and aggressive cancer cells down-regulate CIL in order to transmigrate through endothelial cell linings or to migrate through connective tissue? How can the down-regulation of CIL further increase the migratory and invasive potential of cancer cells?
As CIL prevents the formation of protrusions between cells, cells cultured at high cell density can form lamellipodia only at free edges (Scarpa et al 2013). Thus, cells showing CIL behavior do not crawl over their neighbors leading to the formation of polylayers and to the scattering of single cells. In particular, when two cell clusters show CIL behavior, they remain separated and do not migrate over each other (Carmona-Fontaine et al 2008). There, reversion of CIL is possible. When two cell populations reverse CIL, they collapse their protrusions at the sites of cell–cell adhesions and therefore they stay clearly separated. If one of the cell populations fails to display CIL, it will invade the other tissue population, which leads to an extensive overlap of the two populations. However, invasion assays have been shown to reveal molecules that are functionally involved in CIL signal transduction (Carmona-Fontaine et al 2008, Theveneau et al 2010). Due to the difficult identification of single cells in tissue explants, assays with dissociated cultured cells, such as a simple collision assay on a 2D substrate where cells can freely move, have been used to measure cell velocity before and after cell–cell collisions. In addition, the angle between the direction of migration before and after contact has to be measured, as it is a measure of the cell repolarization. Cell polarization has been reported to facilitate cell invasion through dense 3D collagen matrices (Koch et al 2012). Indeed, the distribution of the angles is biased in the opposite direction of the cell–cell collision (Carmona-Fontaine et al 2008). Another important point that needs to be analyzed is the determination of the contact acceleration indices of free-moving cells and colliding cells (Paddock and Dunn 1986, Astin et al 2010). How does a cell's migration path deviate from a straight line after cell–cell collision? Thus, the contact acceleration index represents the difference between how far the cell has moved in the direction of migration and how far it would have gone without a collision. However, there are limitations of these cell–cell collision measurements in 2D, as the cells can collide at any incoming angle and, as a consequence, cells can collide leading edge to leading edge or leading edge to cell side, which impacts the response (Abercrombie and Dunn 1975). Moreover, the rotation of the cells after the collision as well as fewer cell–cell collisions make the measurement difficult to perform.
Thus, a novel 1D collision assay has been developed where cell migration is confined on straight extracellular matrix protein lines such as fibronectin lines obtained by microcontact printing-based micropatterning (Scarpa et al 2013). The restriction of the cell migration to a single dimension (1D migration path) forces cells to collide leading edge to leading edge. Subsequently, collisions are followed by a complete repolarization and, thus, cell migration is at a fixed 180° angle, which simplifies the CIL response by eliminating the variability due to the redirection angle. However, whether these assays model the 3D invasion assays in a better way than 2D migration assays has to be further investigated.
3.1. Models on the molecular, cellular and tissue scales
Cellular mechanics can be observed at different levels, such as that of the whole organs, particular organs, a tissue type, a compartment of that tissue, a cluster of cells, single cells and subcellular compartments such as organelles, protein aggregates such as fibers or bundles and single proteins such as receptors interacting with their ligands. All of these different observer levels involve the mechanical properties of cells or tissues regulating the invasiveness and transmigration efficiency of cancer cells. The principal modules on the cellular scale for cell motility can be the spatial polarization of signaling pathways, regulation of the actin cytoskeleton and the dynamics of focal adhesions. There are still the mechanical properties of proteins such as the focal adhesion protein vinculin that is involved in facilitating 3D cell invasion (Mierke et al 2010); however, the elongation of the proteins due to external force application is still elusive, but may dramatically alter the invasion-increasing effect of vinculin in 3D microenvironments. There are many models explaining a particular point at a certain level and how this influences cell invasion and transendothelial migration, but an overall view at different levels, including the protein and gene expression levels, is still elusive. Thus, future model development seems to favor solving that problem.
3.2. How can the fluid state of cells help to understand cancer and inflammation?
Until now, this question could not be answered. However, we have seen that the fluid state of cells can serve as a model to predict the function or mechanical properties of cells, such as invasiveness and transendothelial migration efficiency. The approximation of the cells as a fluid seems to be helpful in understanding the squeezing of cancer cells through their microenvironment such as a collagen fiber network or endothelial monolayers of blood or lymph vessels. Until now, it has not been clear whether this model of regarding the cells as a simple fluid may help to understand the mechanical and material properties of cancer and immune cells.
3.3. How similar is the behavior of cancer or immune cells to polymers?
As cancer cells and immune cells consist of polymer networks in their cytoplasm and nucleus, the polymerization may be applied after an adaption to these living cells in order to predict their ability to migrate through a 3D extracellular matrix and transmigrate through a closed endothelial cell monolayer. Many physical models are currently suggested to play a role in cell motility and are as such applied to whole cells and their microenvironments. Cancer cells or immune cells can show partial elastic or partial viscous behavior. In particular, the elastic behavior of cancer or immune cells seems to be determined by the elastic properties of the actin stress fiber network, the intermediate filament network and the microtubule network. However, cancer cells and immune cells display viscoelastic behavior as they are not purely elastic and not purely viscous (Mierke et al 2010). Moreover, the cells' microenvironment shows viscoelastic behavior. However, all of the principles observed in physics can be applied to cancer cells, immune cells and their microenvironment in order to describe the complex situation with simple and robust models. This reduction on the mechanical properties may then reveal the important pathways and mechanisms needed to understand cell invasion in 3D and transendothelial migration.
