
Abstract
Anaplastic astrocytoma (AA, WHO grade III) is, second to Glioblastoma, the most common and most malignant type of adult CNS tumour. Since survival for patients with AA varies markedly and there are no known useful prognostic or therapy response indicators, the primary purpose of this study was to examine whether knowledge of the known genetic abnormalities found in AA had any clinical value. The survival data on 37 carefully sampled AA was correlated with the results of a detailed analysis of the status of nine genes known to be involved in the development of astrocytic tumours. These included three genes coding for proteins in the p53 pathway (TP53, p14ARFand MDM2), four in the Rb1 pathway (CDKN2A, CDKN2B, RB1 and CDK4) and PTEN and EGFR. We found that loss of both wild-type copies of any of the three tumour suppressor genes CDKN2A, CDKN2B and RB1 or gene amplification of CDK4, disrupting the Rb1 pathway, were associated with shorter survival (P=0.009). This association was consistent in multivariate analysis, including adjustment for age (P=0.013). The findings suggest that analysis of the genes coding for Rb1 pathway components provides additional prognostic information in AA patients receiving conventional therapy.
Similar content being viewed by others


Main
Adult astrocytomas are classified by WHO (Kleihues and Cavanee, 2000) into Diffuse Astrocytoma malignancy grade II (AII), Anaplastic Astrocytoma malignancy grade III (AA) and Glioblastoma malignancy grade IV (GB). Standard treatment of both AA and GB is surgery followed by radiotherapy. Anaplastic Astrocytoma patients have variable survival with a median survival of approximately 3 years – intermediate between AII (median approximately 7 years) and GB (8–12 months) (Kleihues and Cavanee, 2000; Herfarth et al, 2001; Backlund et al, 2003). The histological criteria for an AA diagnosis are also intermediate between the AII and the GB, the histological findings in AA ranging from tumours that may be difficult to separate from an AII to those that have to be distinguished from a GB. The fact that AA may develop from an AII or may arise de novo and that both have a tendency to develop into a GB also provides us with diagnostic difficulties. Adequate histological sampling is essential to exclude a GB. As with all astrocytic tumours, young age has been generally accepted as a positive prognostic factor for AA (Perry et al, 1999; Smith et al, 2001; Rasheed et al, 2002). However, no dependable histological criteria have been found to provide accurate prognostic indicators permitting a subdivision of AA into good and bad prognostic groups.
More than a dozen genes have been documented as being aberrant in astrocytic tumours and these abnormalities generally accumulate in a particular order in the different malignancy grades. TP53 is the most commonly aberrant gene in AII and AA – approximately 50% of these tumours have no wild-type TP53 and a further 10–15% have one mutated and one wild-type allele (Rasheed et al, 1994; Ichimura et al, 2000; Kato et al, 2000). Glioblastomas that develop from AII or AA also have frequent TP53 mutations, while the incidence of TP53 mutations in primary or de novo GB is lower – around 35% (Watanabe et al, 1996). Yet, primary GBs show disruption of the p53 pathway at similar levels to AII and AA due to homozygous deletions of p14ARF or amplification and overexpression of MDM2 (Ichimura et al, 2000). The majority of GBs also have genetic abnormalities disrupting the Rb1 pathway, that is, by one of the following: loss of wild-type CDKN2A (generally together with loss of wild-type CDKN2B by homozygous deletion) or loss of wild-type RB1 or amplification and overexpression of CDK4. In addition, losses of wild-type PTEN occur in almost one-half of GB and amplification of EGFR in approximately one-third (Ueki et al, 1996; Rasheed et al, 1997; Schmidt et al, 1999; Ichimura et al, 2000). Anaplastic Astrocytoma also occasionally show losses of both wild-type copies of the three tumour suppressor genes (TSGs) located on 9p, CDKN2A, CDKN2B, p14ARF (Ichimura et al, 2000), simultaneously disrupting both the Rb1 and p53 pathways, as well as loss of both wild-type copies of the PTEN gene on 10q (Schmidt et al, 1999). Loss of both wild-type RB1 has not been reported in AA (Ueki et al, 1996; Ichimura et al, 2000). Amplification of proto-oncogenes occurs almost exclusively in GB. In AA, CDK4 / MDM2 amplification is rarely seen and EGFR amplification occurs in <10% of the cases (Bigner et al, 1988; Ichimura et al, 2000). Thus, AAs as defined by histological criteria appear to consist of a small fraction showing genetic changes similar to GB (e.g. loss of wild-type CDKN2A, CDKN2B, p14ARF and PTEN; amplification of CDK4 and EGFR) and a majority showing abnormalities similar to AII (i.e. loss of wild-type TP53). In AA, the frequency of hemizygous deletions of significant gene loci is greater than that found in AII, but less than that found in GB and in most cases the retained allele is wild type (Schmidt et al, 1994; Nishizaki et al, 1998; Ichimura et al, 2000).
