
Abstract
Affecting about 1 in 12 Americans annually, depression is a leading cause of the global disease burden. While a range of effective antidepressants are now available, failure and relapse rates remain substantial, with intolerable side effect burden the most commonly cited reason for discontinuation. Thus, understanding individual differences in susceptibility to antidepressant therapy side effects will be essential to optimize depression treatment. Here we perform genome-wide association studies (GWAS) to identify genetic variation influencing susceptibility to citalopram-induced side effects. The analysis sample consisted of 1762 depression patients, successfully genotyped for 421K single-nucleotide polymorphisms (SNPs), from the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study. Outcomes included five indicators of citalopram side effects: general side effect burden, overall tolerability, sexual side effects, dizziness and vision/hearing side effects. Two SNPs met our genome-wide significance criterion (q<0.1), ensuring that, on average, only 10% of significant findings are false discoveries. In total, 12 additional SNPs demonstrated suggestive associations (q<0.5). The top finding was rs17135437, an intronic SNP within EMID2, mediating the effects of citalopram on vision/hearing side effects (P=3.27 × 10−8, q=0.026). The second genome-wide significant finding, representing a haplotype spanning ∼30 kb and eight genotyped SNPs in a gene desert on chromosome 13, was associated with general side effect burden (P=3.22 × 10−7, q=0.096). Suggestive findings were also found for SNPs at LAMA1, AOX2P, EGFLAM, FHIT and RTP2. Although our findings require replication and functional validation, this study demonstrates the potential of GWAS to discover genes and pathways that potentially mediate adverse effects of antidepressant medications.
Similar content being viewed by others
Introduction
Associated with substantial co-morbidity1 and increased mortality,2 major depressive disorder (MDD) imposes immense costs in human suffering and economic productivity. While the development of multiple classes of antidepressant medications has greatly improved MDD treatment in recent years,3, 4 a substantial proportion of patients (∼50%) fail to attain adequate response to their initial antidepressant therapy.5, 6 Compounding the difficulty of identifying successful treatment, antidepressant use is frequently associated with adverse drug reactions, with the inability to tolerate side effects being the most common reason for discontinuing antidepressant therapy.7, 8, 9, 10 Clinicians currently have no way to predict individual efficacy and side effect profiles, and trial and error switching often leaves MDD patients in psychological distress for weeks or months. Clearly, improved methods of patient–antidepressant matching to minimize side effects would greatly improve depression treatment.
Previous studies have indicated that antidepressant response is substantially heritable,11, 12, 13, 14 suggesting pharmacogenomics research as a promising avenue toward individualizing antidepressant treatment. Preliminary pharmacogenetic research has, for instance, suggested an important role for genes related to serotonin function in antidepressant side effects.15, 16, 17 The serotonergic system is involved in the regulation of physiological functions that are often disturbed in antidepressant treatment, including neuroendocrine mechanisms regulating reproductive events such as spermatogenesis, ovulation and sexual behavior.15, 16, 18, 19, 20 However, to date, robust consistent evidence associating any specific candidate gene or polymorphism to antidepressant side effect response has been rare. This is due, at least in part, to the inherent shortcomings of candidate gene approaches, which are sharply restricted by current limitations in knowledge of depression neurobiology and antidepressant mechanism of action. Methods that systematically screen variants across the whole genome for association with antidepressant side effects are therefore critical to discover novel variants. Such approaches have recently begun to yield tangible results, with the number of replicated marker–disease associations increasing dramatically since the introduction of genome-wide association studies (GWAS),21 and some initial applications in the context of psychiatric pharmacogenomics.22, 23, 24, 25, 26, 27
In this paper, we use a GWAS approach to search for genetic variation affecting the susceptibility for citalopram-induced side effects. Our study sample consists of the 1762 MDD patients in the level 1 citalopram medication trial of the Sequenced Treatment Alternatives to Relieve Depression (STAR*D).28 Analyses were performed on the five primary side effect dimensions indicated by factor analyses of a battery of side effect indicators.28
Materials and methods
Study design and subjects
Subjects came from the STAR*D study, which has been described in detail elsewhere.28 In short, STAR*D is a multistage trial of different treatment options for patients with nonpsychotic MDD. STAR*D enrolled a total of 4041 outpatients with MDD.29 In level 1, all patients were given only citalopram. Those patients who did not have an efficacious response or could not tolerate side effects to citalopram were then randomized to other treatment options in other levels, 2a or 2–4. Other treatment options included, but were not limited to, sertraline, buproprion, buspirone, cognitive therapy, various combinations of these treatments and using one of these treatments in combination with citalopram. To maximize power, this study focuses exclusively on the 1762 patients participating in the initial, level 1, citalopram-only trial, for whom genome-wide genotype data were collected.
