
Abstract
Background:
Classical galactosemia is caused by severe galactose-1-phosphate uridyltransferase deficiency. Despite life-long galactose-restriction, many patients experience long-term complications. Intoxication by galactose and its metabolites as well as over-restriction of galactose may contribute to the pathophysiology. We provided temporary low-dose galactose supplements to patients. We assessed tolerance and potential beneficial effects with clinical monitoring and measurement of biochemical, endocrine, and IgG N-glycosylation profiles.
Methods:
We enrolled 26 patients (8.6 ± 1.9 y). Thirteen were provided with 300 mg of galactose/day followed by 500 mg for 2 wk each (13 patient controls).
Results:
We observed no clinical changes with the intervention. Temporary mild increase in galactose-1-phosphate occurred, but renal, liver, and bone biochemistry remained normal. Patients in the supplementation group had slightly higher leptin levels at the end of the study than controls. We identified six individuals as “responders” with an improved glycosylation pattern (decreased G0/G2 ratio, P < 0.05). There was a negative relationship between G0/G2 ratio and leptin receptor sOb-R in the supplementation group (P < 0.05).
Conclusion:
Temporary low-dose galactose supplementation in children over 5 y is well tolerated in the clinical setting. It leads to changes in glycosylation in “responders”. We consider IgG N-glycan monitoring to be useful for determining individual optimum galactose intake.
Similar content being viewed by others


Main
Classical galactosemia (OMIM 230400) is caused by a profound deficiency of galactose-1-phosphate uridyltransferase (GALT) enzyme activity (EC 2.7.7.12). The most common mutation in the GALT gene in affected patients is c.563A>G in exon 6 (p.Q188R). We have demonstrated that 92.9% of Irish patients harbor this mutation in a homozygous state (1). Despite newborn screening and early treatment with a life-long galactose-restricted diet, a high percentage of patients experience long-term complications, including cognitive impairment, speech and language abnormalities, tremor, ataxia, osteopenia, and over 80% of females have premature ovarian insufficiency (2,3,4). The pathophysiology is complex and comprises toxic build-up of galactose and its metabolites galactose-1-phosphate (Gal-1-P) and galactitol, altered glycoprotein and glycolipid production, disruption of multiple cell signaling pathways, and gene dysregulation (5,6).
Our group recently described glycosylation abnormalities in IgG N-glycans of patients with galactosemia along with the potential application of IgG N-glycans as biomarkers for patients (6,7). Glycosylation is a widespread post-translational modification of proteins and deregulation of glycosylation is associated with a wide range of diseases (8). Along these lines, we have previously described galactose incorporation ratios as a method of studying the presence of N-glycan processing defects in children with galactosemia (7). In earlier and on-going studies, we have identified that galactosylation is abnormal in patients with galactosemia and may play a role in the neurological manifestations by means of impaired galactosylation of cerebrosides and glycoproteins in the brain (5,7 and unpublished data).
We have also demonstrated that the metabolic hormone leptin is lower in girls and boys with galactosemia than in healthy controls when adjusted for gender and pubertal stage (9). Leptin, a member of the cytokine family, is predominantly produced in adipocytes but to a much lesser extent also found in stomach, kidney, liver, and other cells, a process which can be stimulated by, for example, insulin and corticosteroids (10). It has an impact on food intake, energy homeostasis, and a variety of metabolic functions through its effects on the central nervous system and peripheral organs (11). Leptin is an important factor for reproduction, including initiation and progression of puberty (12) and it is implicated in many other physiological processes such as bone formation (13). Hypoleptinaemia has been described in catabolic conditions such as anorexia nervosa and diabetic ketoacidosis (12,14). Conversely, hyperleptinaemia has been found in obesity, hyperinsulinaemia, and nonalcoholic fatty liver disease (15,16).
Intracellular leptin actions are mediated through the long isoform of the leptin receptor (Ob-R), a member of the class I cytokine receptor family. Essentially, it is found in different isoforms, all of which derive from the leptin receptor gene but only the long form of Ob-R with a 302 amino acids intracellular domain contains a motif for activation of the Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway (11). There is potential divergence in its signaling between different regions in the brain, as Ob-R signaling can also activate phosphatidylinositol-3-kinase (PI3-K) and extracellular signal-regulated kinase-1 and 2 (ERK1/2) (17). The soluble isoform sOb-R is considered to be important to maintain physiological leptin concentrations in the blood-stream (18).
