
Abstract
Aberrant methylation of the adenomatous polyposis coli (APC) gene promoter occurs in about 40% of breast tumours and has been correlated with reduced APC protein levels. To what extent epigenetic alterations of the APC gene may differ according to specific breast cancer phenotypes, remains to be elucidated. Our aim was to explore the role of APC methylation in the inflammatory breast cancer (IBC) phenotype. The status of APC gene promoter hypermethylation was investigated in DNA from normal breast tissues, IBC and non-IBC by both conventional and real-time quantitative methylation-specific PCR (MSP). APC methylation levels were compared with APC mRNA and protein levels. Hypermethylation of the APC gene promoter was present in 71% of IBC samples (n=21) and 43% of non-IBC samples (n=30) by conventional MSP (P=0.047). The APC gene also showed an increased frequency of high methylation levels in IBC (in 74% of cases, n=19) vs non-IBC (in 46% of cases, n=35) using a qMSP assay (P=0.048). We observed no significant association between APC methylation levels by qMSP and APC mRNA or protein expression levels. In conclusion, for the first time, we report the association of aberrant methylation of the APC gene promoter with the IBC phenotype, which might be of biological and clinical importance.
Similar content being viewed by others


Main
The adenomatous polyposis coli (APC) gene, mapped to chromosome 5q21 (Kinzler et al, 1991), plays a prominent role in the development of colorectal cancer, both in the autosomal dominant inherited familial APC syndrome (Bodmer et al, 1987; Groden et al, 1991; Joslyn et al, 1991; Kinzler et al, 1991; Nishisho et al, 1991) and in sporadic colorectal cancer (Fearon and Vogelstein, 1990; Miyoshi et al, 1992; Powell et al, 1992). An impaired function of APC, most often attributable to mutations within the coding sequence of the gene, leads to a lack of degradation and nuclear accumulation of β-catenin, which acts as a transcriptional activator, causing loss of cell growth control (Sparks et al, 1998). Moreover, APC functions in pathways counteracting metastasis by mediating intercellular adhesion and stabilising the cytoskeleton (Fearnhead et al, 2001).
Similar to findings in colorectal cancers, it has been suggested that disruption of the APC/β-catenin pathway may be involved in breast cancer. Loss of APC expression and upregulation of β-catenin have been described in human breast cancer and breast cancer cells (Ho et al, 1999; Jonsson et al, 2000; Schlosshauer et al, 2000). Somatic APC mutations are reported in only a minority of breast cancers (Furuuchi et al, 2000), despite high rates of allelic loss at chromosome locus 5q21 (Thompson et al, 1993; Medeiros et al, 1994). Nevertheless, epigenetic inactivation of APC due to DNA methylation is frequently present in both breast cancer cell lines and breast cancer tissue. In most cultured breast cancer cells, there is a complete concordance between APC promoter methylation and silencing of its transcript (Virmani et al, 2001). Cellular APC expression can be restored after demethylation with 5-aza-2′-deoxycytidine treatment. APC promoter methylation also occurs in a significant number of primary breast tumours (ranging from 28 to 53% of cases, depending on the applied technology) (Jin et al, 2001; Virmani et al, 2001; Liu et al, 2007). The frequency of APC methylation in primary breast tumours increases with tumour stage and size (Virmani et al, 2001; Roa et al, 2004; Chen et al, 2007; Liu et al, 2007). Moreover, hypermethylation of APC can be detected in breast aspirate fluid DNA (Lee et al, 2004) and serum DNA from patients with pre-invasive and early-stage breast cancer (Dulaimi et al, 2004a). DNA methylation of APC in serum of early breast cancer patients who had not undergone adjuvant systemic treatment appeared to be an independent prognostic marker for overall survival (Muller et al, 2003, 2004). These findings indicate a potential for the use of this epigenetic marker, alone or in combination with other markers, both for early detection of breast cancer and for clinical, routine risk assessment in patients with breast cancer.
Epigenetic inactivation due to hypermethylation is well established for APC in breast carcinoma but as yet, few studies have addressed whether epigenetic alterations of the APC gene might characterise specific breast cancer phenotypes. Because of their similar treatment, inflammatory breast cancer (IBC) has been rarely studied separately from other forms of locally advanced breast cancer in the past, despite differences in age-specific incidence rates, clinical presentation, histology, hormone receptor status and, finally, prognosis (Lerebours et al, 2005). Recent reports, however, have revealed a unique molecular profile of IBC (Bertucci et al, 2004; Van Laere et al, 2005, 2007), shedding light on its unique ability to rapidly invade and metastasise. This has led us to consider the possibility of a specific methylation pattern for this breast cancer phenotype. Notably, biological processes that are altered in IBC include cell motility, cell adhesion, regulation of the cell cycle, invasion and angiogenesis (see reference Dirix et al, 2006 and references therein). An important role for epigenetic silencing of APC in IBC might be expected based on previous studies from our group that showed low expression of this gene in IBC compared to non-IBC (unpublished data).