Clinical measures
Side effect presence and tolerance was measured by the Patient-Rated Inventory of Side Effects (PRISE).28 For each of the eight biological systems assessed (gastrointestinal, heart, skin, nervous system, vision/hearing, genital/urinary, sleep and sexual functioning), patients were asked two types of questions, one relating to specific side effects (for example, dizziness, anorgasmia) and one relating to the overall tolerability of side effects for a given biological system. For the specific side effects, patients were asked to indicate whether the side effect was present (0=no side effect; 1=side effect present). There were four items for the gastrointestinal system, three for heart, four for skin, four for the nervous system, two for vision/hearing system, four for the genital/urinary system, two for sleep and three for sexual functioning. In addition, an overall score of side effect tolerability was given for each of the eight biological systems as a trichotomous item, scored: 0=no side effects; 1=tolerable side effects; or 2=distressing side effects.29
Specifying side effect phenotypes
Specifying side effect phenotypes for GWAS proceeded in two steps. The first was to condense the 34 side effect indicators of the PRISE using factor analysis, thereby improving measurement through identifying the latent phenotypic constructs underlying the side effect indicator.30, 31 The second was to analyze longitudinal change in side effect phenotypes, to derive treatment effects for each drug based on the obtained factor scores.32
Identifying valid and reliable side effect measures was essential for optimizing power in the GWAS. This required data reduction, as using individual PRISE items as GWAS outcomes would provide minimal statistical power, given the presence of substantial measurement error in self-report measures combined with the inherently weak statistical power of dichotomous outcomes.30, 33 Given the limitations of using individual items as GWAS outcomes, collapsing the items into symptom class sums corresponding to clinical experience (for example, central nervous system, gastrointestinal) may seem an appealing, intuitive approach. However, preliminary analyses indicated that the internal consistency of many of these symptom class measures was poor, despite their intuitive appeal (for example, Cronbach's α=0.61 for gastrointestinal items and =0.66 for central nervous system items). Thus, we employed a more rigorous psychometric approach to identify robust latent side effect constructs.
Using Mplus 6.0,34 exploratory factor analyses were conducted to empirically examine the factor structure of the 34 side effect indicators of the PRISE. Of the several potential factors emerging from the exploratory analysis, five were retained based on overall fit to the data, interpretability and having a Cronbach's α >0.70, which indicates good reliability for the factor.35 The five factors were: (1) a general side effect burden factor in which all of the 26 symptom measures served as factor indicators; (2) an overall tolerability factor based on the eight system-specific tolerability indicators; (3) a sexual factor; (4) a dizziness factor; and (5) a factor relating to visionhearing (that is, ocularauricular) side effects. Once the optimal factor structure was determined, factor scores for each of the five factors were calculated using the standard regression scoring approach. The factor loadings and Cronbach's α for each factor are displayed in Table 1.
Factor analysis results presented in Table 1 were based on all observations, irrespective of the time spent on a treatment. Given that side effects sometimes vary by duration of treatment with a specific drug,36 additional factor analyses were performed, which divided the STAR*D sample based on the number of days on a given treatment. This was carried out to confirm that the factor solution was robust across treatment duration and not artifactual because of heterogeneity across time (see Supplementary Tables A1–A5). Another potentially confounding issue in evaluating side effects to antidepressant treatment is the presence of somatoform symptoms, which commonly co-occur with depression and may influence perception of the presence and severity of side effects, independently of actual side effects.37, 38 Thus, the factor analyses discussed above were repeated controlling for baseline somatoform and hypochondriasis diagnoses, assessed using the Psychiatric Diagnostic Screening Questionnaire39 (see Supplementary Tables A6–A7). Comparable factors emerged with similar patterns of loadings when the sample was stratified by the number of days on drug and when controlling for somatoform symptoms, supporting the robustness of the factor structures.
Generating treatment effect measures
To maximize power for pharmacogenomic GWAS, we developed a method to estimate medication treatment effects from all available information32 using mixed modeling.40, 41 Our method first determines the optimal functional form of overtime drug response, then screens many possible covariates to select those that improve the precision of the treatment effect estimates, and finally generates the individual treatment effect estimates based on the best fitting model using best linear unbiased predictors.42 As this approach takes advantage of all available information in STAR*D, it results in more precise estimates than traditional approaches that estimate treatment effects using only two assessments (for example, subtracting pre- from post-treatment observations).43 In addition, as treatment effects are based on mixed-model trajectory slopes, they are more robust to early dropout/discontinuation than competing approaches.44, 45
To determine the optimal model of overtime drug response for each side effect outcome, we fit a series of models specifying linear change for a given number of days on drug and flat thereafter. This series began with a model assuming that maximal drug response was achieved at day 1. Each subsequent model specified an incrementally longer duration until maximal drug response was achieved, with the final model assuming that the drug effect did not plateau (that is, linear change throughout the trial). The function produced by the log likelihoods of this series was then optimized to determine the best estimate of the average number of days until maximal drug response. This duration varied across side effect outcomes with dizziness plateauing earliest (36 days on drug), and general side effect burden and overall tolerability plateauing latest (137 days on drug) (Supplementary Figure B1).
After determining the optimal functional form of overtime drug response, 55 covariates collected during STAR*D were screened to identify those that improved the precision of the treatment effect estimates, using a criterion based on reduction in residual error variance relative to treatment random effect variance.23, 32 Screened covariates consisted of trial design characteristics, socio-demographic measures, clinical information, health-care access and reason for study exit (Supplementary Table B1 for full list). The number of selected covariates was 6 for vision/hearing and sexual side effects, 7 for general side effect burden and overall tolerability and 8 for dizziness. Design characteristics and concurrent psychiatric diagnoses (particularly drug abuse and hypochondriasis) comprised the vast majority of selected covariates (Supplementary Table B2 for full list).