Attempts to correlate the genetic findings in AA with patient outcome have to-date not provided any convincing correlations with survival (Galanis et al, 1998; Olson et al, 1998; James et al, 1999; Smith et al, 2001; Rasheed et al, 2002).
The primary purpose of this study was to explore potential associations between somatic mutations and survival in a series of 37 AA, since the identification of genetic prognostic indicators would be of clinical value. We have studied three genes involved in the p53 pathway (TP53, MDM2 and p14ARF), four genes involved in the Rb1 pathway (RB1, CDKN2A, CDKN2B and CDK4), PTEN and EGFR. With the exception of TP53, all these genes are more commonly altered in GB than in AA. Clinical data were also considered and tested for associations with survival and when appropriate used to adjust for potential confounders. Our findings suggest that patients with well-sampled tumours fulfilling the WHO criteria for AA (Kleihues and Cavanee, 2000) having genetic abnormalities affecting any of the genes coding for the components of the Rb1 pathway have a poor outcome.
Materials and methods
Patients and tumour tissue
A total of 37 AA patients operated at the Karolinska Hospital, Stockholm, or Sahlgrenska University Hospital, Gothenburg between 1989 and 1994 were included in the study. All tumours were extensively sampled for histology and were reassessed and classified as AA according to the histological criteria in the 2000 WHO classification (Kleihues and Cavanee, 2000). End of follow-up of the patients was set to 1st October 2003, providing a minimum follow-up time of 10.3 years. The clinical data were collected from the patient records at the hospitals where they were treated and / or followed up. The data included age and sex of the patient, tumour localisation, duration and type of symptoms before diagnosis, date of operation(s), postoperative radiotherapy and chemotherapy and date of death. Data were missing from a maximum of one case for any clinical parameter. All operations were gross total. Thus, no cases diagnosed on biopsy only were included. The tumour tissue was particularly well sampled – often all tissue was processed with adjacent pieces alternatively frozen for molecular analysis and fixed for histology (up to 15 blocks). The criteria for an AA diagnosis were strictly according to WHO (Kleihues and Cavanee, 2000). The tumours were moderately to highly cellular and the tumour cells showed a high nuclear : cytoplasmic ratio, distinct nuclear and cytoplasmic pleomorphism with coarse nuclear chromatin and mitotic activity. There was no notable microvascular proliferation and no necrotic foci found anywhere in the material from any case. All patients gave informed consent and ethics committee approval for the project was obtained at all sites.
Genetic analysis
Most of the genetic data on this tumour series has been previously published (Reifenberger et al, 1994; Ichimura et al, 1996; Schmidt et al, 1999; Ichimura et al, 2000; Liu et al, 2000). Briefly, tumour DNA was isolated from tumour pieces histopathologically judged to contain more than 70% tumour cells. Each tumour was genetically analysed and compared with the individual patient's white blood cell DNA. RB1, CDKN2A, CDKN2B, p14ARF, TP53 and PTEN were analysed for deletions and mutations. The allelic status was assessed by studying paired samples of each patients’ blood and tumour DNA by either densitometry on Southern blotting using 32P-labelled gene-specific probes or microsatellite analysis using 33P-labelled PCR in loci in or around each gene by means of a PhosphorImager analysis (for details, see (Schmidt et al, 1994; Ichimura et al, 1996; Schmidt et al, 1999; Ichimura et al, 2000)). In addition, multiplex PCR was used for assessment of homozygous deletion of PTEN, as other methods would be complicated by the pseudogene of PTEN (Schmidt et al, 1999). Mutation screening of RB1, CDKN2A, CDKN2B, p14ARF, TP53 and PTEN was carried out using either single-strand conformational polymorphism (SSCP), denaturing gradient gel electrophoresis (DGGE) or direct sequencing to identify somatic mutations in retained alleles (Ichimura et al, 1996; Schmidt et al, 1999; Ichimura et al, 2000) (see Figure 1). The presence or absence of amplification of the proto-oncogenes CDK4, MDM2 and EGFR was determined using microsatellite analysis and Southern blotting with densitometry (Reifenberger et al, 1994; Ichimura et al, 2000; Liu et al, 2000). The genetic data were complete. In Table 2 each single patient is presented with an individual publication number, the same as used in previous publications (Reifenberger et al, 1994; Schmidt et al, 1994; Ichimura et al, 1996; Ichimura et al, 1998; Schmidt et al, 1999; Ichimura et al, 2000; Liu et al, 2000).

