
Abstract
Gene mutations independent of BCR::ABL1 have been identified in newly diagnosed patients with chronic myeloid leukemia (CML) in chronic phase, whereby mutations in epigenetic modifier genes were most common. These findings prompted the systematic analysis of prevalence, dynamics, and prognostic significance of such mutations, in a clinically well-characterized patient population of 222 CML patients from the TIGER study (CML-V) by targeted next-generation sequencing covering 54 myeloid leukemia-associated genes. In total, 53/222 CML patients (24%) carried 60 mutations at diagnosis with ASXL1 being most commonly affected (n = 20). To study mutation dynamics, longitudinal deep sequencing analysis of serial samples was performed in 100 patients after 12, 24, and 36 months of therapy. Typical patterns of clonal evolution included eradication, persistence, and emergence of mutated clones. Patients carrying an ASXL1 mutation at diagnosis showed a less favorable molecular response to nilotinib treatment, as a major molecular response (MMR) was achieved less frequently at month 12, 18, and 24 compared to all other patients. Patients with ASXL1 mutations were also younger and more frequently found in the high risk category, suggesting a central role of clonal evolution associated with ASXL1 mutations in CML pathogenesis.
Similar content being viewed by others

Introduction
Chronic myeloid leukemia (CML) is characterized by the BCR::ABL1 gene fusion, which codes for a constitutively active tyrosine kinase [1]. Inhibition of this aberrant kinase via imatinib – a selective tyrosine kinase inhibitor (TKI), revolutionized the treatment of CML [2]. Second-generation TKIs such as nilotinib improved treatment further, resulting in earlier and higher response rates while offering lower risk of disease progression [3]. Whilst some patients show resistance to TKI treatment and are at high risk of progression, for a large number of patients living without treatment has become a realistic aim if durable deep molecular remission under TKI treatment has been achieved [4, 5]. According to previous studies, myeloid-leukemia associated mutations occur in addition to BCR::ABL1 in approximately 30% of newly diagnosed CML patients [6,7,8]. Mutations were most commonly identified in epigenetic modifier genes such as ASXL1, DNMT3A, and TET2. Two retrospective studies suggested an inferior response to TKI of CML patients harboring mutations of epigenetic regulator genes [7, 9]. Large population studies revealed an increased prevalence of such variants also in elderly people lacking hematological disorders [10,11,12], suggesting a rate of 10% in people older than 65 years but only 1% in people younger than 50 years of age [11]. However, our recent findings of ASXL1 mutations in children and young adults with CML indicate that the existence of such mutations can be disease- rather than age-related in CML patients [13].
To advance our understanding of the role of mutations in addition to BCR::ABL1, the study presented here aimed to evaluate their prevalence and dynamics in a large, well-characterized patient population in an ongoing prospective clinical trial. Furthermore, the clinical impact of such mutations was investigated to understand their effect on molecular response during TKI treatment.
Patients and methods
Patients
The patient cohort consisted of 222 randomly selected chronic phase CML patients treated within the prospective German TIGER study (CML-V; NCT01657604). All patients received nilotinib based TKI therapy. Patients’ characteristics are provided in Table 1 and Supplementary Table 1. Median age was 52 years (range, 18–79 years), 140 (63%) were male. 116 patients (52.3%) received nilotinib monotherapy and 106 patients (47.7%) a combination with nilotinib and pegylated interferon alpha 2b (pegIFN) according to the study protocol. All procedures were in accordance with the standards of the ethics committee and the Declaration of Helsinki. Informed consent for participation in the trial and molecular analysis was obtained from all patients.
Sample preparation
Peripheral blood samples of all patients were collected at diagnosis and every three months on treatment. Genomic DNA (gDNA) was isolated from peripheral blood leukocytes after red cell lysis using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s recommendations. Additionally, buccal swab samples were used where available for the isolation of constitutional DNA to verify the somatic origin of mutations. The RNeasy Mini Kit (Qiagen) or TRIzol reagent (Invitrogen, Carlsbad, CA, USA) was used according to the manufacturer’s recommendations to extract total leukocyte RNA after red cell lysis from at least 20 ml of peripheral blood. As previously published, complementary DNA synthesis was performed using random hexamer primers and Moloney murine leukemia virus reverse transcriptase (Invitrogen) [14] or SuperScript IV VILO Master Mix (Thermo Fisher Scientific, Waltham, MA, USA) according to manufacturer’s instructions.