The aim of our study was to explore the role of APC methylation in the IBC phenotype. We therefore investigated independently the methylation status of the APC gene promoter in DNA from inflammatory and non-inflammatory breast tumours, as well as from non-neoplastic breast tissues by using both a conventional methylation-specific PCR (MSP) method and a quantitative real-time MSP (qMSP) approach. Furthermore, we compared APC methylation levels obtained by qMSP with APC mRNA and protein levels.
Materials and methods
Patients and sample collection
A total of 105 snap-frozen or paraffin-embedded human breast tumour samples was retrieved from the tissue archive of the General Hospital Sint-Augustinus, Wilrijk, Belgium. Two different data sets were used in this study (see Table 1). Conventional MSP was performed on 51 paraffin-embedded tumour samples, of which 21 were IBC and 30 were non-IBC. Patient age at diagnosis ranged from 49 to 78 years (mean, 63 years) for non-IBC patients and from 31 to 82 years (mean, 60 years) for IBC patients. In addition, 27 non-neoplastic breast tissues from women who had breast reductive surgery were used. These patient ages ranged from 24 to 62 years (mean, 42 years). qMSP was performed on 54 snap-frozen tumour samples, of which 19 were IBC and 35 were non-IBC. Patient age at diagnosis ranged from 31 to 89 years (mean, 59 years) for non-IBC patients and from 45 to 74 years (mean, 60 years) for IBC patients. In addition, 15 matched normal breast tissues adjacent to a breast tumour and 9 non-neoplastic breast tissues from women who had breast reductive surgery were used in this experiment.
All cases were randomly selected. IBC was diagnosed according to the criteria mentioned in the AJCC (American Joint Committee on Cancer)-TNM staging system (Singletary et al, 2002). All patients with IBC showed diffuse enlargement of the involved breast of sudden onset. There was erythema and oedema of the skin involving more than one-third of the breast. The presence of dermal lymphatic invasion as an isolated observation was not sufficient for the diagnosis of IBC and was not necessary for the diagnosis either. Relevant clinical data at diagnosis, such as age, clinical stage (AJCC TNM staging system), histological grade (Nottingham modification of the Bloom and Richardson histological grading system (Bloom and Richardson, 1957; Elston, 1984)) and hormone receptor status were obtained from medical records. Each patient gave a written informed consent. All protocols were reviewed and approved by the Ethical Committee of the General Hospital Sint-Augustinus.
Conventional nested MSP
Paraffin sections (10 μm in thickness) from the primary breast tumours were cut on a clean blade. Paraffin ribbons were then mounted on membrane-covered metal frame slides (MMI, Glattbrug, Switzerland) and dried overnight. Tissue sections were deparaffinised in xylene and rehydrated through a graded alcohol series to nuclease-free water. Tissue sections were stained with the HistoGene staining solution (Arcturus Engineering Inc., Mountain View, CA, USA), specifically designed to preserve intact nucleic acids from captured cell populations. The SLμCut system (MMI) with a solid-state UV laser was used for microdissection of tumour epithelial cells (average tissue area of 8 mm2).
DNA extractions from microdissected tumour epithelial cells were performed using the QIAamp DNA Micro Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's protocols. Sodium bisulphite conversion of DNA samples was carried out using reagents provided in the EZ DNA Methylation Kit (Zymo Research, Orange, CA, USA). Five hundred ng of DNA were treated with sodium bisulphite following the manufacturer's recommendations.
Bisulphite-treated DNA was used as a template for a conventional nested MSP to determine the presence or absence of methylation of the promoter region of APC. Template amplification by a nested MSP was done essentially as described earlier (House et al, 2003). Step one primers flanked the CpG-rich promoter region of APC. PCR products of step one were diluted 1 : 1000 and subjected to the second step of MSP that incorporated a set of primers (labelled as unmethylated (U) or methylated (M)) that were designed to recognise sodium bisulphite-induced modifications of unmethylated cytosines. The primer sequences are listed elsewhere (House et al, 2003). Both steps of the nested MSP utilised a 25 μl reaction volume, 0.5 μl of Jump Start Red Taq DNA polymerase (Sigma, St Louis, MO, USA) and 4 μl of DNA template. The thermal profile for step one of the nested MSP was as follows: 3 min at 95°C, then 35 repetitive cycles of denaturation (95°C × 30 s), annealing (56°C × 30 s) and extension (72°C × 30 s), followed by a final 4 min extension at 72°C. Step two of the nested MSP was performed in a similar fashion with a few adjustments to the annealing temperature (60°C) to allow for optimal template discrimination, and to the PCR cycle number (25). DNA isolated from normal peripheral lymphocytes from healthy individuals served as a negative methylation control. In vitro methylated DNA was used as the positive methylation control. Each PCR product (10 μl) was loaded onto a 1.0% agarose gel, stained with GelStar nucleic acid stain solution (Cambrex Charles City, IA, USA) and directly visualised under UV illumination.
Quantitative real-time MSP
DNA extractions from snap-frozen tissue specimens were performed using the QIAamp DNA Micro Kit (Qiagen, Valencia, CA, USA) according to the manufacturer's protocols. Sodium bisulphite conversion of DNA samples was carried out using the EZ DNA Methylation Kit (Zymo Research, Orange, CA, USA). Two μg of DNA were treated with sodium bisulphite following the manufacturer's recommendations.