Finally, treatment effects were generated by employing a unique feature of the mixed-model random effects. Briefly, the mixed model estimates two types of parameters, coefficients that describe the predictors' average effects for the full sample (that is, fixed effects) and deviations from the average effects for each subject (that is, random effects). Thus, for each of the antidepressants investigated, we were able to output treatment effects as random drug effects. Intuitively, these treatment effects quantify how much each subject's side effect phenotype changes in response to a given drug, relative to the average effect for all subjects who took the drug. Treatment effects estimated in the manner described here have been published previously both in former GWAS of the currently analyzed STAR*D study24 and several GWAS of the CATIE antipsychotic clinical trial.23, 26, 46, 47
Genotyping and quality control
Details of the STAR*D genotyping and quality control methods have been described previously.27 Briefly, approximately half the sample (N=964) was genotyped on the Affymetrix Human Mapping 500k Array Set, and the other half was genotyped using the Affymetrix Genome-Wide Human Array 5.0 (N=975). Both versions genotype the same set of SNPs, the only difference being that the later 5.0 version accomplishes this with one, rather than two, microarray/s. As preliminary examination indicated differences in data quality between the two platforms, we conducted additional data cleaning routines separately by platform, in PLINK. Subjects were excluded for low (<0.95) genotype call rates (3 and 85 subjects excluded in the 500K and 5.0 arrays, respectively), and SNPs were excluded for low (<0.01) minor allele frequency (14 562 and 12 012 SNPs excluded in the 500K and 5.0 arrays, respectively) and low genotype call rate (19 113 and 54 447 SNPs excluded in the 500K and 5.0 arrays, respectively). SNPs were not excluded based solely on deviations from the Hardy–Weinberg equilibrium given the possibility of informative reasons for departures from the Hardy–Weinberg equilibrium.48, 49 PLINK was also used to identify 89 potential genotype–clinical sex disagreement, which were excluded from analysis. While the default PLINK sex disagreement settings (that is, X-chromosome inbreeding/homozygosity estimate >0.8 for female and <0.2 for male) are conservative, they ensure that no unambiguous data are included in the analysis. After cleaning and merging, our analysis included 421 789 SNPs from 1762 subjects with a successful genotyping rate of 99.6%.
Statistical analyses and multiple testing
All association testing was conducted in PLINK,50 using a linear regression model of additive SNP effects. As STAR*D is an ethnically heterogeneous sample (79% Caucasian, 15% African American and 6% ‘Other’), in the GWAS we adjusted for ancestral background, which can otherwise cause spurious associations due to population stratification. Specifically, we used the multi-dimensional scaling approach implemented in PLINK (http://www.pngu.mgh.harvard.edu/~purcell/plink/strat.shtml#mds), which is essentially equivalent to principal component method implemented in Eigensoft.51 Input data for the multi-dimensional scaling approach were the genome-wide average proportion of alleles shared identical by state between any two individuals. The first multi-dimensional scaling dimension from this genetic similarity matrix captures the maximal variance in the genetic similarity; the second dimension is orthogonal to the first and captures the maximum amount of residual genetic similarity, and so on. We included the first five ancestral multi-dimensional scaling dimensions as covariates in the GWAS, based on analyses showing that additional dimensions neither predicted response nor explained significant additional covariance between SNPS.24
We used a false discovery rate (FDR)52 approach to declare significance. In comparison to controlling a family-wise error rate (for example, Bonferroni correction), FDR (a) provides a better balance between finding true effects versus controlling false discoveries, (b) results in comparable standards for declaring significance across studies because it does not directly depend on the number of tests, and (c) is relatively robust against having correlated tests.53 FDR is commonly used in many high-dimensional applications and has been successfully applied in the context of GWAS.54, 55, 56 We set an FDR threshold of 0.1 for declaring genome-wide significance. This specifies that, on average, 10% of the SNPs declared significant are expected to be false discoveries. In addition, we discuss suggestive associations at an FDR threshold of 0.5 to reduce the probability of Type II statistical errors, while explicitly noting reduced confidence in these associations. Operationally,57 FDR was controlled using q-values. Q-values are FDRs calculated using the P-value of the markers as thresholds for declaring significance,58, 59 and can be described as:

where π0 is an estimate of the proportion of null associations, p is the observed P-value, P is a random normally distributed variable, H0 is the null hypothesis that the SNP–side effect association β=0 and H1 is the alternative hypothesis that β≠0. Thus, the numerator equals the product of the proportion of null associations × the observed P-value, and the denominator is the weighted sum of the probability of obtaining a test statistic at least as extreme as the one observed, given the null hypothesis, and of the probability of obtaining a test statistic at least as extreme as the one observed given the alternative hypothesis, weighted by the proportion of null associations (see Storey58 for formal derivation). While π0 may be empirically estimated, we have assumed the most conservative value π0=1; thus, the formula simplifies to the observed P-value divided by the probability of obtaining a test statistic at least as extreme as the one observed given the null hypothesis.
For the most promising SNPs, we performed a variety of additional analyses to examine the robustness of the signal. First, we tested the SNPs separately in the subjects who self-identified as European Americans (EA) only and African American (AA) only (Supplementary Tables C1 and C2). In addition, for each SNP, we performed haplotype (proxy) analyses that incorporate information from other SNPs in that region (http://www.pngu.mgh.harvard.edu/~purcell/plink/proxy.shtml). Such analyses may provide a technical validation of the single SNP result or point to a particularly informative haplotype.