Real-time quantitative PCR and Sanger sequencing
For this study, patients’ molecular response to therapy was measured after 12, 18, 24, and 36 months of therapy, unless the patient had died or dropped out. In total 657 samples were analyzed. LightCycler technology (Roche Diagnostics, Mannheim, Germany) or the Quantstudio5 (Thermo Fisher) system was employed for BCR::ABL1 transcript quantification as described previously [15, 16]. Molecular response was assessed by RT-qPCR determining the ratio of BCR::ABL1 to ABL1 transcripts and reported on the International Scale (IS) [17]. Treatment response was reported as log level reduction of BCR::ABL1IS according to the European LeukemiaNet (ELN) recommendations: major molecular response (MMR) ≤ 0.1%, MR4 ≤ 0.01%, MR4.5 ≤ 0.0032%, MR5 ≤ 0.001% [18, 19]. Patients with failure criteria according to ELN 2013 recommendations [18] were analyzed by Sanger sequencing for the presence of BCR::ABL1 kinase domain mutations.
Next-generation sequencing
Next-generation sequencing (NGS) of 54 genes frequently mutated in various entities of myeloid malignancies was performed using the TruSight Myeloid Panel on the Mini Seq platform (Illumina, San Diego, CA, USA) with 8 samples per run. In total, 522 samples were analyzed; 222 samples at diagnosis with follow-up analyses for 100 patients after 12, 24, and 36 months of therapy. Mean amplicon coverage ranged from 1762–21,465 reads/amplicon. The collected data was analyzed using the VariantStudio Software (version 2.0; Illumina, San Diego, CA, USA) with the hg19 genome build and a sensitivity level of 5% variant allele frequency (VAF). Variants detected at low VAF (<5%) were only included if the same variant was found with a frequency above 5% in at least one sample of the respective patient. As recommended by the developers, the software’s own PASS filter was used. Synonymous coding mutations and intronic polymorphisms were excluded. Variants with an allele frequency of ≥1% in the general population were also excluded. In accordance to recent recommendations of Branford et al. [8] the Combined Annotation Dependent Depletion (CADD; version1.3) [20] was used with a threshold of ≥20 to filter missense variants; additionally, only those variants predicted to be damaging by at least 3 of 4 prediction tools were included. We used the open access tools Polymorphism Phenotyping v2 (PolyPhen-2) [21] and dbNSFP version 4.0a [22] for SIFT [23], MutationTaster [24] and FATHMM [25]. Missense variants considered to be germline by buccal-swap analysis (n = 5) or due to a VAF of 50% or consistent VAF of 50 or 100% over time (n = 6) which otherwise met the published criteria of clinically relevant mutations [8] were regarded separately. For the predictive analysis, the ASXL1 c.1934dupG mutation was excluded based on frequent sequencing artifacts in this homopolymer area.
Statistics
For statistical analysis, all timepoints were calculated from the day of randomization or from the day of the first visit when trial participation was confirmed. To be allocated to a certain time, a molecular analysis was performed within the ±3-months interval around that time. Any event worse than “no MMR”, that is disease progression or death from any cause within an interval, was rated as “no MMR” for this and any following interval. The probability of MMR was given as the proportion of patients with at least MMR or “no MMR” at 12, 18, and 24 months, according to their mutation status. The association between two categorical variables was assessed with Fisher’s exact test. Group comparisons with regard to continuous variables were performed with the Mann-Whitney U-Test. To investigate the influence of mutations on molecular status at 12, 18, and 24 months under consideration of other baseline variables, multiple logistic regression was used. All statistical tests were explorative. The level of significance was 0.05. Point estimations are given together with their 95% confidence interval (95%-CI). SAS version 9.4 was used for analysis.
Results
Molecular response
BCR::ABL1IS was assessed at months 12, 18, and 24 for all 222 patients of this sub-cohort of the TIGER study. After one year of TKI treatment, 181 patients (82% of 222, 95%-CI: 76–86%) had achieved at least MMR, whilst 49% of patients showed a molecular response of MR4 or better (95%-CI: 43–56%), 29% had at least MR4.5 (95%-CI: 23–35%), and 5% were in MR5 (95%-CI: 3–9%). At 18 months, one patient had been lost to follow-up and three patients (rated as “no MMR”) had died. Thus, of 221 patients, 192 (87%, 95%-CI: 82–91%) had achieved at least MMR, whilst 54% (95%-CI: 48–61%) were assessed as MR4 or better and 32% (95%-CI: 26–39%) had achieved at least MR4.5. MR5 was achieved for 8% (95%-CI: 5–13%) of 221 patients. Before month 24, one more patient was lost to follow-up. At 24 months, of 220 patients, 193 (88%, 95%-CI: 83–91%) had achieved at least MMR. Proportions of patients with at least MR4, at least MR4.5, or MR5 were 59% (95%-CI: 52–65%), 38% (95%-CI: 32–45%), and 14% (95%-CI: 10–19%), respectively.