The real-time PCR-based quantification of APC methylation levels was done essentially as described earlier (Eads et al, 2000). Two sets of primers and probes, designed specifically for sodium bisulphite-converted DNA, were used: a methylated set for APC and a reference set, β-actin (ACTB), to normalise for input DNA. The primers and probes used for APC and ACTB are listed elsewhere (Eads et al, 2000; Usadel et al, 2002). Fluorogenic PCRs were carried out in a reaction volume of 25 μl in 96-well plates in a 7900 Sequence Detector (Applied Biosystems, Foster City, CA, USA). PCR was carried out in separate wells for each primer/probe set, and each sample was run in duplicate. The final reaction mixture consisted of 600 nmol l−1 of each primer (Applied Biosystems), 200 nmol l−1 of probe (Applied Biosystems) and 12.5 μl of Universal Master Mix (Applied Biosystems). Five μl of bisulphite-converted genomic DNA (250 ng) was used in each real-time MSP reaction. Thermal cycling was initiated with a first denaturation step of 95°C for 10 min. The thermal profile for the PCR was 95°C for 15 s and 60°C for 1 min. Data obtained during 50 cycles of amplification were analysed. Each plate included water blanks, a positive and a negative control. DNA isolated from normal peripheral lymphocytes from healthy individuals served as a negative methylation control. In vitro methylated human DNA (Zymo Research) was used as the positive methylation control.
The ratio between the values obtained in the two TaqMan analyses was used as a measure for the degree of methylation of APC. The percentage of fully methylated molecules at the APC locus was calculated by dividing the APC/ACTB ratio of a sample by the APC/ACTB ratio of fully methylated human DNA and multiplying by 100. We use the abbreviation PMR (Percentage of Methylated Reference) to indicate this measurement.
Quantitative real-time RT–PCR
APC methylation levels by qMSP were compared to APC mRNA expression levels by qRT–PCR in 52 frozen breast tumour biopsies (of which 19 were IBC and 33 were non-IBC). For two cases, tissue was not in sufficient quantity for performing both analyses.
RNA isolation and expression analyses were performed as described earlier (Van der Auwera et al, 2004). Briefly, total RNA was isolated with the RNeasy Mini RNA isolation kit (Qiagen, Valencia, CA, USA) and cDNA was synthesised from 1 μg of total RNA with the cDNA Archive kit (Applied Biosystems). Primers and probes were purchased from Applied Biosystems assay-on-demand. All samples were run on a 7900HT Fast Real-time PCR Detector (Applied Biosystems) in duplicate. Quantitative RT–PCR was performed in a reaction volume of 25 μl including 10 μl of cDNA. We averaged the expression of ACTB and 18SrRNA as internal reference genes to normalise input cDNA. The  method was used to compute relative expression values (Livak and Schmittgen, 2001).
method was used to compute relative expression values (Livak and Schmittgen, 2001).
Immunohistochemical staining of TMA
The tissue microarray (TMA) used in this study was described earlier (Van Laere et al, 2007). Briefly, formalin-fixed, paraffin-embedded tissue blocks containing breast cancer were retrieved from the archives of the General Hospital St-Augustinus. Areas of invasive breast carcinoma were identified on corresponding haematoxylin and eosin-stained slides, and the tissue blocks were cored and transferred to a recipient ‘master’ block using a Tissue Microarrayer (Beecher Instruments, Silver Spring, MD, USA). A total cohort of 76 cases of breast cancer was divided between two slides. Four cores were arrayed for each specimen.
TMA slides (4 μm in thickness) were deparaffinised, hydrated and subjected to antigen retrieval by heating slides in 10 mM citrate buffer (sodium citrate, pH 6.0) at 98°C for 30 min, followed by a 30 min cool-down and rinsing in wash buffer. The sections were incubated in 3% H2O2 for 10 min to inhibit endogenous peroxidase. The sections were subsequently incubated at room temperature with antibodies against the C-terminal region of the APC protein (C-20, 1 : 100, 60 min; Santa Cruz Biotechnology, Santa Cruz, CA, USA), after which sections were stained using the DAKO EnVision detection system (Dako, Glöstrup, Denmark). Sections were developed using 3,3′-diaminobenzidine for 10 min, counterstained with haematoxylin and mounted with Aquatex medium (Merck, Darmstadt, Germany).
Stained slides were then analysed for APC expression. Tumours were graded into two categories based on staining pattern: those showing loss of expression and those with expression.
Statistical analysis
APC gene methylation was analysed in two ways: as a continuous variable and as a dichotomous variable (i.e., high or low, using the median PMR value as a threshold). As a continuous variable, the association of APC methylation with type of tumour (inflammatory vs non-inflammatory) and with the different groups of clinical variables was summarised using the Mann–Whitney and Kruskal–Wallis non-parametric tests, according to the number of categories. To summarise the associations between APC methylation (coded as high or low) and tumour type or other clinical variables we used the χ2 statistics. The Fisher's Exact Test was used when one cell had an expected count of less than 5. The level of significance was set to P<0.05. All analyses were carried out using SPSS version 11.0 (SPSS, Chicago, IL, USA).