Results
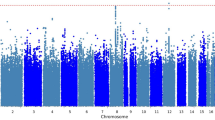
Table 2 provides details on those SNPs that were genome-wide suggestive (q<0.5). Figure 1 shows quantile–quantile plots for all analyses. The plots show that the distribution of P-values from the GWAS are generally on a straight line, indicating the expected P-value distribution under the null hypothesis assuming no effects of the markers. However, in four of these five plots, there is also evidence that markers in the right upper corner have P-values smaller than would be expected under the null hypothesis, suggesting true association between these markers and the outcome variable. The plots also display λ values (that is, the ratio of the median observed P-value of the distribution to the expected P-value under the null hypothesis) approximately equal to 1 (λ=0.996–1.033), indicating no systematic test statistic inflation and suggesting that population stratification was generally well-controlled. The full set of GWAS P-values considered in this study is available for download at: http://www.people.vcu.edu/~ejvandenoord/.

Quantile–quantile (Q–Q) plots for genome-wide association studies (GWAS) results of five citalopram-induced side effect measures. Points represent −log10(P-values) for each single-nucleotide polymorphism (SNP)–side effect outcome association test. Red lines represent the expected P-value distribution under the null hypothesis of no true associations. Blue lines represent 95% confidence intervals for rejecting the null hypothesis at each P-value rank. The genomic inflation parameter (λ) is defined as the ratio of the median observed P-value to the expected median under the null distribution, thus quantifying systematic test statistic inflation.
The top significant finding, rs17135437, exhibited positive association between vision/hearing side effects and minor allele count. This SNP is located in an intron of EMID2 on chromosome 7, as described in Table 2 (P=3.27 × 10−8, q=0.026). Examination of linkage disequilibrium (LD) in the surrounding region showed no other assayed SNPs in LD with this locus (R2<0.06). Consistent with this LD information, proxy haplotype tests showed that none of the adjacent SNPs evidenced more than modest association to citalopram-induced vision/hearing side effects (Figure 2a). Also, the SNP's minor allele frequency (MAF) is low in the study sample (MAF=0.019), and somewhat greater for AA than EA. However, separating the sample by racial/ethnic ancestry and reanalyzing the SNP shows that the direction and magnitude of the effect is consistent between AA and EA. Thus, this association provides mixed evidence of true discovery, with the lack of the availability of SNPs in high LD (to check for possible technical errors) and low MAF suggesting a degree of caution, but the consistency across racial/ethnic groups offering some reassurance.

Regional plots for genome-wide association study (GWAS) results of (a) EMID2, (b) LAMA1, (c) AOX2P and (d) intergenic rs16965962 associations. Points represent −log10(P-values) for association tests and are color coded to denote linkage disequilibrium to the target single-nucleotide polymorphism (SNP) in the HapMap Phase II reference data. Recombination rate is represented by the light blue lines. The plots' bottom panels show the names and locations of genes in the UCSC Genome Browser, with exon positions denoted by cross-hatches and transcription direction by arrows.80
Among the other strongest associations was a pair of SNPs in high LD (R2=0.978) at LAMA1, on chromosome 18. Both of these SNPs showed strong positive associations between overall tolerability to citalopram side effects and minor allele count (rs4398173: P=5.99 × 10−7, q=0.315; rs3810046: P=4.78 × 10−7, q=0.281). MAF was high for both SNPs (>0.45), and exhibited only minor differences across racial/ethnic groups. Consequently, the direction, magnitude and significance of the associations were consistent in racial/ethnic stratified reanalysis. Examination of the LD structure in the study sample showed modest, but significant LD between six assayed SNPs, spanning ∼15 kb. All SNPs identified in PLINK proxy haplotype analyses showed systematic association to the overall tolerability phenotype (P<0.05), but did not approach genome-wide significance, except in the case of the two markers described above. Of note, while the two SNPs approaching genome-wide significance are located slightly downstream (∼10 kb) of LAMA1, the haplotype that they represent clearly overlaps the gene boundary by ∼5 kb, suggesting that the reported hits may be associated with proximate regulatory sequence or a functional variant within the gene's downstream end (Figure 2b).
The strongest signal for citalopram-induced dizziness comprised two highly proximate SNPs (∼4 kb apart) at AOX2P, on chromosome 2. Both of these SNPs showed strong positive associations between dizziness and minor allele count (rs13430864: P=5.12 × 10−7, q=0.181; rs13423450: P=7.57 × 10−7, q=0.228). Racial/ethnic stratified analyses indicated different haplotype structures between EA and AA, with AA evidencing a ∼6 kb proxy haplotype including four assayed SNPs, but no discernable LD in the same region among EA. The association involving rs13430864 demonstrated consistent direction and effect magnitude across EA and AA. However, rs13423450, part of a common haplotype with rs13430864 in AA but not EA, only evidenced association among AA. This combined with moderate differences in MAF by racial/ethnic status suggests variability in the structure and function of this locus—thus, this association should be regarded as tentative, pending further evidence (Figure 2c).
Suggestive associations for general side effect burden included rs7715172, at EGFLAM, on chromosome 5 (P=2.91 × 10−6, q=0.358). Reanalyzing the data separately by race/ethnicity indicated that the association was in the same direction and magnitude in both subsamples. Examination of LD structure indicated the presence of a relatively large haplotype, with seven adjacent assayed SNPs in moderate LD (=0.11–0.59) to the implicated SNP. The haplotype spanned ∼5 kb, and three assayed SNPs in the haplotype were located within the gene boundary of EGFLAM (Supplementary Figure D1). Consistent with this LD structure, proxy haplotype analyses indicated six of the surrounding SNPs to exhibit moderate association to the general side effect burden (P<0.05). However, MAF was relatively low (=0.071) and moderate racial/ethnic difference were evident; thus, this finding should be considered suggestive, conditional on further evidence. Additional suggestive associations were found between general side effect burden and SNPs at RPT2 (rs6764050: P=8.73 × 10−7, q=0.182) and FHIT (rs4502542: P=1.62 × 10−6, q=0.262), as well as several intergenic SNPs (Supplementary Figures D1 and D2). Among the intergenic associations was a genome-wide significant finding at rs16965962, located in a gene desert on chromosome 7 (P=3.22 × 10−7, q=0.096). As shown in the regional plot (Figure 2d), this intergenic SNP represented a distinct susceptibility haplotype spanning ∼30 kb, with eight assayed SNPs showing clear, systematic association general side effect burden.