Mutations at diagnosis
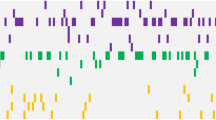
At the time of diagnosis, 53 of 222 patients (24%) carried 60 mutations in addition to BCR::ABL1, affecting the genes ASXL1, ATRX, BCOR, BCORL1, CALR, CBL, CBLB, CUX1, DNMT3A, FBXW7, GATA2, GNAS, IKZF1, JAK2, KDM6A, PHF6, RUNX1, SMC3, TET2, U2AF1, and WT1 (Fig. 1). In detail, 18 distinct nonsense mutations, 13 frameshift variants, 22 missense mutations, two inframe deletions and one inframe insertion were detected. ASXL1 mutations were most frequently identified and were observed in 20 patients (9%). Six patients showed more than one additional mutation. This affected patient #36 (ASXL1, ASXL1), #66 (CALR, FBXW7), #159 (RUNX1, ATRX, PHF6), #171 (RUNX1, GNAS), #177 (ASXL1, BCOR) and #180 (ASXL1, TET2). Median age of patients carrying additional mutations to BCR::ABL1 at diagnosis was 54 years (range, 19–78 years). Buccal swab samples were available for 13 patients and showed no or lower-level mutation frequencies, hence indicating a somatic origin of the respective mutations.

Mutations were found in 53/222 patients (24%) with ASXL1 being the most commonly affected gene (n = 20). Patients and genes are displayed in columns and rows, respectively. A unique color is assigned to each mutated gene. Bisected cell represents two variants in the same gene and patient.
Apart from the mutations described above, 10 distinct missense variants were detected in 14 patients that were considered to be germline but otherwise met the published criteria of clinically relevant mutations [8] (Supplementary Table 1). The affected genes were ASXL1, ATRX, CBL, IDH2, KDM6A, RUNX1, TET2, TP53, and ZRSR2. These variants were found in all analyzed samples of the respective patients with a VAF of approximately 50% (n = 8) or 100% (n = 2) and/or were confirmed to be germline via buccal swab analysis if available (n = 5). Four patients not analyzed in follow-up carried a TET2 variant with a VAF of approximately 50% that was otherwise considered clinically relevant.
Mutation dynamics
Investigating follow-up samples of 100 randomly chosen patients of the cohort described above, we observed three mutational patterns: (I) eradication, (II) persistence, (III) emergence of mutations (Table 2a–c). Regarding mutation prevalence at month 12, 24, and 36 please refer to Fig. 2A–C.

Longitudinal deep sequencing analysis of serial samples after (A) 12, (B) 24, and (C) 36 months of therapy. DNMT3A mutations were most frequently identified. Patients and genes are displayed in columns and rows, respectively. A unique color was assigned to each mutated gene. A bisected cell represents two variants in the same gene and patient. Asterisks stand for low allele variants (<5% variant allele frequency).
Most mutations were eradicated together with the BCR::ABL1 clone under nilotinib therapy (pattern I, Table 2a). Nineteen patients showed this type of mutation pattern, most commonly affecting ASXL1 with n = 12 detections. For most patients, the mutation was detected at diagnosis only and was undetectable during treatment. However, in patient #36 two ASXL1 mutations detected at diagnosis were also found at month 12. After a year of nilotinib treatment the mutations had been suppressed to low VAFs of 2.7% and 2.3%, respectively (Table 2a). At this point the patient showed a BCR::ABL1IS transcript level of 2.8%. Analysis of subsequent follow-up samples showed that BCR::ABL1 as well as both ASXL1 mutations were cleared which indicates that the ASXL1 mutations were located in Ph+ cells. At month 24, patient #21 showed re-emergence of an ASXL1 mutation initially detected at diagnosis (the month 12 sample was tested negative) with a BCR::ABL1IS transcript level of 13% at that time (Table 2a). Another follow-up sample was analyzed at month 36. The ASXL1 variant increased in frequency compared to month 24 and a BCR::ABL1IS transcript level of 35% was assessed at this point. The patient underwent allogeneic stem cell transplantation. Though not included in NGS follow-up analysis, patient #194, who presented with an ASXL1 mutation at diagnosis, showed an E255K BCR::ABL1 mutation at months 12 and 18 as detected by Sanger sequencing (Supplementary Table 1).