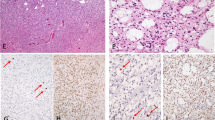
Genetic analysis of AA18. (A) A Southern blot hybridised with probes for CDKN2A and CDKN2B showing homozygous deletion of both genes. The control locus hybridisation (D2S44) on the same blot gives a clear signal from the tumour lane. (B) Microsatellite analysis at D17S796, the locus approximately 1.3 Mb telomeric to TP53. The densitometric profile indicates LOH in tumour DNA (arrow). (C) Sequencing of TP53 exon 8. G at position 824 is mutated to T (arrow), resulting in Cys to Phe missense mutation at codon 275. B, blood DNA; T, tumour DNA.
Statistical analysis and definitions of genes and pathways as ‘normal’ or ‘abnormal’
The molecular information was restricted to genetic data. All genes studied were in each tumour classified as either ‘normal’ or ‘abnormal’. The TSGs (CDKN2A, CDKN2B, RB1, TP53, p14ARF and PTEN) were categorised as ‘abnormal’ when there was no wild-type allele present, either due to homozygous deletion or deletion of one allele and mutation of the other. A comparison of survival in cases with all genes categorised as ‘normal’ with those having any gene(s) categorised as ‘abnormal’ was also carried out.
For the proto-oncogenes (EGFR, CDK4 and MDM2), gene amplification (as in previous publications defined as a minimum average of five copies per genome) (Ichimura et al, 2000; Liu et al, 2000) was categorised as ‘abnormal’. Cellular pathways (p53 and Rb1 pathways) were categorized in a similar manner. A pathway was classified as ‘abnormal’ if any gene coding for a protein component of the pathway was classified as ‘abnormal’.
Of the 37 tumours analysed, 25 (68%) were from the patients’ first operation. They are referred to as ‘primary’ cases. A total of 12 patients (32%) had a prior operation and a histological diagnosis of either AII (six cases) or AA (six cases). The six cases that had a recurrent AA are referred to as ‘recurrent’ cases, while the six cases that had progressed from an AII are referred to as ‘progressed’ cases. The starting point for patient follow-up was the date of the operation from which the tumour tissue used in the genetic analysis was collected, survival being calculated from this date as we do not know the genetic status of the tumour removed at earlier operations. To address the potential bias of including the six ‘recurrent’ patients calculating survival as above, separate, additional correlation analysis was performed, with survival of the six ‘recurrent’ patients calculated from the date of their first AA operation, whenever a statistically significant association with survival was found.
To study the potential influence of single clinical or genetic factors on patient survival, univariate analysis using Wilcoxon–Gehan statistic (Gehan, 1965) was performed, with the patients divided into two groups, except for the age and duration of symptoms factors. When analysing age and survival, with age as a continuous variable, Cox Regression analysis was used. In multivariate analysis of the genetic factors and survival, Cox Regression analysis was carried out adjusting for age and whether the tumour studied was from a ‘primary’ or not (with the patients divided into two groups: ‘primary’ or ‘recurrent’ / ‘progressed’). All statistical analyses were performed using SPSS (Statistical Package for the Social Sciences) for Windows, release 12.0.1.