We have demonstrated in adult patients with galactosemia that serum IgG N-glycan profiles showed consistent individual alterations in response to a partial and transient diet liberalization over 16 wk (6). In detail, the individual profiles were improved for all but one adult patient, at a galactose intake of 1,000 mg/day, with decreases in agalactosylated (G0) and increases in digalactosylated (G2) N-glycans (6).
This study, which builds on our previous work, aims at establishing if classical galactosemia is a modifiable, multi-systemic glycosylation defect in children. The objective of this study was to mildly relax strict galactose restriction in children with galactosemia aged 5–12 y by giving temporary low-dose oral galactose supplements using limited quantities of cow’s milk. We aimed to assess, firstly, tolerance and potential beneficial effects of galactose supplementation and, secondly, to progress the development and validation of novel biochemical and endocrine markers as prognostic indices in children with galactosemia.
Results
Demographics
Clinical data are presented in Table 1 . The study cohort comprised 9 females (35%) and 17 males (65%) aged 8.6 ± 1.9 y (range 5.1–11.2 y). All patients had a normal body weight and height with a BMI (kg/m2) between 9th and 91st centile (median 25th centile, mean 36th centile). SDS of BMI between our galactose supplementation group and patient controls were not significantly different (−0.36 vs. −0.34, NS). Only prepubertal patients (Tanner stage 1) were recruited for the study. Median age at diagnosis was 2 d (mean ± SD 3 ± 3, range 1–9). All patients were on a strict lactose-free diet since the diagnosis was made following newborn screening.
Tolerance and Safety
We observed no clinical changes in the galactose supplementation group after 2 wk of 300 mg of galactose intake and after 2 subsequent weeks of 500 mg of galactose. An eye review immediately after completing the 500 mg galactose supplementation trial did not reveal any changes, particularly no early signs of cataracts ( Table 1 ), and systemic exam was unremarkable. Galactose supplements were given with breakfast and well tolerated. There were no adverse clinical indicators such as gastrointestinal discomfort or abdominal pain and no symptoms suggestive of pseudotumor cerebri, including headaches or blurred vision. One patient with a history of atopic eczema developed a skin infection and received hydrocortisone cream and antibiotics. This nonserious adverse event was considered mild-to-moderate in severity and not related to galactose supplementation. A patient in the control group developed a mild gastroenteritis toward the end of the study.
Data Completeness
Two patients missed a single blood test each for unavoidable reasons. All participants had an endpoint assessment. Completeness of data was 92%.
Gal-1-P
Mean Gal-1-P levels increased only slightly in the galactose supplementation group (baseline on lactose-free diet 2.5 vs. 2.9 mg/dl after 2 wk of 300 mg of galactose (P < 0.05), and 3.1 after 500 mg (P < 0.01 vs. baseline, Figure 1 ). Gal-1-P levels reverted to baseline at approximately 2 wk after discontinuation of galactose supplementation. Mean Gal-1-P levels in galactosemia patient controls on a strict lactose-free diet were 2.5 and 2.7 mg/dl (NS), respectively.

Galactose-1-phosphate concentrations in packed erythrocytes of children with galactosemia on a strict lactose-free diet (Pre, patient controls 1st, Co1, and 2nd sample, Co2) and on a partial and transient galactose liberalization for a period of 2 wk each (300 mg, 500 mg) and thereafter (Post) (*P < 0.05 and **P< 0.01, Wilcoxon matched-pairs signed rank test). White boxes depict results obtained from the galactose supplementation group and grey boxes depict results from patient controls. Data are given as a box-and-whisker diagram. The box extends from the 25th to the 75th centile with a line in the middle which is plotted at the median. The whiskers extend from minimum to maximum.
Biochemistry
There were no statistically significant differences between the groups in the patients’ renal, liver, and bone biochemistry profiles or coagulation studies ( Table 2 ). In detail, there were no changes observed in the galactose supplementation group following 300 or 500 mg of galactose intake for renal, liver, and bone biochemical profiles and coagulation tests ( Table 2 ), (data for electrolytes, alkaline phosphatase, albumin, urea, total protein, full blood count, and fibrinogen not shown).
Leptin and Leptin Receptor
As expected, there was a hyperbolic relationship between serum leptin and soluble leptin receptor sOb-R in the entire study group (data not shown). Serum leptin concentrations are presented in Figure 2 ; all patients had leptin levels below 10 ng/ml. There were no statistically significant differences between the groups at baseline, i.e., between the data sets Pre and Co1. As a trend, patients in the galactose supplementation group had slightly higher leptin levels at the end of the study than patient controls (P < 0.05). There were no significant differences between leptin levels at 300 and 500 mg of galactose supplementation.