In five patients mutations persisted (pattern II; Table 2b). Follow-up samples also revealed a reduction of the allele burden of four of these mutations (DNMT3A in all cases) compared to the level at diagnosis whereas the CALR mutation displayed a slightly increased VAF in follow up analyses. Since VAF was significantly above BCR::ABL1 transcript levels in all patients, the BCR::ABL1 translocation has most likely occurred on top of the preexisting mutation. Median age of these patients was 59 years (range, 42–71 years) at diagnosis.
Fifteen variants in 13/100 (13%) patients newly emerged in follow-up that had not been detected in the corresponding diagnostic samples (pattern III; Table 2c). Affected genes were ASXL1, BCOR, DNMT3A, IKZF1, JAK2, STAG2, TET2, and TP53 with the highest prevalence in DNMT3A (n = 5). Since VAFs of these variants were significantly above BCR::ABL1IS transcript levels in all patients, these mutated clones are considered to be BCR::ABL1 negative. Patients gaining a mutation in follow-up had a median age of 56 years (range, 32–73 years) at diagnosis.
Data further suggests different mutational dynamics in follow-up samples in patients with more than one mutation. In patients #19 and #36 both mutations followed the same mutation patterns of eradication and emergence, respectively (Table 2a, c). Non-parallel mutation dynamics were observed in patients #11 (BCOR eradication; TET2 emergence), #21 (ASXL1 eradication/re-emergence, ASXL1 and STAG2 emergence), #66 (FBXW7 eradication, CALR persistence) and #71 (ASXL1 eradication, DNMT3A emergence) (Table 2a–c).
Clinical impact of mutations
In patients without mutation at diagnosis, the probabilities of achieving MMR or better were 85% (95%-CI: 78–89%) at 12 months, 89% (95%-CI: 84–93%) at 18 months, and 89% (95%-CI: 84–93%) at 24 months. With 82% (95%-CI: 66–91%) at 12 months, 91% (95%-CI: 76–97) at 18 months, and 94% (95%-CI: 80–98%) at 24 months the probabilities of MMR in the patients with mutations (excluding ASXL1) were not significantly different. In contrast, patients carrying an ASXL1 mutation at diagnosis achieved significantly lower MMR probabilities than patients without any mutation (Fig. 3), showing 55% (95%-CI: 34–74%) at 12 months (p = 0.0036), 60% (95%-CI: 39–78%) at 18 months (p = 0.0018), and 65% (95%-CI: 43–82%) at 24 months (p = 0.0076). Investigating the categorizations “at least MR4“, “at least MR4.5”, and “at least MR5“, no statistically significant and clinically relevant difference between “no mutations” and either “ASXL1” or “other mutations” was found.

Patients carrying an ASXL1 mutation at diagnosis achieved significantly lower MMR probabilities than patients without any mutation, showing 55% (95%-CI: 34–74%) at 12 months (p = 0.0036), 60% (95%-CI: 39–78%) at 18 months (p = 0.0018), and 65% (95%-CI: 43–82%) at 24 months (p = 0.0076). Black vertical lines describe the 95% confidence intervals around the proportions indicated by the upper end of the bars. MMR Major molecular response.
Using multiple logistic regression, apart from mutation status, no additional variable reported in Table 1 had a significant influence on the achievement of the molecular status “MMR or better” at 12, 18, or 24 months. The odds ratios of ASXL1 as compared with “no mutations” were 0.222 (95%-CI: 0.084–0.589), 0.179 (95%-CI: 0.065–0.496), and 0.223 (95%-CI: 0.079–0.631) with increasing MMR proportions and time.