4. Discussion of the physical understanding of cell invasion, basement membrane crossing and transendothelial migration
The physical understanding of cell invasion and transmigration through basement membranes or endothelial cell linings is still rarely investigated, but comes more in the current focus of physically based cancer research. Whether cell sensitivity to its mechanical microenvironment occurs on a molecular or a cellular scale is still an open question and needs to be further investigated. There are some computational studies investigating the effect of the mechanical properties of the microenvironment, such as stiffness, shear stress and adhesivity on cell motility and gene expression, to describe the mechanistic pathways regulating cancer progression (Katira et al 2013, Mitchell and King 2013). The propagation of a mechanical signal within a focal adhesion has been shown to take only a few hundred milliseconds (Na et al 2008). This excludes the possibility of a diffusion process or a chemical reaction that would require at least minutes and not seconds. Thus, a fast propagation of the mechanical signal seems to be possible, such as through elastic waves that involve the deformation of the focal adhesion proteins such as Src (Wang et al 2005b). In particular, this elastic wave propagation may include the activation of Src and the integrins. Moreover, contractile forces that do not involve the actomyosin cytoskeleton regulate the response to external stress evoked by the extracellular matrix or the rigidity of the microenvironment. In addition, cells can also pull on the extracellular matrix that is independent of the elastic properties of the extracellular matrix (Trichet et al 2012). However, the value of stress is shown to adapt to the rigidity of the extracellular matrix that emphasizes the existence of a large-scale mechanosensitive apparatus regulating the amount of stress used by the cell to pull on the extracellular matrix.
Many mechanical details regarding these three essential steps of cancer progression are still not understood—in particular, the mechanical properties of the basement membrane and the extracellular matrix during cancer cell transmigration and the mechanical properties of the endothelial cell lining of vessels during the transendothelial migration of cancer cells. However, the interplay and the force-feedback or in general the mechanical feedback mechanisms are not well understood. Taken together, the biochemical assays and gene expression profile analyses need to be coordinates in an experimental approach.
5. Conclusions and outlook
There is still much to investigate in the novel and state-of-the-art field of the physical view of cancer research. What has turned out to be necessary is that the classical eight hallmarks of cancer need to be refined in terms of mechanical properties and their impact on malignant cancer progression. The same is true for the hallmarks of inflammation, as they also omit the mechanical properties of leukocytes and their microenvironment in inflammation. However, the biomechanical interactions of cancer cells with their microenvironment during cancer metastasis seem to play a key role revealing the spreading of aggressive and invasive cancer cells from the primary tumor and their ability to transmigrate endothelial cell linings in blood or lymph vessels. In addition, the analysis of the mechanical properties may also help us to predict the overall survival rate of the patient more accurately, as it may then be possible to predict more accurately whether a certain tumor will metastasize. In particular, the physical and material properties of cancer cells facilitate their migratory behavior and their transport through the human body after they have transmigrated into blood or lymph vessels and, hence, may support or inhibit metastasis. External forces from the microenvironment may additionally regulate the motility of cancer cells of epithelial origin in the structurally complex extracellular matrix scaffold during the invasion, intravasation and extravasation of cancer cells in and of the vascular system (extra- and intravasation, respectively). Thus, insights into the role of the physical and mechanical processes regulating the complex process of metastasis are a prerequisite for the development of new approaches for cancer diagnosis and treatment. Additionally, the mechanical properties of immune cells upon inflammation may also help to avoid a strong immune reaction, if not needed and to support tissue healing.
Taken together, besides providing effective prognostic and diagnostic tools for therapies inhibiting metastasis and revealing the prerequisites for proper wound healing after inflammation, the knowledge of the role of biomechanics in cell motility may also inspire inverse strategies to promote wound healing in terms of connective tissue regeneration after injuries. The impact of the key mechanical properties of the tumor microenvironment such as mechanical forces, stiffness, pore sizes and steric hindrances on cancer progression as well as the mechanical properties of stromal cells and endothelial cells on cancer cell invasion in general and after the use of therapeutic drugs have to be explored systematically and in detail to reveal the underlying mechanisms.
However, cutting-edge genetic or biochemical approaches should be combined with novel and state-of-the-art biophysical approaches to unravel the mechanics of cancer and immune cells and the mechanical properties of the local tissue microenvironment.
Finally, the combination of physics, molecular biology and biochemistry may provide the strength to reduce the divergent effects of potential cancer drugs on cellular or organ responses in animal cancer disease models and cancer patients and, subsequently, may lead to efficient and straightforward cancer treatments. The novel field 'the physics of cancer' is currently headed by biological physics and soft matter physics, but is still less represented in biology, immunology and medical cancer research. However, five years ago a network between biologists and physicists was established under the NIH Physical Science–Oncology Centers investigating the physical aspects of cancer. The physical approach for cancer research provides not simply a source sink for novel techniques for oncologists to investigate functional properties of cancer cells, it rather discovers novel aspects that help to understand cancer progression in more detail, to distinguish between malignant and benign tumors, and to refine the functional pathways involved in cancer disease progression in order to inhibit them specifically.
Acknowledgments
This work was supported by the Deutsche Krebshilfe (Grant No 109432) and the ESF-SAB (No 100147954) founded young scientist research group. I thank Thomas M L Mierke for proof-reading, helping with the references and for helpful discussions.