Results
APC methylation analysis by conventional MSP
The APC promoter region was analysed for presence or absence of methylation in formalin-fixed and paraffin-embedded tissue samples of invasive breast cancer (including 30 cases of non-IBC and 21 cases of IBC) and of non-neoplastic breast tissue from unaffected women (n=27) for control purposes. Hypermethylation of the APC gene promoter was observed in 28 of 51 (55%) breast tumours. A low frequency of methylation (11%) was also present in non-neoplastic breast tissues (obtained from reduction mammoplasty specimens, n=27). When we compared methylation frequencies of breast tumours, a higher frequency of APC promoter methylation was found in IBC (71%) when compared to non-IBC (43%) (P=0.047; χ2). Representative examples of the MSP products of bisulphite-treated samples using primers for specific unmethylated and methylated sequences are shown in Figure 1.

Results of conventional MSP in human breast cancer samples and normal breast tissues. The presence of a PCR product in lanes marked M indicates methylated APC promoter, whereas a product in lanes marked U indicates unmethylated promoter. IBC cases T11, T13 and T18, non-IBC case T16 and normal breast tissues N8 and N9 have both methylated and unmethylated promoters, whereas IBC case T17, non-IBC cases T12, T14, T15 and T19 and normal breast tissue N10 show only unmethylated promoters.
It was reported earlier that gene methylation increases with age (Issa, 2000). Therefore, the high incidence of aberrant methylation in breast carcinoma specimens compared with normal breast tissue could be, at least in part, because of the older age of the average patient with breast cancer. However, a statistical analysis revealed no correlation between patient age and frequency of hypermethylation in our series of normal breast specimens (n=27) (P=0.583; Mann–Whitney test). We compared breast carcinoma specimens from patients 50 years old or younger vs specimens from patients older than 50 years and again, we did not observe a significant difference in frequency of APC methylation between these two patient groups (P=0.349; χ2).
There was a significant correlation of APC promoter hypermethylation with the histological grade of the tumour (P=0.034; χ2) and with tumour stage (P=0.026; χ2) in our series of breast carcinomas (n=51). No associations were found with other tumour characteristics (tumour size, presence of axillary metastasis, expression of ER and PR and HER2 or p53 status).
APC methylation analysis by quantitative real-time MSP
A real-time MSP assay was used to quantify the relative number of methylated alleles in frozen breast tumour specimens from 35 patients with non-IBC and 19 patients with IBC. In addition, the methylation analysis was performed on DNA from matched non-neoplastic breast tissues from cancer patients (n=15) and from non-neoplastic breast tissues from unaffected women (n=9).
APC promoter methylation was detectable in 53 of 54 (98%) of breast carcinoma samples. In seven of nine (78%) non-neoplastic breast tissues from unaffected women, APC promoter methylation could be detected, albeit at a very low level. The median PMR value was 0.03 (range, 0.00–0.18) for non-neoplastic breast tissue (n=9), 0.68 (range, 0.00–94.31) for non-IBC (n=35) and 9.78 (range, 0.01–68.04) for IBC (n=19) (Figure 2). Comparison between non-neoplastic (n=9) and cancer tissue (n=54) revealed statistically significant results (P=0.006; Mann–Whitney test). Comparison of PMR values between IBC and non-IBC revealed no significant results. However, when cases were coded based on methylation levels (using as a threshold the median PMR value), the frequency of IBC samples with a high APC methylation status (74%) was significantly increased when compared to non-IBC (46%) (P=0.048; χ2).

Promoter methylation levels of APC in non-neoplastic breast tissue (n=9), matched normal breast tissue adjacent to a breast tumour (n=15) and biopsies from patients with non-IBC (n=35) and patients with IBC (n=19). Box plots show median, upper and lower quartiles. Median PMR values were 0.03 (range, 0–0.18) for non-neoplastic breast tissue, 0.29 (range, 0–26.35) for matched normal breast tissue adjacent to a breast tumour, 0.68 (range, 0.00–94.31) for non-IBC and 9.78 (range, 0.01–68.04) for IBC.
Furthermore, we compared APC methylation levels in matched normal appearing breast tissue and primary breast tumour. Despite the small sample size (for only 15 of 54 patients both normal and cancerous tissue was available for analysis), we did observe a trend towards a correlation between PMR values in normal appearing adjacent tissue and carcinoma cells (r=0.502, P=0.057) (Figure 3). PMR values for APC were significantly elevated in tumour compared with matched normal breast tissue (P=0.022; Wilcoxon signed-rank test). The median level of APC methylation was 3.27 (range, 0.01–94.31) in tumour compared with 0.29 (range, 0–26.35) in matched normal tissue.

The methylation levels were compared between matched benign and breast cancer tissues from 15 patients with breast cancer. y-axis, the percentages of fully methylated reference in each patient sample as obtained by qMSP. x-axis, benign (normal) or breast cancer (cancerous) tissues.
Overall, there were no statistical differences between PMR values in tumour tissue and clinicopathological factors (tumour size and stage, presence of axillary metastasis, histological grade, expression of ERs and HER2 or p53 status), with the exception of the expression of PR, for which a statistical trend toward higher methylation levels in PR-negative breast tumours was observed (P=0.075; Mann–Whitney). No association was found between PMR values in tumour tissue and patient age.