Table 1 displays patient characteristics distinguished by “no mutations” and “all mutations”, the latter separated in “ASXL1 mutations” and “other mutations”. Any mutations at diagnosis occurred in 46/203 (22.7%) low risk patients according to the EUTOS score and in 7/19 (36.8%) high risk patients (n.s.). However, all mutations other than ASXL1 were only identified in low-risk patients and accordingly, all seven mutations identified in 19 high-risk patients were ASXL1 mutations. ASXL1 mutations at diagnosis occurred in 13/203 (6.4%) low risk patients and 7/19 (38.8%) high risk patients (p = 0.0004). Patients with ASXL1 mutation were more frequently at high risk than patients with other (p = 0.0005) or no mutations (p = 0.0012). Moreover, patients with ASXL1 mutation were younger than patients with other mutations (p = 0.0380). No further associations between mutations and other patient characteristics were observed.
Discussion
This study provides a prospective analysis of leukemia-associated gene mutations in newly diagnosed CML patients treated within a controlled clinical trial. Several retrospective studies found that mutations in leukemia-associated genes in addition to BCR::ABL1 are not only a phenomenon seen in advanced or blast crisis CML but also in chronic phase CML [6,7,8,9]. As CML is primarily diagnosed in chronic phase, a better understanding of clonal molecular evolution may aid therapeutic decision-making, risk-stratification and management in the future.
At the time of diagnosis, patients most frequently carried ASXL1 mutations (n = 20). ASXL1 mutations are also commonly detected in other myeloid malignancies and are generally associated with a poor clinical outcome. As an epigenetic and transcriptional regulator, ASXL1 is involved in chromatin modifications and interacts with polycomb complex proteins as well as transcriptional activators and suppressors [26]. Recently, Yang et al. showed a gain-of-function in mice carrying ASXL1 exon 12 mutations yielding gene truncation. The truncation resulted in more open chromatin and a dysregulated expression of genes critical for the self-renewal and differentiation of hematopoietic stem cells [27]. Moreover, as a component of the BAP1 histone H2A deubiquitinase complex, ASXL1 truncation increases its stability, strengthening the association of the BAP1 complex with chromatin, driving oncogenic gene expression [28].
ASXL1 mutations have previously been observed in CML in adults [6,7,8] and also in children [13] where ASXL1 was the only gene found to be mutated which may indicate a relevant role of such mutations in some cases. The study presented here provides novel evidence for an adverse response to TKI treatment of CML patients carrying mutant ASXL1 as the presence of an ASXL1 mutation at diagnosis was associated with a worse response to nilotinib treatment as measured by MMR achievement at month 12, 18, and 24. Mutations other than ASXL1 mutations at diagnosis did not affect molecular response during follow-up. Moreover, patients with ASXL1 mutations were younger than patients with other mutations and more frequently at high risk than patients with other or no mutations. All mutations other than ASXL1 were only identified in low risk patients. These findings indicate that ASXL1 mutations may have an active role in CML pathogenesis and clonal evolution.
Our data confirm previous findings which show that ASXL1 mutations are rarely detected during MMR suggesting eradication of the mutation in most patients by TKI treatment [7]. However, we found a slower treatment response in CML patients carrying an ASXL1 mutation at diagnosis despite mutation clearance in 11/12 of the affected patients in which follow-up samples were analyzed. This suggests that clonal evolution with ASXL1 mutations on top of BCR::ABL1 influences TKI susceptibility of leukemic cells and thus response to therapy. The molecular mechanism behind this phenomenon needs to be explored further. Recently, Wang et al. [28] identified a small molecule inhibitor which targets aberrant BAP1 catalytic activity in ASXL1-mutated leukemia, reversing oncogenic gene expression. In the future, combined or sequential treatment strategies could help CML patients with ASXL1 mutations achieve better molecular responses.
Whilst the presence of multiple mutations per patient are commonly detected in blast crisis CML, acute myeloid leukemia (AML) and myelodysplastic neoplasms (MDS) [29,30,31], 47/53 (89%) of all affected patients in this study showed only one mutation in addition to BCR::ABL1 at diagnosis indicating that the clonal architecture in CML might be less complex compared to other myeloid malignancies (Fig. 1). However, five patients of the cohort investigated in this study (#36, #66, #171, #177 and #180) showed two mutations and one patient (#159) showed three distinct mutations in addition to BCR::ABL1 at diagnosis. Patient #159 also carried an Y253H BCR::ABL1 mutation (month 12) as detected by Sanger sequencing and was the only patient in this cohort to develop blast crisis. The patient died 21 months after treatment initiation. As CML samples are increasingly sequenced to investigate causes of resistance, persistence, and relapse, future analyses may confirm that the number of additional mutations has clinical consequences, as seen in AML and chronic myelomonocytic leukemia (CMML), where the number of mutations detected is associated with a worse prognosis [32, 33]. The current study is based on targeted NGS, including a panel of 54 leukemia-associated genes, whereas whole exome or genome analyses might reveal additional mutations. As clinical datasets combined with sequencing information expand (e.g., within the HARMONY Plus Alliance; www.harmony-alliance.eu), a clearer picture of the impact of additional mutations may emerge.