Discussion
To maximize the benefit of antidepressant therapy to the individual patient, not only should the most efficacious drug be prescribed at the time of first presentation, but also the drug with the minimal side effect profile. Understanding individual differences in the development of side effects following antidepressant therapy is therefore essential to personalizing the treatment of depression. In this study, we performed GWAS on five side effect factors, including general side effect burden, overall tolerability, sexual adverse reactions, dizziness and vision/hearing-related side effects. We detected two SNPs, which according to our pre-identified criteria (FDR controlled at q<0.1) can be considered genome-wide significant.
Our top genome-wide finding involved rs17135437, an SNP within the gene EMID2, with minor allele count positively associated with the severity of citalopram vision/hearing side effects. EMID2 encodes the protein collagen α-1(XXVI) chain in humans,60 which has been found to regulate corneal collagen fibrillogenesis.61 Variation in the gene has also been associated with ataxia in neurodegenerative disorders.62 Moreover, EMID2 has shown evidence of pharmacogenetic side effect moderation, albeit to a pharmacodynamically distinct phenotype, aspirin-induced asthma.63 On balance, considering the strength of association, links with ocular and neurocognitive functions and tentative evidence of pharmacogenetic activity, rs17135437, may be considered a reasonable candidate in future pharmacogenomic studies of selective serotonin-reuptake inhibitors.
The associations of two high-LD LAMA1 SNPs to overall tolerability also suggest credible biological mechanisms. LAMA1 encodes a vital component of laminin proteins—laminin subunit α-1. Binding to cells via a high-affinity receptor, laminin is thought to mediate cell attachment, migration and differentiation of cells into tissues during embryonic development by interacting with other extracellular matrix components, as well as maintenance of tissue phenotype and promotion of tissue survival.64, 65 This relatively frequently studied gene has been linked to several cancers including non-small-cell lung and colorectal,66, 67 as well as cardiovascular pathophysiology.68 LAMA1 has also been associated with pharmacogenetic moderation of thiazolidinedione-induced side effects to diabetes treatment and,69 more relevantly, with moderating the effects of antidepressant buproprion on smoking cessation.70 Thus, robust GWAS support and prior evidence of antidepressant moderation suggest LAMA1 as a plausible candidate for future pharmacogenetic and functional analyses.
The two high-LD candidates at pseudogene AOX2P, on chromosome 2, are relatively poorly characterized, but variants within the pseudogene have been identified and analyzed in two bioinformatic studies based on a targeted nonsynonymous SNP approach,71 and a large-scale identification and characterization of putative alternative promoters of human genes.72 Given that pseudogenes, by definition, lack protein-coding function, this finding should be viewed with a degree of skepticism. However, considering that functional genes have been misclassified as pseudogenes,73 pseudogene transcripts have demonstrated trans-regulation of homologous coding genes,74 and some endogenous small interfering RNA are derived from pseudogene transcription,75 it would be premature to dismiss this result as a false positive.
Similarly, we view the association of variants in EGFLAM to citalopram-induced general side effect burden as tentative because of MAF differences between racial/ethnic groups. However, there are potential side effect mechanisms involving EGFLAM suggested by previous research, including studies of ocular structure and function (for example, murine eye pathophysiology76 and photoreceptor ribbon synapse formation).77 Thus, this gene remains a plausible candidate for future consideration. Finally, the second genome-wide significant SNP (rs16965962) was located in a gene desert on chromosome 7. While the lack of involvement in protein-coding sequence somewhat diminishes this marker's prior probability of being a true discovery, there is precedent for such intergenic associations to replicate in independent samples, such as found with a region on chromosome 9 in Type 2 diabetes mellitus.78 Furthermore, the association signal here was notably robust, including eight assayed SNPs spanning a ∼30 kb haplotype (Figure 2d). Thus, this finding may be evidence of long-range regulatory elements, consistent with recent research identifying gene desert enhancer hotspots associated with coronary artery disease.79
Beyond individual findings, it is also worth noting that all top associations exhibited positive effects of minor allele count and side effect severity, rather than the expected mix of positive and negative effects (Table 2). Further inspection of the GWAS output confirmed the legitimacy of this finding, and demonstrated that, as expected, negative and positive coefficients were equally likely in the full GWAS results. The pattern was largely supported in the ethnically stratified analyses as well, where all coefficients, except general side effect burden—rs16965962, were positive. Although the binomial probability of arriving at this result by chance is small, the lack of precedent in previous pharmacogenomic research suggests that the finding may be due to sampling error. However, it is also possible that the result is due to uncommon polymorphisms having an increased probability of deleterious effect. Future research will be required to adjudicate between these possibilities.
Currently, it is premature to suggest direct clinical applications of these findings for prescribing antidepressants. On the contrary, actualizing the promise of pharmacogenomics and translating academic findings into clinical applications will require a cumulative process of aggregating and jointly considering large bodies of evidence using meta-analytic and data integration techniques. Thus, it is crucial to conduct and rapidly disseminate GWAS results from large, well-designed clinical trials with genomic data, such as STAR*D. To facilitate this process, we provide all P-values (http://www.people.vcu.edu/~ejvandenoord/) as a resource for investigators with the requisite samples to carry out replication or meta-analysis.