Results
Clinical data
In Table 1, the clinical data for all cases are summarised with median survival and some statistical comparisons. In Table 2, the patients have been placed in one of three groups: (a) ‘primary’ AA; (b) ‘progressed’ AA (progressed from A) or (c) ‘recurrent’ AA. Postoperative survival in the whole series varied from 4 months to at least 13.9 years with a median of 4.3 years. Nine of the 37 patients were still alive at the end of follow-up, with a minimum follow-up time of 10.3 years. The fraction of males was uncommonly small in the series, with a male : female ratio of 1 : 1.5. There was no significant difference in survival between the sexes, or between the 33 (89%) patients with supratentorial and the four (11%) with infratentorial tumours. Seven (28%) of the 25 patients with ‘primary’ AA were alive at the end of follow-up as well as one (17%) of the six patients from each of the ‘recurrent’ and ‘progressed’ groups. The tumours were equally distributed to the left and right sides, with the patients with the tumour on the left side having shorter median survival, but only of borderline significance (P=0.078).
The mean age at the operation from which material was obtained was 38.8 years. The mean age at first diagnosis of AA in all the 37 patients was 37.3. Dividing all the 37 patients into three approximately equally large age groups (see Table 1 and Figure 2) demonstrates that the oldest age group (>45 years) had the shortest median survival (1.8 years), although with a wide range (220 days to 8.5 years). The association between age and survival was borderline significant both in univariate analysis of the three groups (P=0.096), and in Cox regression analysis with age as a continuous variable (P=0.093).

Survival curves in three different age groups: 8–30 years (n=12); 31–45 years (n=14); 46–68 years (n=11). Univariate analysis: P=0.096.
The most common début symptom in all cases indicating the disease process was epilepsy, described in 65% (24 out of 37) of the patients. The duration between these presenting symptoms and the operation for AA ranged in the ‘primary’ cases from a few days to one patient (AA59) with a history of epilepsy for 6.5 years. As shown in Table 1, dividing the ‘primary’ cases according to the duration of symptoms before operation revealed no significant survival differences.
In the following text, the cases will be referred to in the order that they appear in Table 2:
The 25 ‘primary’ AA had a median survival of 5.6 years and a mean age of 41 years. In all, 15 of them received postoperative radiotherapy (RT) at a total dose of 50–54 Gy given in 28–30 fractions over 6 weeks. One patient (AA29) was given a course of 37 Gy over 2.5 weeks and survived 1.9 years. Eight patients (AA59, AA76, AA26, AA65, AA73, AA105, AA94 and AA57) were never irradiated. In one case (AA106), it was not possible to access the postoperative notes. As shown in Table 1, the median postoperative survival of the eight ‘primary’ nonirradiated AA patients (6.3 years) was slightly, but not significantly, longer than for the 16 irradiated patients (5.1 years). Four of the 25 ‘primary’ AA patients (AA69, AA37, AA94, AA7) received chemotherapy postoperatively, one of which received polychemotherapy (AA69). Six of the 25 patients (AA102, AA69, AA100, AA79, AA37 and AA93) were later reoperated upon due to relapse and three of them (AA69, AA100 and AA93) then had evidence of progression to GB. The average time to progression in these three cases was 1.9 years.
Six patients had progressed from an AII to an AA (average time to progression to AA was 4.4 years). Their mean age was 32 years and median survival following the AA operation was 2.1 years. Three of them were irradiated after the AA operation (AA104, AA50 and AA3), while the other three (AA17, AA18 and AA86) had received postoperative RT after their first operation for AII. AA86 received postoperative chemotherapy and AA104 was reoperated again after 19 months at which time the tumour had progressed to a GB. AA3 was still alive at the end of follow-up (survival >13.4 years after AA and >13.8 years after the AII diagnosis).
The six ‘recurrent’ cases had had AA at the first operation and their mean age at that time was 28.5 years. Median survival, counted from that first AA operation, was 10.8 years while median survival from the operation for the recurrence was 1.6 years. Of the six ‘recurrent’ cases, three (AA2, AA87, AA51) had had postoperative RT after the first operation. AA53 received RT after the relapse and two patients (AA81 and AA13) did not receive RT at any time. AA81 was given postoperative chemotherapy after the recurrence, while AA51 had received chemotherapy after the first operation. Three patients (AA51, AA53, AA87) were reoperated a third time and none showed evidence of progression to GB.