Concordance of conventional MSP and quantitative real-time MSP
As a validation, the results of qMSP were compared with those obtained by conventional MSP in 18 patient samples (including nine cases of non-IBC and nine cases of IBC). There was a substantial agreement between results obtained by quantitative real-time MSP and those obtained by conventional MSP (κ=0.658, P=0.005). Nine of 10 cases (90%) negative for APC hypermethylation by conventional MSP were also negative by quantitative real-time MSP whereas six of eight cases (75%) positive for APC hypermethylation by conventional MSP were also positive by quantitative real-time MSP.
Correlation of APC methylation levels by qMSP and APC mRNA expression
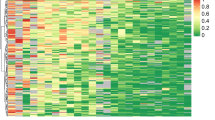
We compared promoter methylation status of APC defined by qMSP to loss of mRNA expression assessed by qRT–PCR in 52 of 54 (96%) breast tumour samples (including 19 IBC and 34 non-IBC) (Figure 4). In two breast cancer cases, frozen tissue was not sufficient in quantity for performing both analyses. The expression of APC appeared to be unrelated to the associated levels of APC methylation in breast cancer samples (n=52). The median APC mRNA expression level was 58.22 (range, 29.84–82.67) in samples with a high methylation status and 54.12 (range, 20.72–105.25) in samples with a low methylation status (P=0.600, Mann–Whitney).

Comparison of APC promoter methylation, APC mRNA expression and APC protein expression in 54 breast tumours. (Upper) Methylation status of the APC locus as determined by qMSP. (Lower) APC mRNA expression levels measured by real-time quantitative RT–PCR (n=52). APC protein status is indicated by circles located between the two charts (n=34). An open circle indicates APC positivity, whereas a black circle denotes APC negativity, as determined by immunohistochemistry (IHC).
Correlation of APC methylation levels by qMSP and APC protein expression
To determine whether APC promoter hypermethylation affects protein expression in breast tumours, we assessed the association between APC methylation levels by qMSP and loss of APC protein expression by immunohistochemistry in 34 of 54 (63%) breast tumours (including 22 cases of non-IBC and 12 cases of IBC) (Figure 4). Intensive cytoplasmic expression of the APC protein was found in 28 of 34 cases (82.3%) in our series, whereas the other six cases (17.6%) showed loss of the APC protein (Figure 5). The APC protein expression in breast cancer was independent of the APC methylation level. Of the 10 breast cancers with low APC methylation status by qMSP, eight (80%) expressed APC protein. Of the 24 cases of invasive breast cancer with high APC methylation status by qMSP, loss of APC protein was seen in four (16.7%).

APC expression in human breast cancer. (A) Breast cancer with normal APC expression. (B) Breast cancer with loss of APC protein expression.
Discussion
Epigenetic CpG-island hypermethylation of the promoter region has been proposed as an alternative way to inactivate the APC tumour suppressor gene. It has been shown that APC gene hypermethylation can be detected in ductal carcinoma in situ, lobular carcinoma in situ, and invasive ductal and lobular tumours of all pathological grades and stages, which indicates that hypermethylation of APC can be a relatively early event in breast tumorigenesis (Dulaimi et al, 2004a). It remains to be determined to what extent epigenetic alterations of the APC gene might characterise specific breast cancer phenotypes. This study represents the first comprehensive comparison of the frequencies of APC gene promoter hypermethylation in the inflammatory and non-inflammatory breast cancer phenotype. For this purpose, the APC gene was analysed independently by two different techniques: a conventional MSP and a quantitative real-time MSP.
We performed laser capture microdissection of archival histological sections to collect relatively pure, or at least considerably enriched, populations of tumour epithelial cells. Because of the inherent difficulties (i.e., DNA degradation as a result of the formalin fixation and the typical small sample size) of assaying for methylation in DNA recovered from microdissected tissues, we used the nested variant of the classical MSP to analyse these samples. This technique, originally developed by Palmisano et al (2000), incorporates a two-stage PCR approach that allows a more sensitive (one methylated allele in >50 000 unmethylated alleles) detection of methylation in clinical samples harbouring small amounts of poor quality DNA. The frequency of APC methylation in our series of primary breast cancers was consistent with that recently reported by others (Jin et al, 2001; Virmani et al, 2001; Dulaimi et al, 2004b; Liu et al, 2007). Notably, specimens from patients with IBC had the highest frequency of methylation. Although the P-value was only just significant, IBC samples showed 1.6 times more APC hypermethylation than non-IBC samples. APC methylation was associated with histological grade and tumour stage but this should be confirmed on a larger data set.