Serum concentration of leptin in the galactose supplementation group (white boxes) and galactosemia patient controls (gray boxes). As a trend, patients in the galactose supplementation group had slightly higher leptin levels at the end of the study (Post) than patient controls (Co2) (*P < 0.05, Mann-Whitney test).
Figure 3 demonstrates data for sOb-R in serum. In the supplementation group, sOb-R levels at 300 mg of galactose supplementation were slightly lower than at baseline (P < 0.05), which is in keeping with the leptin data at this point. In addition, sOb-R levels at 500 mg of galactose supplementation were slightly higher than at 300 mg of galactose supplementation (P < 0.05). For patient controls, there were no significant changes in sOb-R serum levels in the course of this study. As a trend, patients in the galactose supplementation group had slightly higher sOb-R levels at the end of the study than patient controls (P < 0.05).

Serum concentration of sOb-R in the galactose supplementation group (white boxes) and galactosemia patient controls (gray boxes). In the galactose supplementation group, sOb-R levels at 300 mg of galactose supplementation were slightly lower than at baseline (*P < 0.05) and sOb-R levels at 500 mg of galactose supplementation were slightly higher than at 300 mg (*P < 0.05, Wilcoxon matched-pairs signed rank test). Patients in the galactose supplementation group had slightly higher sOb-R levels at the end of the study (Post) than patient controls (Co2) (*P < 0.05, Mann-Whitney test).
IgG N-glycan Analysis
Figure 4 presents the N-glycan ratio G0/G2 for the patients with galactose supplements and patient controls at baseline and at the end of the study. Within the galactose supplementation group, we identified six individuals as “responders” with a decrease in the G0/G2 ratio, i.e., higher amounts of relative digalactosylated structures as a quantitative measure of galactose incorporation into glycoproteins (P < 0.05, Wilcoxon matched-pairs signed rank test). Only for the subgroup of “responders” a significant decrease in the G0/G2 ratio was detected during the course of the study (P < 0.05, ANOVA) but not for “nonresponders” and patient controls.

IgG N-glycan G0/G2 ratio for galactosemia patients with galactose supplements (white circles) and galactosemia patient controls (gray circles) both at baseline and at the end of the study. Within the galactose supplementation group, we identified six individuals as “responders” with a decrease in the G0/G2 ratio, i.e., higher amounts of digalactosylated structures as a quantitative measure of galactose incorporation into glycoproteins (*P < 0.05, Wilcoxon matched-pairs signed rank test). Data (mean ± SD) for healthy pediatric controls (black circle) have been previously published by our group (7).
In the patient control group, there was only one patient with a slight improvement in his G0/G2 ratio over the course of the study.
The different response to galactose supplements and differential levels of galactosylation given as G0/G2 ratio in the galactose supplementation group were independent of sex. For both sexes, there was a trend toward a negative correlation between the G0/G2 ratio and sOb-R levels, however, it was not significant for the subgroups probably due to the small numbers. In detail, for the girls in the galactose supplementation group, we found Pearson r = −0.787, P = 0.063 (NS), and for the boys Pearson r = −0.279, P = 0.077 (NS). We identified a negative relationship between the G0/G2 ratio and sOb-R levels when we tested all patient samples in the galactose supplementation group which was not found for in the galactosemia patient control group ( Figure 5 ).

A negative relationship between IgG N-glycan G0/G2 ratio and sOb-R levels (linear regression analysis) is demonstrated for all patient samples in the galactose supplementation group but not for patient controls. Unfilled symbols represent data from patients with galactose supplementation (Y = −17.08*X + 58.69, r2 = 0.1220, P < 0.05, solid line). Filled symbols represent data from patient controls (Y = 4.660*X + 22.60, r2 = 0.0201, NS, dashed line).
For the G0/G1 ratio, however, the differences noted between the galactose supplementation group and patient controls, and also within groups, both at baseline and the end of the study were found to be not statistically significant (galactose supplementation group 0.899 ± 0.176 and 0.899 ± 0.150, patient controls 0.930 ± 0.119 and 0.938 ± 0.130, respectively).