Genetic data
The genetic data for each individual tumour is presented in Table 2, with mutation nomenclature according to recommendations of the Nomenclature Working Group whenever applicable (Antonarakis, 1998). In Table 3, the results of univariate and multivariate analysis of the potential influence of single genetic factors on patient survival are given. In these analyses, shorter median survival was always seen in cases categorised as ‘abnormal’ as compared to those categorised as ‘normal’.
As presented in Table 3, the only single genetic aberration showing a significant negative association with survival was CDK4 amplification. The statistical significance was borderline in univariate analysis (P=0.058) but significant in multivariate analysis adjusting for age and whether the tumour was ‘primary’ or ‘recurrent’ / ‘progressed’ (P=0.019). Disruption of the Rb1 pathway was negatively associated with survival in univariate (P=0.009) as well as in multivariate analysis (P=0.013). The survival curves for the Rb1 pathway groups ‘normal’ and ‘abnormal’ are shown in Figure 3.

Survival curves for cases with loss of both wild-type alleles of any TSG coding for a component of the Rb1 pathway (CDKN2A, CDKN2B, RB1) or amplification of CDK4, that is, Rb1 pathway classified as ‘abnormal’ (n=8; median survival 1.4 years) and cases coded as ‘normal’ (n=29; median survival 5.8 years), respectively. Univariate analysis: P=0.009.
A total of 15 cases (AA15, AA59, AA76, AA34, AA102, AA26, AA69, AA100, AA61, AA105, AA37, AA52, AA104, AA50, AA81) with all genes classified as ‘normal’ survived longer than those with at least one of the genes classified as ‘abnormal’. This was statistically significant in univariate analysis (P=0.013).
To avoid bias in including the survival data counting survival for the six ‘recurrent’ patients with the ‘recurrent’ operation as starting point, separate survival analyses were carried out when a statistically significant association between the genetic results and survival was found. As described in Materials and Methods, this was done calculating survival for these six cases from the date of their first AA operation. Similar P-values as those reported above were seen in each additional univariate and multivariate analysis. For the main finding, the association between Rb1 pathway and survival, the P-value in multivariate analysis was 0.014 (compared to P=0.013).
Discussion
We have focused exclusively on AA as defined by the latest edition of the WHO classification of tumours of the nervous system (Kleihues and Cavanee, 2000). Each tumour was extensively sampled for histology and no tumours with the histological characteristics of GB or AII were accepted for inclusion. Each piece of tissue used for the genetic analysis was histologically characterised to ensure that tumour tissue was studied. This was confirmed by the genetic analysis of the tumour tissue which, when compared with the individual patients’ white blood cell DNA, showed genetic abnormalities in all cases (e.g. loss of heterozygosity (LOH) at a number of sites in addition to point mutations) (Ichimura et al, 1998; Miyakawa et al, 2000). The age range of the patients and the location of the tumours are comparable to similar studies, while the sex distribution is slightly unusual showing comparatively high number of female subjects (Kleihues and Cavanee, 2000). A linear relationship between young age and longer survival was not possible to demonstrate with the limited number of patients in this study, but when the patients were divided up in three equally large groups, the subjects in the oldest group had the worst and the youngest age group the best outcome. The survival data overall in this series is in accord with previous studies (Perry et al, 1999; Herfarth et al, 2001; Lin et al, 2003) showing a huge variation from a few months to more than 13 years, widely overlapping that observed for both AII and GB. Our minimum follow-up here was 10.3 years, which is necessary in studies where survival can be long.
In a situation where no curative treatment is available, as is the case for AA, therapy is often modified individually unless all patients have been included in a particular clinical trial. Most, but far from all, patients received postoperative RT of 50–54 Gy. This variation in treatment is further complicated by the ‘recurrent’ and ‘progressed’ tumours, where the patients have received radiotherapy at their first diagnosis and were therefore not eligible for further irradiation. In addition, it is not unusual to use cytotoxic drugs at recurrences.