The strictly qualitative detection of methylation by the conventional MSP is hampered by some shortcomings (Ogino et al, 2006): (a) conventional MSP cannot reliably distinguish low levels of methylation from high levels of methylation; (b) the frequency of hypermethylation might be overestimated (Aggerholm and Hokland, 2000; Brakensiek et al, 2004); and (c) assay performance characteristics are difficult to assess. To validate the acquired conventional MSP data and, also, to obtain additional information on the fraction of methylated alleles in IBC and non-IBC, we combined our conventional MSP results with those obtained by a quantitative real-time MSP assay. This assay was performed separately on DNA samples purified from frozen breast tumour biopsies collected at the time of surgery. All samples were evaluated by a pathologist to assess the presence of malignant cells. Although for only 18 samples (nine IBC and nine non-IBC), snap-frozen and formalin-fixed and paraffin-embedded biopsies were available in parallel, the quantitative real-time MSP results corroborated the observations by conventional MSP. PMR values were not significantly different in IBC and non-IBC specimens (which might reflect variation in the tumour cell content of biopsies). However, high APC methylation levels were 1.6 times more frequently present in IBC specimens than in non-IBC specimens, which was just statistically significant.
The methylation levels of APC in samples from non-neoplastic tissue adjacent to a breast tumour correlated with that observed in samples of the corresponding tumour tissue (trend for statistical significance). Notably, APC methylation levels in ‘normal’ tissue from cancer patients were significantly higher than in breast tissue from unaffected women and in some cases values as high as those observed in breast tumour tissue were measured. In a recent comprehensive study of methylation of RASSF1A promoter in breast tissue samples, it was uncovered that primary tumours had significantly higher promoter methylation than control reduction mammoplasty tissue, with adjacent normal samples having intermediate levels (Yan et al, 2006). Interestingly, global profiling of DNA methylation revealed more methylated genes in normal adjacent samples than in normal donor control samples. There are several possible explanations for this observation. The intermediate levels of methylation might reflect the infiltration of neoplastic cells in histologically ‘normal’ surrounding breast tissue. Alternatively, the hypermethylation of genes in samples from normal tissue adjacent to a breast tumour could be explained by field cancerisation. This concept was originally proposed by Slaughter et al (1953) to explain the development of multiple primary tumours and locally recurrent cancer (Slaughter et al, 1953). Previous studies have confirmed that genetic abnormalities exist in histologically normal breast tissues immediately adjacent to invasive cancers (Deng et al, 1996). Now, it has also been suggested that the primary tumour might serve as an epicentre from which methylation density progressively diffuses outwards to surrounding tissues (Yan et al, 2006).
We found no significant difference in APC mRNA and protein levels among tissues with low or high APC methylation status. Possible explanations for these results are: (i) samples with APC promoter methylation may have only one allele affected, allowing expression from the unaltered allele (Esteller et al, 2000); (ii) gene silencing is not a single event, but instead a series of events that begin with a marked drop in transcription and ends with its complete cessation (Turker, 2002); (iii) APC methylation patterns among the tumour cells that constitute a given sample might be heterogeneous and (iv) APC expression might be inactivated by gene mutations or allelic losses and not by methylation of CpG sites in the promoter region.
To conclude, we have shown that aberrant methylation of the APC gene promoter characterises the IBC phenotype. It should be emphasised that more investigation is required to determine to what extent the epigenetic inactivation of APC affects the biological behaviour of these tumours. Our study was limited not only by the small sample size, but also by the use of a candidate gene approach that was based on the observation of low gene expression. The analysis of more IBC cases and the consideration of additional genes, which is facilitated by several recently described techniques, such as methylation-sensitive arbitrarily primed PCR, restriction landmark genomic sequencing and CpG-island microarrays (see reference (Ushijima, 2005) and references therein), could present a great opportunity for enriching our knowledge of IBC biology.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Aggerholm A, Hokland P (2000) DAP-kinase CpG island methylation in acute myeloid leukemia: methodology vs biology? Blood 95: 2997–2999
Bertucci F, Finetti P, Rougemont J, Charafe-Jauffret E, Nasser V, Loriod B, Camerlo J, Tagett R, Tarpin C, Houvenaeghel G, Nguyen C, Maraninchi D, Jacquemier J, Houlgatte R, Birnbaum D, Viens P (2004) Gene expression profiling for molecular characterization of inflammatory breast cancer and prediction of response to chemotherapy. Cancer Res 64: 8558–8565