As with any genetic associations, our findings will require replication and functional validation. However, this study shows the potential of GWAS to discover genes and pathways that mediate adverse effects of antidepressant medication. A better understanding of these mechanisms and the roles of specific polymorphisms will facilitate the development of improved biomarker-based approaches to personalize antidepressant therapy. It is hoped that this research will eventually contribute, however incrementally, to reducing the global health burden of depression, facilitating efficient prescription of the most efficacious and least toxic antidepressant medication to MDD patients.
References
Kessler RC, Chiu WT, Demler O, Walters EE . Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry 2005; 62: 617–627.
Mykletun A, Bjerkeset O, Dewey M, Prince M, Overland S, Stewart R . Anxiety, depression, and cause-specific mortality: The HUNT study. Psychosom Med 2007; 69: 323–331.
Berton O, Nestler EJ . New approaches to antidepressant drug discovery: beyond monoamines. Nat Rev Neurosci 2006; 7: 137–151.
Vaswani M, Linda FK, Ramesh S . Role of selective serotonin reuptake inhibitors in psychiatric disorders: a comprehensive review. Progr Neuro-Psychopharmacol Biol Psychiatry 2003; 27: 85–102.
Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006; 163: 28–40.
Turner EH, Matthews AM, Linardatos E, Tell RA, Rosenthal R . Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med 2008; 358: 252–260.
Bull SA, Hu XH, Hunkeler EM, Lee JY, Ming EE, Markson LE et al. Discontinuation of use and switching of antidepressants—influence of patient–physician communication. JAMA 2002; 288: 1403–1409.
Bull SA, Hunkeler EM, Lee JY, Rowland CR, Williamson TE, Schwab JR et al. Discontinuing or switching selective serotonin-reuptake inhibitors. Ann Pharmacother 2002; 36: 578–584.
Mitchell AJ . High medication discontinuation rates in psychiatry—how often is it understandable? J Clin Psychopharmacol 2006; 26: 109–112.
Maddox JC, Levi M, Thompson C . The compliance with antidepressants in general practice. J Psychopharmacol 1994; 8: 48–53.
Franchini L, Serretti A, Gasperini M, Smeraldi E . Familial concordance of fluvoxamine response as a tool for differentiating mood disorder pedigrees. J Psychiatr Res 1998; 32: 255–259.
Malhotra AK, Murphy Jr GM, Kennedy JL . Pharmacogenetics of psychotropic drug response. Am J Psychiatry 2004; 161: 780–796.
Oreilly RL, Bogue L, Singh SM . Pharmacogenetic response to antidepressants in a multicase family with affective disorder. Biol Psychiatry 1994; 36: 467–471.
Pare CMB, Mack JW . Differentiation of 2 genetically specific types of depression by response to antidepressant drugs. J Med Genet 1971; 8: 306–309.
Zobel A, Maier W . Pharmacogenetics of antidepressive treatment. Eur Arch Psychiatry Clin Neurosci 2010; 260: 407–417.
Kato M, Serretti A . Review and meta-analysis of antidepressant pharmacogenetic findings in major depressive disorder. Mol Psychiatry 2010; 15: 473–500.
Porcelli S, Drago A, Fabbri C, Gibiino S, Calati R, Serretti A . Pharmacogenetics of antidepressant response. J Psychiatry Neurosci 2011; 36: 87–113.
Ayala ME . Brain serotonin, psychoactive drugs, and effects on reproduction. Cent Nerv Syst Agents Med Chem 2009; 9: 258–276.
Segraves RT . Effects of psychotropic drugs on human erection and ejaculation. Arch Gen Psychiatry 1989; 46: 275–284.
Pollack MH, Reiter S, Hammerness P . Genitourinary and sexual adverse effects of psychotropic medication. Int J Psychiatry Med 1992; 22: 305–327.
Altshuler D, Daly MJ, Lander ES . Genetic mapping in human disease. Science 2008; 322: 881–888.
Aberg K, Adkins DE, Bukszar J, Webb BT, Caroff SN, Miller del D et al. Genomewide association study of movement-related adverse antipsychotic effects. Biol Psychiatry 2010; 67: 279–282.
Adkins DE, Aberg K, McClay JL, Bukszar J, Zhao Z, Jia P et al. Genomewide pharmacogenomic study of metabolic side effects to antipsychotic drugs. Mol Psychiatry 2011; 16: 321–332.
Adkins DE, Aberg K, McClay JL, Hettema JM, Kornstein SG, Bukszar J et al. A genomewide association study of citalopram response in major depressive disorder—a psychometric approach. Biol Psychiatry 2010; 68: e25–e27.
McClay JL, Adkins DE, Aberg K, Bukszar J, Khachane AN, Keefe RS et al. Genome-wide pharmacogenomic study of neurocognition as an indicator of antipsychotic treatment response in schizophrenia. Neuropsychopharmacology 2011; 36: 616–626.
McClay JL, Adkins DE, Aberg K, Stroup S, Perkins DO, Vladimirov VI et al. Genome-wide pharmacogenomic analysis of response to treatment with antipsychotics. Mol Psychiatry 2011; 16: 76–85.