The primary aim of this study was to test for potential associations between mutations and survival in a clinically, histopathologically and genetically well-defined series of AA. There are few studies of this type where only AA is analysed. Most studies combine AA with the much more malignant GB under the terms ‘malignant astrocytomas’ or ‘malignant gliomas’. The study of Smith et al (2001) analysed AA separately and found a positive association between TP53 mutation and survival and a negative association between loss of wild-type PTEN and survival. They included a large number of AA diagnosed with biopsy only, making a reliable histological classification more difficult. We did not find any clear associations between TP53 mutation and survival in any of the comparisons. Since TP53 mutations can have a dominant negative effect (Willis et al, 2004), we even performed an analysis with cases with TP53 mutation and retention of one wild-type allele coded as ‘abnormal’, and this, too, showed no association with survival (P=0.165). It is notable that the commonest point mutation of TP53 (while differing in base substitution) occurred at codon 273 in 27% of the tumours (Table 2). Similar findings have been reported in GB recently (Ohgaki et al, 2004).
As seen in Table 2, some tumours had loss of one wild-type allele of some TSGs. In fact, for all studied TSGs, this was relatively frequent. We generally do not know the biological significance of TSGs losing only one allele. It may result in lowered molar levels of the gene product providing a growth advantage. The concept of haploinsufficiency has been well documented for relatively few genes, but might be of relevance, for example, CDKN2A and p14ARF (see review by Quon and Berns, 2001, and references therein) (Quon and Berns, 2001). Epigenetic mechanisms, such as methylation, could also be of importance. Although the sensitivity of SSCP and DGGE as mutation screening methods for point mutations is high, we cannot exclude the possibility that we have missed mutations in single cases, since sequencing analysis was only performed in cases where abnormalities were detected by SSCP or DGGE.
Although 22 of 37 AAs had loss of one PTEN allele, only one (AA29) had lost both wild-type PTEN copies (loss of one copy and point mutation of the stop codon of the other resulting in the addition of eight amino acids to the C-terminal). The biological consequence of this PTEN mutation is unclear as all critical regions of the protein were wild type. Survival was relatively short in this case (1.9 years). In a previous study of 129 GB, we found that a combination of aberrations of Rb1 pathway-related genes by loss of both wild-type alleles of TSGs or amplification of proto-oncogenes (almost always combined with p53 pathway abnormalities) and loss of both wild-type copies of PTEN was associated with particularly poor outcome (Backlund et al, 2003). It is important to note that none of the AAs here had this genetic profile.
The major finding of the present study is that patients with AA who have a somatic mutation profile resulting in loss of both wild-type copies of TSGs or amplification of oncogenes involved in the Rb1 pathway (as found frequently in GB), have a worse outcome as compared to AAs without these abnormalities. There was two out of 25 (8%) ‘primary’ AAs with this type of Rb1 pathway abnormality. The ‘recurrent’ and ‘progressed’ cases showed this type of Rb1 pathway abnormality in 50%. TP53 mutations, generally with loss of the second allele, are much more common in our cohort of AA (about two-third of all cases regardless of whether ‘primary’ or ‘recurrent’) than we found in our GB series (45 out of 129 or 35%) (Backlund et al, 2003), a finding that is in line with previous reports (Rasheed et al, 1994; Kato et al, 2000).