Bloom HJ, Richardson WW (1957) Histological grading and prognosis in breast cancer; a study of 1409 cases of which 359 have been followed for 15 years. Br J Cancer 11: 359–377
Bodmer WF, Bailey CJ, Bodmer J, Bussey HJ, Ellis A, Gorman P, Lucibello FC, Murday VA, Rider SH, Scambler P (1987) Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature 328: 614–616
Brakensiek K, Langer F, Kreipe H, Lehmann U (2004) Low level of DAP-kinase DNA methylation in myelodysplastic syndrome. Blood 104: 1586–1587
Chen YL, Xie YT, Wen XZ, Deng DJ (2007) Aberrant methylation of APC and Bikunin CpG islands in sporadic breast carcinomas. Zhonghua Yu Fang Yi Xue Za Zhi 41 (Suppl): 17–19
Deng G, Lu Y, Zlotnikov G, Thor AD, Smith HS (1996) Loss of heterozygosity in normal tissue adjacent to breast carcinomas. Science 274: 2057–2059
Dirix LY, Dam PV, Prove A, Vermeulen PB (2006) Inflammatory breast cancer: current understanding. Curr Opin Oncol 18: 563–571
Dulaimi E, Hillinck J, de Caceres II, Al Saleem T, Cairns P (2004a) Tumor suppressor gene promoter hypermethylation in serum of breast cancer patients. Clin Cancer Res 10: 6189–6193
Dulaimi E, Hillinck J, de Caceres II, Al Saleem T, Cairns P (2004b) Tumor suppressor gene promoter hypermethylation in serum of breast cancer patients. Clin Cancer Res 10: 6189–6193
Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, Danenberg PV, Laird PW (2000) MethyLight: a high-throughput assay to measure DNA methylation. Nucl Acids Res 28: e32
Elston CW (1984) The assessment of histological differentiation in breast cancer. Aust N Z J Surg 54: 11–15
Esteller M, Sparks A, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Gonzalez S, Tarafa G, Sidransky D, Meltzer SJ, Baylin SB, Herman JG (2000) Analysis of adenomatous polyposis coll promoter hypermethylation in human cancer. Cancer Res 60: 4366–4371
Fearnhead NS, Britton MP, Bodmer WF (2001) The ABC of APC. Hum Mol Genet 10: 721–733
Fearon ER, Vogelstein B (1990) A genetic model for colorectal tumorigenesis. Cell 61: 759–767
Furuuchi K, Tada M, Yamada H, Kataoka A, Furuuchi N, Hamada J, Takahashi M, Todo S, Moriuchi T (2000) Somatic mutations of the APC gene in primary breast cancers. Am J Pathol 156: 1997–2005
Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M (1991) Identification and characterization of the familial adenomatous polyposis coli gene. Cell 66: 589–600
Ho KY, Kalle WH, Lo TH, Lam WY, Tang CM (1999) Reduced expression of APC and DCC gene protein in breast cancer. Histopathology 35: 249–256
House MG, Guo M, Iacobuzio-Donahue C, Herman JG (2003) Molecular progression of promoter methylation in intraductal papillary mucinous neoplasms (IPMN) of the pancreas. Carcinogenesis 24: 193–198
Issa JP (2000) CpG-island methylation in aging and cancer. Curr Top Microbiol Immunol 249: 101–118
Jin Z, Tamura G, Tsuchiya T, Sakata K, Kashiwaba M, Osakabe M, Motoyama T (2001) Adenomatous polyposis coli (APC) gene promoter hypermethylation in primary breast cancers. Br J Cancer 85: 69–73
Jonsson M, Borg A, Nilbert M, Andersson T (2000) Involvement of adenomatous polyposis coli (APC)/beta-catenin signalling in human breast cancer. Eur J Cancer 36: 242–248
Joslyn G, Carlson M, Thliveris A, Albertsen H, Gelbert L, Samowitz W, Groden J, Stevens J, Spirio L, Robertson M (1991) Identification of deletion mutations and three new genes at the familial polyposis locus. Cell 66: 601–613
Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D (1991) Identification of FAP locus genes from chromosome 5q21. Science 253: 661–665
Lee A, Kim Y, Han K, Kang CS, Jeon HM, Shim SI (2004) Detection of tumor markers including carcinoembryonic antigen, APC, and cyclin D2 in fine-needle aspiration fluid of breast. Arch Pathol Lab Med 128: 1251–1256
Lerebours F, Bieche I, Lidereau R (2005) Update on inflammatory breast cancer. Breast Cancer Res 7: 52–58
Liu Z, Yang L, Cui DX, Liu BL, Zhang XB, Ma WF, Zhang Q (2007) Methylation status and protein expression of adenomatous polyposis coli (APC) gene in breast cancer. Ai Zheng 26: 586–590
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25: 402–408
Medeiros AC, Nagai MA, Neto MM, Brentani RR (1994) Loss of heterozygosity affecting the APC and MCC genetic loci in patients with primary breast carcinomas. Cancer Epidemiol Biomarkers Prev 3: 331–333
Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y (1992) Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet 1: 229–233
Muller HM, Fiegl H, Widschwendter A, Widschwendter M (2004) Prognostic DNA methylation marker in serum of cancer patients. Ann N Y Acad Sci 1022: 44–49
Muller HM, Widschwendter A, Fiegl H, Ivarsson L, Goebel G, Perkmann E, Marth C, Widschwendter M (2003) DNA methylation in serum of breast cancer patients: an independent prognostic marker. Cancer Res 63: 7641–7645
Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, Koyama K, Utsunomiya J, Baba S, Hedge P (1991) Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science 253: 665–669