Sequencing analyses of diagnosis samples and corresponding annual follow-up samples in 100 selected patients revealed various mutation patterns under therapy, including eradication (pattern I), persistence (pattern II), and emergence (pattern III). Regarding the eradication group, in 19/100 (19%) patients (at least one) additional mutation was detected at diagnosis that was subsequently undetectable under nilotinib treatment (Table 2a). As these mutations were not detectable in subsequent BCR::ABL1 negative follow-up samples, it may be concluded that only BCR::ABL1 positive cells harbored these additional mutations. One patient showed re-occurrence of a variant at month 24 with a concurrently increased BCR::ABL1IS transcript level of 1.1%, supporting the notion that these additional mutations are located in BCR::ABL1 cells. To further strengthen these findings, future analyses could employ error-corrected NGS to increase sensitivity further and confirm mutation concomitance.
Fifteen variants in 13/100 (13%) patients were only detected in follow-up samples. One of these mutations was the JAK2 V617F mutation in patient #76 which has been previously described as uncommon in CML [34]. In some of these rare cases the diagnosis of a JAK2 V617F-positive disease preceded the acquisition of the Ph-chromosome [35, 36]; in others the second myeloproliferative disease emerged in the remission phase of CML [37,38,39]. In the case presented here, the JAK2 V617F mutation was not detected at diagnosis, indicating that it was either initially masked by the predominantly existing Ph+ cells or arose during nilotinib treatment. It is known that there are multiple pathways in which expression of the BCR::ABL1 tyrosine kinase causes genetic instability with subsequently emerging mutations. The resulting adverse response to treatment may no longer depend on the BCR::ABL1 kinase itself [40].
In a minority of patients (5/100; 5%) mutations in addition to BCR::ABL1 persisted over all time points analyzed and therefore seem to precede the BCR::ABL1 translocation indicating a multistep pathogenesis in CML as suggested previously [6, 41, 42]. One mutation affecting CALR displayed slightly increased VAFs in follow-up samples despite successful suppression of the BCR::ABL1 clone. CALR mutations are known drivers of myeloproliferative neoplasms [43] and may therefore contribute to a clonal advantage and expansion even after the BCR::ABL1 clone has been eradicated. Four DNMT3A mutations showed stable lower VAFs in follow up samples compared to the respective diagnosis sample. A suppression of the mutation, but not eradication despite good response, indicates the existence of a preleukemic clone that subsequently acquired BCR::ABL1. DNMT3A is known to be frequently mutated with increasing age in the general population as part of a phenomenon termed clonal hematopoiesis of indeterminate potential (CHIP) [10,11,12]. In elderly people without hematologic disorders DNMT3A mutant clones are unlikely to expand over time [44], which supports the finding of stable VAFs under TKI therapy in CML patients. Age-related clonal hematopoiesis is associated with an increased risk of developing hematologic cancer [11]. In the study presented here, median age of patients carrying additional mutations at diagnosis was 54 years (range, 22–78 years) whilst mutation frequency was 24%, thus exceeding the previously established age-related mutation prevalence of <5% within the healthy population in this age range [11]. Whilst most detected mutations are likely a direct feature of CML pathology, the DNMT3A mutations in particular may have occurred as age-related events which might predispose individuals to CML. The precise implication of preleukemic clonal hematopoiesis for CML has been analyzed in a few studies but will undoubtedly be better understood as sequencing efforts expand [6,7,8]. Similarly, in 14 patients germline mutations were detected which met the published criteria of clinically relevant mutations [8]. Five of the affected patients also carried at least one somatic mutation. This indicates that germline mutations may also function as predisposing factors to CML.
Conclusion
The study presented here aimed to evaluate the prevalence, dynamics and clinical impact of BCR::ABL1 independent gene mutations in chronic phase CML patients treated with nilotinib in an ongoing prospective clinical trial. Mutations in addition to BCR::ABL1 were frequently identified at diagnosis and were found to vary in their dynamics during treatment. ASXL1 mutations were most common and affected patients showed MMR less frequently at month 12, 18, and 24 compared to all other patients. ASXL1 may, after further validation, serve as an additional prognostic factor for molecular response in newly diagnosed CML patients.