Full-scale IQ Score
85% of patients (11 in either group) underwent psychology review including a baseline IQ assessment. Age at IQ testing was 7.1 ± 2.6 y for the study group and 7.3 ± 1.7 y for the patient control group (NS). Three out of four patients who did not receive a formal assessment had a history of mild speech delay. The average FSIQ (±SD) in the galactose supplementation group was 70 ± 16 (range 43–91) and 71 ± 8 (50–79) in the patient control group (NS). For the subgroup of galactose supplementation “responders” with an improved glycosylation ratio (see below) the average FSIQ score was 79 ± 9, whereas the “non-responders” had an average FSIQ score of 59 ± 17, however, the number of patients in both subgroups (n = 6 and 7, respectively) was too small to detect significant differences (P = 0.08, NS). In the entire patient cohort, there was no correlation between FSIQ and G0/G2 glycosylation ratio (Pearson r = −0.031, NS).
Discussion
In previous studies, we and others have shown that a less strict lactose-free diet in patients with classical galactosemia beyond infancy does not change long-term outcome in a negative way. There are case reports of patients with galactosemia (p.Q188R homozygous) with a mild phenotype who have been taking a liberalized, normal diet later in life (19,20). We have found in our own cohort that patients with a slightly higher galactose intake did not experience an increased incidence of complications; conversely, patients who were very compliant with a very galactose-restricted diet did not have more favorable outcomes (2). This is in line with the data published on Estonian patients with galactosemia aged 7–14 y (21). A Dutch study on three adolescent patients (p.Q188R homozygous) with increasing doses of galactose intake to a maximum of 600 mg per day for a period of 6 wk detected no significant change in metabolites levels (22). The authors concluded that excluding trace amounts of galactose from the diet may no longer be justified but once the diet is made more liberal, appropriate follow-up would be mandatory.
We also published data on adult patients with galactosemia aged 18–25 y on various amounts of galactose intake and revealed different galactose tolerance levels in patients with identical genotypes and even in siblings (6). The individual N-glycan profiles improved for four out of five adults at a galactose intake of 1,000 mg per day, with an unfavorable biochemical response in one adult patient, which underlines the need for applying this technology as a new biomarker for galactosemia patients (6). Given the complex pathophysiology, it is not possible to identify a specific biosynthetic pathway as many cellular glycoproteins and glycolipids can be affected, along with associated signaling pathways, accessory salvage pathways, and also modifier genes.
Physiologically, the relative amount of agalactosylated structures (G0) in glycoproteins such as serum IgG exhibits a parabolic function with a decrease from infancy to a minimum at 25 y of age followed by an increase beyond that age (23). Conversely, the relative incidence of digalactosylated structures (G2) changes inversely to G0 whereas monogalactosylated structures are constant in both sexes (23). A high G0/G1 or G0/G2 ratio indicates a higher level of agalactosylated structures relative to mono- or diagalactosylated N-glycans. In our cohort, the G0/G2 ratio was found to be more informative than G0/G1 ratio. In principle, variations in IgG galactosylation can be caused by genetic and epigenetic factors, including environmental factors, galactose availability, and expression of the major enzyme involved in glycan processing, galactosyltransferase (B4GALT1). Along these lines, we have recently demonstrated that the expression of B4GALT1 was decreased by twofold in adults with galactosemia vs. healthy controls in DNA microarray analysis (24). We could also demonstrate that this finding remained significant for the subgroup of adult patients with neurological impairment (24).
We here used 300 and 500 mg of daily galactose supplements in our study patients as this amount is close to the range of the endogenous galactose synthesis which is 0.53–1.05 mg/kg body weight per hour (25). Essentially, the endogenous galactose production rate in adult patients is approximately 13 mg/kg per day; however, it is higher for galactosaemic newborns with 41 mg/kg per day (26). Berry et al. (27) have shown that a 3-wk supplementation of 200 mg galactose per day in adult patients did not lead to any changes in erythrocyte Gal-1-P levels. We here speculate that the threshold for mildly elevated levels of galactose metabolites, such as Gal-1-P, in patients on a lactose-restricted diet is probably in the range of 200–300 mg galactose per day.
As leptin dysregulation can occur in individuals with galactosemia (9), we here test our hypothesis that its soluble receptor sOb-R might be altered due to significant N-glycosylation abnormalities. In principle, the human leptin receptor is a glycoprotein with an abundance of N-glycosylation sites (28). In detail, the long form of the leptin receptor (Ob-Rb) contains 20 potential N-glycosylation sites that regulate Ob-Rb function, and alterations of N-linked glycans in Ob-Rb may contribute to altered protein conformation and reduced thermodynamic stability (29). The molecular mechanism for the regulation of Ob-R are unclear, however, it has been shown that endoplasmic reticulum stress suppresses sOb-R generation (30). Essentially, sOb-R represents the main leptin binding activity in human blood and modulates the bioavailability of free leptin (31).