Ogino S, Kawasaki T, Brahmandam M, Cantor M, Kirkner GJ, Spiegelman D, Makrigiorgos GM, Weisenberger DJ, Laird PW, Loda M, Fuchs CS (2006) Precision and performance characteristics of bisulfite conversion and real-time PCR (MethyLight) for quantitative DNA methylation analysis. J Mol Diagn 8: 209–217
Palmisano WA, Divine KK, Saccomanno G, Gilliland FD, Baylin SB, Herman JG, Belinsky SA (2000) Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res 60: 5954–5958
Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW (1992) APC mutations occur early during colorectal tumorigenesis. Nature 359: 235–237
Roa JC, Anabalon L, Tapia O, Martinez J, Araya JC, Villaseca M, Guzman P, Roa I (2004) Promoter methylation profile in breast cancer. Rev Med Chil 132: 1069–1077
Schlosshauer PW, Brown SA, Eisinger K, Yan Q, Guglielminetti ER, Parsons R, Ellenson LH, Kitajewski J (2000) APC truncation and increased beta-catenin levels in a human breast cancer cell line. Carcinogenesis 21: 1453–1456
Singletary SE, Allred C, Ashley P, Bassett LW, Berry D, Bland KI, Borgen PI, Clark G, Edge SB, Hayes DF, Hughes LL, Hutter RV, Morrow M, Page DL, Recht A, Theriault RL, Thor A, Weaver DL, Wieand HS, Greene FL (2002) Revision of the American Joint Committee on Cancer staging system for breast cancer. J Clin Oncol 20: 3628–3636
Slaughter DP, Southwick HW, Smejkal W (1953) Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 6: 963–968
Sparks AB, Morin PJ, Vogelstein B, Kinzler KW (1998) Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res 58: 1130–1134
Thompson AM, Morris RG, Wallace M, Wyllie AH, Steel CM, Carter DC (1993) Allele loss from 5q21 (APC/MCC) and 18q21 (DCC) and DCC mRNA expression in breast cancer. Br J Cancer 68: 64–68
Turker MS (2002) Gene silencing in mammalian cells and the spread of DNA methylation. Oncogene 21: 5388–5393
Usadel H, Brabender J, Danenberg KD, Jeronimo C, Harden S, Engles J, Danenberg PV, Yang S, Sidransky D (2002) Quantitative adenomatous polyposis coli promoter methylation analysis in tumor tissue, serum, and plasma DNA of patients with lung cancer. Cancer Res 62: 371–375
Ushijima T (2005) Detection and interpretation of altered methylation patterns in cancer cells. Nat Rev Cancer 5: 223–231
Van der Auwera I, Van Laere SJ, Van den Eynden GG, Benoy I, van Dam P, Colpaert CG, Fox SB, Turley H, Harris AL, Van Marck EA, Vermeulen PB, Dirix LY (2004) Increased angiogenesis and lymphangiogenesis in inflammatory versus noninflammatory breast cancer by real-time reverse transcriptase-PCR gene expression quantification. Clin Cancer Res 10: 7965–7971
Van Laere S, Van der Auwera I, Van den Eynden G, Van Hummelen P, van Dam P, Van Marck E, Vermeulen PB, Dirix L (2007) Distinct molecular phenotype of inflammatory breast cancer compared to non-inflammatory breast cancer using Affymetrix-based genome-wide gene-expression analysis. Br J Cancer 97: 1165–1174
Van Laere S, Van der Auwera I, Van den Eynden GG, Fox SB, Bianchi F, Harris AL, van Dam P, van Marck EA, Vermeulen PB, Dirix LY (2005) Distinct molecular signature of inflammatory breast cancer by cDNA microarray analysis. Breast Cancer Res Treat 93: 237–246
Virmani AK, Rathi A, Sathyanarayana UG, Padar A, Huang CX, Cunnigham HT, Farinas AJ, Milchgrub S, Euhus DM, Gilcrease M, Herman J, Minna JD, Gazdar AF (2001) Aberrant methylation of the adenomatous polyposis coli (APC) gene promoter 1A in breast and lung carcinomas. Clin Cancer Res 7: 1998–2004
Yan PS, Venkataramu C, Ibrahim A, Liu JC, Shen RZ, Diaz NM, Centeno B, Weber F, Leu YW, Shapiro CL, Eng C, Yeatman TJ, Huang THM (2006) Mapping geographic zones of cancer risk with epigenetic biomarkers in normal breast tissue. Clin Cancer Res 12: 6626–6636
Acknowledgements
Ilse Van der Auwera is a research assistant of the Fund for Scientific Research Flanders. Steven Van Laere is a predoctoral assistant of the University of Antwerp. This work was supported by the Fund for Scientific Research Flanders Grant No G.0430.03. We thank Manon van Engeland for the collaboration and support in the DNA methylation experiments. Furthermore, we thank the technical staff of the Laboratories of Pathology from the General Hospital Sint-Augustinus and the University of Antwerp for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Van der Auwera, I., Van Laere, S., Van den Bosch, S. et al. Aberrant methylation of the Adenomatous Polyposis Coli (APC) gene promoter is associated with the inflammatory breast cancer phenotype. Br J Cancer 99, 1735–1742 (2008). https://doi.org/10.1038/sj.bjc.6604705
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6604705
Keywords
This article is cited by
-
Mechanisms of tRNA-derived fragments and tRNA halves in cancer treatment resistance
Biomarker Research (2020)
-
Gap Junctions and Wnt Signaling in the Mammary Gland: a Cross-Talk?
Journal of Mammary Gland Biology and Neoplasia (2019)
-
Inflammatory Breast Cancer: Diagnostic, Molecular and Therapeutic Considerations
Current Breast Cancer Reports (2019)
-
Epigenetic Downregulation of PTEN in Gallbladder Cancer
Journal of Gastrointestinal Cancer (2017)
-
Genetic analysis of intestinal polyp development in Collaborative Cross mice carrying the Apc Min/+ mutation
BMC Genetics (2016)