References
Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172–83.
Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med. 2017;376:917–27.
Kantarjian HM, Hughes TP, Larson RA, Kim DW, Issaragrisil S, le Coutre P, et al. Long-term outcomes with frontline nilotinib versus imatinib in newly diagnosed chronic myeloid leukemia in chronic phase: ENESTnd 10-year analysis. Leukemia. 2021;35:440–53.
Saussele S, Richter J, Guilhot J, Gruber FX, Hjorth-Hansen H, Almeida A, et al. Discontinuation of tyrosine kinase inhibitor therapy in chronic myeloid leukaemia (EURO-SKI): a prespecified interim analysis of a prospective, multicentre, non-randomised, trial. Lancet Oncol. 2018;19:747–57.
Etienne G, Guilhot J, Rea D, Rigal-Huguet F, Nicolini F, Charbonnier A, et al. Long-term follow-up of the french stop imatinib (STIM1) study in patients with chronic myeloid leukemia. J Clin Oncol. 2017;35:298–305.
Schmidt M, Rinke J, Schafer V, Schnittger S, Kohlmann A, Obstfelder E, et al. Molecular-defined clonal evolution in patients with chronic myeloid leukemia independent of the BCR-ABL status. Leukemia. 2014;28:2292–9.
Kim T, Tyndel MS, Kim HJ, Ahn JS, Choi SH, Park HJ, et al. Spectrum of somatic mutation dynamics in chronic myeloid leukemia following tyrosine kinase inhibitor therapy. Blood. 2017;129:38–47.
Branford S, Kim DDH, Apperley JF, Eide CA, Mustjoki S, Ong ST, et al. Laying the foundation for genomically-based risk assessment in chronic myeloid leukemia. Leukemia. 2019;33:1835–50.
Nteliopoulos G, Bazeos A, Claudiani S, Gerrard G, Curry E, Szydlo R, et al. Somatic variants in epigenetic modifiers can predict failure of response to imatinib but not to second-generation tyrosine kinase inhibitors. Haematologica. 2019;104:2400–9.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl J Med. 2014;371:2488–98.
Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl J Med. 2014;371:2477–87.
Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20:1472–8.
Ernst T, Busch M, Rinke J, Ernst J, Haferlach C, Beck JF, et al. Frequent ASXL1 mutations in children and young adults with chronic myeloid leukemia. Leukemia. 2018;32:2046–9.
Cross NC, Feng L, Bungey J, Goldman JM. Minimal residual disease after bone marrow transplant for chronic myeloid leukaemia detected by the polymerase chain reaction. Leuk Lymphoma. 1993;11:39–43.
Emig M, Saussele S, Wittor H, Weisser A, Reiter A, Willer A, et al. Accurate and rapid analysis of residual disease in patients with CML using specific fluorescent hybridization probes for real time quantitative RT-PCR. Leukemia. 1999;13:1825–32.
Muller MC, Erben P, Saglio G, Gottardi E, Nyvold CG, Schenk T, et al. Harmonization of BCR-ABL mRNA quantification using a uniform multifunctional control plasmid in 37 international laboratories. Leukemia. 2008;22:96–102.
Cross NC, White HE, Colomer D, Ehrencrona H, Foroni L, Gottardi E, et al. Laboratory recommendations for scoring deep molecular responses following treatment for chronic myeloid leukemia. Leukemia. 2015;29:999–1003.
Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122:872–84.
Hochhaus A, Baccarani M, Silver RT, Schiffer C, Apperley JF, Cervantes F, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020;34:966–84.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D894.
Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;7:20.
Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat. 2011;32:894–9.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2.
Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34:57–65.
Gelsi-Boyer V, Brecqueville M, Devillier R, Murati A, Mozziconacci MJ, Birnbaum D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol. 2012;5:12.
Yang H, Kurtenbach S, Guo Y, Lohse I, Durante MA, Li J, et al. Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood. 2018;131:328–41.
Wang L, Birch NW, Zhao Z, Nestler CM, Kazmer A, Shilati A, et al. Epigenetic targeted therapy of stabilized BAP1 in ASXL1 gain-of-function mutated leukemia. Nat Cancer. 2021;2:515–26.
Grossmann V, Kohlmann A, Zenger M, Schindela S, Eder C, Weissmann S, et al. A deep-sequencing study of chronic myeloid leukemia patients in blast crisis (BC-CML) detects mutations in 76.9% of cases. Leukemia. 2011;25:557–60.
Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74.
Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–7.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21.
Patnaik MM, Tefferi A. Chronic Myelomonocytic leukemia: 2020 update on diagnosis, risk stratification and management. Am J Hematol. 2020;95:97–115.
Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, Zhang L, et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005;106:2162–8.
Bornhäuser M, Mohr B, Oelschlaegel U, Bornhäuser P, Jacki S, Ehninger G, et al. Concurrent JAK2(V617F) mutation and BCR-ABL translocation within committed myeloid progenitors in myelofibrosis. Leukemia. 2007;21:1824–6.
Bocchia M, Vannucchi AM, Gozzetti A, Guglielmelli P, Poli G, Crupi R, et al. Insights into JAK2-V617F mutation in CML. Lancet Oncol. 2007;8:864–6.
Krämer A, Reiter A, Kruth J, Erben P, Hochhaus A, Müller M, et al. JAK2-V617F mutation in a patient with Philadelphia-chromosome-positive chronic myeloid leukaemia. Lancet Oncol. 2007;8:658–60.
Inami M, Inokuchi K, Okabe M, Kosaka F, Mitamura Y, Yamaguchi H, et al. Polycythemia associated with the JAK2V617F mutation emerged during treatment of chronic myelogenous leukemia. Leukemia. 2007;21:1103–4.
Hussein K, Bock O, Seegers A, Flasshove M, Henneke F, Buesche G, et al. Myelofibrosis evolving during imatinib treatment of a chronic myeloproliferative disease with coexisting BCR-ABL translocation and JAK2V617F mutation. Blood. 2007;109:4106–7.
Skorski T. BCR/ABL regulates response to DNA damage: the role in resistance to genotoxic treatment and in genomic instability. Oncogene. 2002;21:8591–604.
Raskind WH, Ferraris AM, Najfeld V, Jacobson RJ, Moohr JW, Fialkow PJ. Further evidence for the existence of a clonal Ph-negative stage in some cases of Ph-positive chronic myelocytic leukemia. Leukemia. 1993;7:1163–7.
Fialkow PJ, Martin PJ, Najfeld V, Penfold GK, Jacobson RJ, Hansen JA. Evidence for a multistep pathogenesis of chronic myelogenous leukemia. Blood. 1981;58:158–63.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Midic D, Rinke J, Perner F, Muller V, Hinze A, Pester F, et al. Prevalence and dynamics of clonal hematopoiesis caused by leukemia-associated mutations in elderly individuals without hematologic disorders. Leukemia. 2020;34:2198–205.
Acknowledgements
The excellent technical assistance of Ms. Anja Waldau is gratefully acknowledged. We thank all colleagues who actively participated in the trial.
Funding
The study was supported by the HARMONY Plus Alliance through the European LeukemiaNet Foundation and by Novartis through the European Treatment and Outcome Study (EUTOS) for CML. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
TE and AHo designed the study; AHo coordinated the TIGER trial; AHo, THB, AB, PLC, SWK, SS participated in the steering board of the trial and treated patients; LS, JR, AHi, SNN, VS, TE performed the molecular work; FS and MP did the statistical analysis; TS, CF and TE coordinated the collection of patients’ data; LS, JR, AHo and TE wrote the manuscript; all authors edited the manuscript and agreed on its content.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schönfeld, L., Rinke, J., Hinze, A. et al. ASXL1 mutations predict inferior molecular response to nilotinib treatment in chronic myeloid leukemia. Leukemia 36, 2242–2249 (2022). https://doi.org/10.1038/s41375-022-01648-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-022-01648-4
This article is cited by
-
BCR/ABL-Positive Chronic Myeloid Leukemia in Children: Current Treatment Approach
Current Oncology Reports (2024)
-
Management and outcome of patients with chronic myeloid leukemia in blast phase in the tyrosine kinase inhibitor era – analysis of the European LeukemiaNet Blast Phase Registry
Leukemia (2024)
-
European LeukemiaNet laboratory recommendations for the diagnosis and management of chronic myeloid leukemia
Leukemia (2023)
-
Chronische myeloische Leukämie
Die Onkologie (2023)
-
Children with chronic myeloid leukaemia treated with front-line imatinib have a slower molecular response and comparable survival compared with adults: a multicenter experience in Taiwan
British Journal of Cancer (2023)