Garriock HA, Kraft JB, Shyn SI, Peters EJ, Yokoyama JS, Jenkins GD et al. A genomewide association study of citalopram response in major depressive disorder. Biol Psychiatry 2010; 67: 133–138.
Rush AJ, Fava M, Wisniewski SR, Lavori PW, Trivedi MH, Sackeim HA et al. Sequenced treatment alternatives to relieve depression (STAR*D): rationale and design. Control Clin Trials 2004; 25: 119–142.
Wisniewski SR, Rush AJ, Balasubramani GK, Trivedi MH, Nierenberg AA . Self-rated global measure of the frequency, intensity, and burden of side effects. J Psychiatr Pract 2006; 12: 71–79.
Bollen KA . Structural Equations with Latent Variables. Wiley: New York, 1989.
Loehlin JC . Latent Variable Models: An Introduction to Factor, Path, and Structural Equation Analysis. Lawrence Erlbaum Associates: Mahwah, NJ, 2004.
van den Oord EJ, Adkins DE, McClay J, Lieberman J, Sullivan PF . A systematic method for estimating individual responses to treatment with antipsychotics in CATIE. Schizophr Res 2009; 107: 13–21.
Cohen J . The cost of dichotomization. Appl Psychol Meas 1983; 7: 249–253.
Muthén LK, Muthén BO . Mplus User's Guide, 6th edn. Muthén & Muthén: Los Angeles, CA, 1998–2010.
Nunnally JC, Bernstein IH . Psychometric Theory, 3rd edn. McGraw-Hill: New York, 1994, p xxiv, 752pp.
Demyttenaere K, Albert A, Mesters P, Dewe W, De Bruyckere K, Sangeleer M . What happens with adverse events during 6 months of treatment with selective serotonin reuptake inhibitors? J Clin Psychiatry 2005; 66: 859–863.
Barbee JG . Mixed symptoms and syndromes of anxiety and depression: diagnostic, prognostic, and etiologic issues. Ann Clin Psychiatry 1998; 10: 15–29.
Regier DA, Rae DS, Narrow WE, Kaelber CT, Schatzberg AF . Prevalence of anxiety disorders and their comorbidity with mood and addictive disorders. Br J Psychiatry 1998; 173: 24–28.
Zimmerman M, Mattia JI . The reliability and validity of a screening questionnaire for 13 DSM-IV Axis I disorders (the psychiatric diagnostic screening questionnaire) in psychiatric outpatients. J Clin Psychiatry 1999; 60: 677–683.
Goldstein H . Multilevel Statistical Models. Arnold: London, 1995.
Searle SR, Casella G, McCuloch CE . Variance Components. Wiley: New York, 1992.
Pinheiro JC, Bates DM . Mixed-Effects Models in S and S-plus. Springer: New York, NY, 2000.
Willett JB, Singer JD, Martin NC . The design and analysis of longitudinal studies of development and psychopathology in context: Statistical models and methodological recommendations. Dev Psychopathol 1998; 10: 395–426.
Laird NM, Ware JH . Random-effects models for longitudinal data. Biometrics 1982; 38: 963–974.
Siddiqui O, Hung HMJ, O’Neill R . MMRM vs LOCF: a comprehensive comparison based on simulation study and 25 NDA datasets. J Biopharmaceut Statist 2009; 19: 227–246.
Aberg K, Adkins DE, Liu Y, McClay JL, Bukszar J, Jia P et al. Genome-wide association study of antipsychotic-induced QTc interval prolongation. Pharmacogenom J 2012; 12: 165–172.
McClay JL, Adkins DE, Aberg K, Bukszar J, Khachane AN, Keefe RSE et al. Genome-wide pharmacogenomic study of neurocognition as an indicator of antipsychotic treatment response in schizophrenia. Neuropsychopharmacology 2011; 36: 616–626.
Wigginton JE, Cutler DJ, Abecasis GR . A note on exact tests of Hardy–Weinberg equilibrium. Am J Hum Genet 2005; 76: 887–893.
Wittke-Thompson JK, Pluzhnikov A, Cox NJ . Rational inferences about departures from Hardy–Weinberg equilibrium. Am J Hum Genet 2005; 76: 967–986.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D . Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006; 38: 904–909.
Benjamini Y, Hochberg Y . Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc Ser B 1995; 57: 289–300.
Brown BW, Russell K . Methods correcting for multiple testing: operating characteristics. Statist Med 1997; 16: 2511–2528.
Lei SF, Yang TL, Tan LJ, Chen XD, Guo Y, Guo YF et al. Genome-wide association scan for stature in Chinese: evidence for ethnic specific loci. Hum Genet 2009; 125: 1–9.
Liu YZ, Guo YF, Wang L, Tan LJ, Liu XG, Pei YF et al. Genome-wide association analyses identify SPOCK as a key novel gene underlying age at menarche. PLoS Genet 2009; 5: e1000420.
Beecham GW, Martin ER, Li YJ, Slifer MA, Gilbert JR, Haines JL et al. Genome-wide association study implicates a chromosome 12 risk locus for late-onset Alzheimer disease. Am J Hum Genet 2009; 84: 35–43.
Black MA . A note on the adaptive control of false discovery rates. J R Stat Soc Ser B 2004; 66: 297–304.
Storey J . The positive false discovery rate: a Bayesian interpretation and the q-value. Ann Stat 2003; 31: 2013–2035.
Storey JD, Tibshirani R . Statistical significance for genomewide studies. Proc Natl Acad Sci USA 2003; 100: 9440–9445.