The material examined and the findings of this study are complex. The numerous statistical tests performed in a retrospective study like this have to be interpreted with caution. A larger series of AA will have to be examined to confirm and extend these results. New developments are providing the technologies that will permit high throughput genetic analysis on larger series of uniformly treated patients. Hopefully, the findings in this study will stimulate further efforts in this regard leading to improved analysis of AA, providing clinicians with important prognostic information and eventually targets for molecular therapy.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Antonarakis SE (1998) Recommendations for a nomenclature system for human gene mutations. Nomenclature Working Group. Hum Mutat 11: 1–3
Backlund LM, Nilsson BR, Goike HM, Schmidt EE, Liu L, Ichimura K, Collins VP (2003) Short postoperative survival for glioblastoma patients with a dysfunctional Rb1 pathway in combination with no wild-type PTEN. Clin Cancer Res 9: 4151–4158
Bigner SH, Burger PC, Wong AJ, Werner MH, Hamilton SR, Muhlbaier LH, Vogelstein B, Bigner DD (1988) Gene amplification in malignant human gliomas: clinical and histopathologic aspects. J Neuropathol Exp Neurol 47: 191–205
Galanis E, Buckner J, Kimmel D, Jenkins R, Alderete B, O’Fallon J, Wang CH, Scheithauer BW, James CD (1998) Gene amplification as a prognostic factor in primary and secondary high-grade malignant gliomas. Int J Oncol 13: 717–724
Gehan EA (1965) A generalized two-sample Wilcoxon test for doubly censored data. Biometrika 52: 650–653
Herfarth KK, Gutwein S, Debus J (2001) Postoperative radiotherapy of astrocytomas. Semin Surg Oncol 20: 13–23
Ichimura K, Bolin MB, Goike HM, Schmidt EE, Moshref A, Collins VP (2000) Deregulation of the p14ARF / MDM2 / p53 pathway is a prerequisite for human astrocytic gliomas with G1–S transition control gene abnormalities. Cancer Res 60: 417–424
Ichimura K, Schmidt EE, Goike HM, Collins VP (1996) Human glioblastomas with no alterations of the CDKN2A (p16INK4A, MTS1) and CDK4 genes have frequent mutations of the retinoblastoma gene. Oncogene 13: 1065–1072
Ichimura K, Schmidt EE, Miyakawa A, Goike HM, Collins VP (1998) Distinct patterns of deletion on 10p and 10q suggest involvement of multiple tumor suppressor genes in the development of astrocytic gliomas of different malignancy grades. Genes Chromosomes Cancer 22: 9–15
James CD, Galanis E, Frederick L, Kimmel DW, Cunningham JM, Atherton-Skaff PJ, O’Fallon JR, Jenkins RB, Buckner JC, Hunter SB, Olson JJ, Scheithauer BW (1999) Tumor suppressor gene alterations in malignant gliomas: histopathological associations and prognostic evaluation. Int J Oncol 15: 547–553
Kato H, Kato S, Kumabe T, Sonoda Y, Yoshimoto T, Han SY, Suzuki T, Shibata H, Kanamaru R, Ishioka C (2000) Functional evaluation of p53 and PTEN gene mutations in gliomas. Clin Cancer Res 6: 3937–3943
Kleihues P, Cavanee WK (2000) WHO Classification of Tumours, Pathology and Genetics of the Nervous System. Lyon: IARC Press
Lin CL, Lieu AS, Lee KS, Yang YH, Kuo TH, Hung MH, Loh JK, Yen CP, Chang CZ, Howng SL, Hwang SL (2003) The conditional probabilities of survival in patients with anaplastic astrocytoma or glioblastoma multiforme. Surg Neurol 60: 402–406, discussion 406
Liu L, Ichimura K, Pettersson EH, Goike HM, Collins VP (2000) The complexity of the 7p12 amplicon in human astrocytic gliomas: detailed mapping of 246 tumors. J Neuropathol Exp Neurol 59: 1087–1093
Miyakawa A, Ichimura K, Schmidt EE, Varmeh-Ziaie S, Collins VP (2000) Multiple deleted regions on the long arm of chromosome 6 in astrocytic tumours. Br J Cancer 82: 543–549
Nishizaki T, Ozaki S, Harada K, Ito H, Arai H, Beppu T, Sasaki K (1998) Investigation of genetic alterations associated with the grade of astrocytic tumor by comparative genomic hybridization. Genes Chromosomes Cancer 21: 340–346
Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, Burkhard C, Schuler D, Probst-Hensch NM, Maiorka PC, Baeza N, Pisani P, Yonekawa Y, Yasargil MG, Lutolf UM, Kleihues P (2004) Genetic pathways to glioblastoma: a population-based study. Cancer Res 64: 6892–6899