Leimeister C, Steidl C, Schumacher N, Erhard S, Gessler M . Developmental expression and biochemical characterization of Emu family members. Dev Biol 2002; 249: 204–218.
Rada JA, Cornuet PK, Hassell JR . Regulation of corneal collagen fibrillogenesis in vitro by corneal proteoglycan (Lumican and Decorin) core proteins. Exp Eye Res 1993; 56: 635–648.
Lim J, Hao T, Shaw C, Patel AJ, Szabo G, Rual JF et al. A protein–protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell 2006; 125: 801–814.
Pasaje CF, Kim JH, Park BL, Cheong HS, Kim MK, Choi IS et al. A possible association of EMID2 polymorphisms with aspirin hypersensitivity in asthma. Immunogenetics 2011; 63: 13–21.
Yurchenco PD, Patton BL . Developmental and pathogenic mechanisms of basement membrane assembly. Curr Pharm Design 2009; 15: 1277–1294.
Haralson MA, Hassell JR . Extracellular Matrix: A Practical Approach. IRL Press/Oxford University Press: Ithaca, NY/Oxford, 1995.
Yoon KA, Park JH, Han J, Park S, Lee GK, Han JY et al. A genome-wide association study reveals susceptibility variants for non-small cell lung cancer in the Korean population. Hum Mol Genet 2010; 19: 4948–4954.
Gylfe AE, Sirkia J, Ahlsten M, Jarvinen H, Mecklin JP, Karhu A et al. Somatic mutations and germline sequence variants in patients with familial colorectal cancer. Int J Cancer 2010; 29: 29.
Akasaka H, Katsuya T, Saitoh S, Sugimoto K, Ohnishi H, Congrains A et al. A promoter polymorphism of lamin A/C gene is an independent genetic predisposition to arterial stiffness in a Japanese general population (the Tanno and Sobetsu study). J Atheroscler Thromb 2009; 16: 404–409.
Bailey SD, Xie C, Do R, Montpetit A, Diaz R, Mohan V et al. Variation at the NFATC2 locus increases the risk of thiazolidinedione-induced edema in the Diabetes REduction Assessment with ramipril and rosiglitazone Medication (DREAM) study. Diabet Care 2010; 33: 2250–2253.
Uhl GR, Liu QR, Drgon T, Johnson C, Walther D, Rose JE et al. Molecular genetics of successful smoking cessation: convergent genome-wide association study results. Arch Gen Psychiatry 2008; 65: 683–693.
Flachsbart F, Franke A, Kleindorp R, Caliebe A, Blanche H, Schreiber S et al. Investigation of genetic susceptibility factors for human longevity—a targeted nonsynonymous SNP study. Mutat Res 2010; 694: 13–19.
Kimura K, Wakamatsu A, Suzuki Y, Ota T, Nishikawa T, Yamashita R et al. Diversification of transcriptional modulation: large-scale identification and characterization of putative alternative promoters of human genes. Genome Res 2006; 16: 55–65.
Long MY, Langley CH . Natural-selection and the origin of jingwei, a chimeric processed functional gene in Drosophila. Science 1993; 260: 91–95.
Hirotsune S, Yoshida N, Chen A, Garrett L, Sugiyama F, Takahashi S et al. An expressed pseudogene regulates the messenger-RNA stability of its homologous coding gene. Nature 2003; 423: 91–96.
Tam OH, Aravin AA, Stein P, Girard A, Murchison EP, Cheloufi S et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature 2008; 453: U534–U538.
Geisert EE, Lu L, Freeman-Anderson NE, Templeton JP, Nassr M, Wang X et al. Gene expression in the mouse eye: an online resource for genetics using 103 strains of mice. Mol Vis 2009; 15: 1730–1763.
Sato S, Omori Y, Katoh K, Kondo M, Kanagawa M, Miyata K et al. Pikachurin, a dystroglycan ligand, is essential for photoreceptor ribbon synapse formation. Nat Neurosci 2008; 11: 923–931.
Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 316: 1336–1341.
Harismendy O, Notani D, Song XY, Rahim NG, Tanasa B, Heintzman N et al. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature 2011; 470: 264–268.
Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 2010; 26: 2336–2337.
Acknowledgements
This project was supported by R01MH078069 to Dr van den Oord and K01MH093731 to Dr Adkins. We thank researchers involved in the initial STAR*D clinical trial (NIMH N01-MH90003), led by study director A John Rush, MD, the STAR*D Genetics project (NIMH R01-MH072802), led by Steven P Hamilton, MD PhD, and STAR*D participants.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Translational Psychiatry website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Adkins, D., Clark, S., Åberg, K. et al. Genome-wide pharmacogenomic study of citalopram-induced side effects in STAR*D. Transl Psychiatry 2, e129 (2012). https://doi.org/10.1038/tp.2012.57
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2012.57
Keywords
This article is cited by
-
Investigating genetic variants for treatment response to selective serotonin reuptake inhibitors in syndromal factors and side effects among patients with depression in Taiwanese Han population
The Pharmacogenomics Journal (2023)
-
Exploratory genome-wide analyses of cortical inhibition, facilitation, and plasticity in late-life depression
Translational Psychiatry (2023)
-
Genome-wide analysis suggests the importance of vascular processes and neuroinflammation in late-life antidepressant response
Translational Psychiatry (2021)
-
The Positive and Negative Experiences of 342 Antidepressant Users
Community Mental Health Journal (2020)
-
Pharmakogenetik in der Psychiatrie: eine Standortbestimmung
Der Nervenarzt (2018)