Olson JJ, Barnett D, Yang J, Assietti R, Cotsonis G, James CD (1998) Gene amplification as a prognostic factor in primary brain tumors. Clin Cancer Res 4: 215–222
Perry A, Jenkins RB, O’Fallon JR, Schaefer PL, Kimmel DW, Mahoney MR, Scheithauer BW, Smith SM, Hill EM, Sebo TJ, Levitt R, Krook J, Tschetter LK, Morton RF, Buckner JC (1999) Clinicopathologic study of 85 similarly treated patients with anaplastic astrocytic tumors. An analysis of DNA content (ploidy), cellular proliferation, and p53 expression. Cancer 86: 672–683
Quon KC, Berns A (2001) Haplo-insufficiency? Let me count the ways. Genes Dev 15: 2917–2921
Rasheed A, Herndon JE, Stenzel TT, Raetz JG, Kendelhardt J, Friedman HS, Friedman AH, Bigner DD, Bigner SH, McLendon RE (2002) Molecular markers of prognosis in astrocytic tumors. Cancer 94: 2688–2697
Rasheed BK, McLendon RE, Herndon JE, Friedman HS, Friedman AH, Bigner DD, Bigner SH (1994) Alterations of the TP53 gene in human gliomas. Cancer Res 54: 1324–1330
Rasheed BK, Stenzel TT, McLendon RE, Parsons R, Friedman AH, Friedman HS, Bigner DD, Bigner SH (1997) PTEN gene mutations are seen in high-grade but not in low-grade gliomas. Cancer Res 57: 4187–4190
Reifenberger G, Reifenberger J, Ichimura K, Meltzer PS, Collins VP (1994) Amplification of multiple genes from chromosomal region 12q13–14 in human malignant gliomas: preliminary mapping of the amplicons shows preferential involvement of CDK4, SAS, and MDM2. Cancer Res 54: 4299–4303
Schmidt EE, Ichimura K, Goike HM, Moshref A, Liu L, Collins VP (1999) Mutational profile of the PTEN gene in primary human astrocytic tumors and cultivated xenografts. J Neuropathol Exp Neurol 58: 1170–1183
Schmidt EE, Ichimura K, Reifenberger G, Collins VP (1994) CDKN2 (p16 / MTS1) gene deletion or CDK4 amplification occurs in the majority of glioblastomas. Cancer Res 54: 6321–6324
Smith JS, Tachibana I, Passe SM, Huntley BK, Borell TJ, Iturria N, O’Fallon JR, Schaefer PL, Scheithauer BW, James CD, Buckner JC, Jenkins RB (2001) PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst 93: 1246–1256
Ueki K, Ono Y, Henson JW, Efird JT, von Deimling A, Louis DN (1996) CDKN2 / p16 or RB alterations occur in the majority of glioblastomas and are inversely correlated. Cancer Res 56: 150–153
Watanabe K, Tachibana O, Sata K, Yonekawa Y, Kleihues P, Ohgaki H (1996) Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol 6: 217–223, discussion 23–24
Willis A, Jung EJ, Wakefield T, Chen X (2004) Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 23: 2330–2338
Acknowledgements
We thank the Departments of Neurosurgery and Pathology at Karolinska and Sahlgrenska University Hospitals, and the many other hospitals in Sweden who helped us obtain follow-up data. Dr Dan Grandér is acknowledged for his helpful critical review of the manuscript. This work was supported by grants from the Swedish Cancer Society, Stockholm's Cancer Society, King Gustaf V Jubilee Fund, Funds of Karolinska Institutet, CAMPOD, Cancer Research UK, The Ludwig Institute for Cancer Research and Jacqueline Serousse Memorial Foundation for Cancer Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Bäcklund, L., Nilsson, B., Liu, L. et al. Mutations in Rb1 pathway-related genes are associated with poor prognosis in Anaplastic Astrocytomas. Br J Cancer 93, 124–130 (2005). https://doi.org/10.1038/sj.bjc.6602661
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6602661
Keywords
This article is cited by
-
Deep DNA sequencing of MGMT, TP53 and AGT in Mexican astrocytoma patients identifies an excess of genetic variants in women and a predictive biomarker
Journal of Neuro-Oncology (2023)
-
Therapeutic vulnerability to PARP1,2 inhibition in RB1-mutant osteosarcoma
Nature Communications (2021)
-
Clinicopathologic Characteristics of Brain Tumors are Associated with the Presence and Patterns of TP53 Mutations: Evidence from the IARC TP53 Database
NeuroMolecular Medicine (2014)
-
The Cdk inhibitor flavopiridol enhances temozolomide-induced cytotoxicity in human glioma cells
Journal of Neuro-Oncology (2013)
-
Growth factor receptors signaling in glioblastoma cells: therapeutic implications
Journal of Neuro-Oncology (2009)