Human embryonic kidney (HEK) 293 cells incubated with tunicamycin, an inhibitor of N-glycosylation and inducer of endoplasmic reticulum, show decreased sOb-R generation along with downregulation of membrane Ob-R and impaired leptin signaling (30). Tunicamycin is a synthetic antibiotic but its underlying mechanism of inhibiting N-glycosylation of glycoproteins, thereby increasing endoplasmic reticulum protein load and dysfunction and eventually endoplasmic reticulum stress, has been described in the context of conditions such as nonalcoholic fatty liver disease and inflammation (32).
Leptin signaling has a well-characterized role in the hypothalamic-pituitary-ovarian axis and also a potential neuroprotective role. Along these lines, the hippocampus, in particular, expresses high levels of leptin receptors and its signaling cascade (33). The hippocampal formation is also a prime site for many neurodegenerative processes, and disruption of leptin signaling may have a role in cognitive defects and, possibly, neurological impairment in adults with galactosemia (24)
In this study, we only studied prepubertal children (Tanner stage 1) over a short period of time to minimize the effects of sex hormone-related changes on the leptin axis. During puberty, the sOb-R levels decrease with increasing Tanner stages for girls and boys (12). Conversely, leptin increases during puberty and adolescence; in girls, there is a gradual increase whereas in boys peak values are found in the first phase of puberty followed by a relative decline (12).
It was interesting to see in our galactose supplementation group that sOb-R levels varied at 300 and 500 mg of galactose supplementation. This could be due to the changes in serum leptin concentrations or due to a glycoprotein modification. We speculate that certain subjects with galactosemia can tolerate varying degrees of limited galactose intakes later in childhood as they may have the ability to utilize auxiliary pathways of galactose metabolism and that sOb-R might be among the modifiable glycoproteins. Our subgroup of “non-responders” may need a different dietetic approach that would enable them to use limited amounts of galactose through accessory biochemical pathways.
However, it is important to emphasize that elimination of dietary galactose is the only available treatment for patients with galactosemia and that it is life-saving, especially in the neonate. Several limitations of our study should be mentioned, including relatively small sample size and the use of milk as source of galactose supplement.
In summary, we have shown that transient galactose supplementation close to the range of the endogenous galactose production rate in prepubertal children aged 5–12 y is safe in the clinical setting. We have identified changes in the serum IgG N-glycosylation status in our subgroup of “responders” that are theoretically beneficial. Our data provide new insight into ongoing and potentially treatable N-glycan processing defects in children with galactosemia. This study also suggests IgG N-glycan monitoring for determining the optimum galactose intake for each patient on an individual basis. However, larger-scale clinical studies are needed.
Methods
Patients
This study was approved by the Research and Ethics Committees of Children’s University Hospital Temple Street, Dublin. The patients’ parents gave full informed written consent prior to enrolment, and the assent of the minor was obtained as appropriate. We recruited 26 children with galactosemia who were being treated at the National Centre for Inherited Metabolic Disorders, Dublin. The study cohort comprised 9 females (35%) and 17 males (65%) aged 8.6 ± 1.9 y (range 5.1–11.2 y) ( Table 1 ). All patients were homozygous for the common GALT mutation c.563A>G in exon 6 (p.Q188R). Residual GALT enzyme activity GALT in erythrocytes was <0.5 μmol substrate (Gal-1-P) converted per hour per gram hemoglobin confirming classical galactosemia (normal range 18–28). Clinical data and blood samples were compiled during outpatient visits.
Study Design
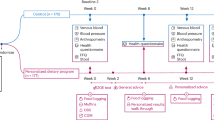
Four time points were chosen in the galactose supplementation group: baseline (Pre), after 2 wk of 300 mg of galactose (300 mg), after 2 subsequent weeks of 500 mg of galactose (500 mg), and at the end of the study after 2 wk back on a strict lactose-free diet (Post). In the patient control group on a strict lactose-free diet, only two time points were chosen: baseline (Co1) and at the end of the study, i.e., 6 wk later (Co2). The study was planned as a fixed size pilot study based on our previous studies (6,7,9). We used a modified Zelen design where participants are randomized prior to giving consent to enrol a representative cohort of our patients. We included careful analysis of the patients’ clinical data to ensure that there was no consent bias along with a selection bias. The trial was registered on the ClinicalTrials.gov website (identification number NCT02218632).
Lactose-Free Diet
All patients had been on a lactose-free diet since diagnosis. Patient controls remained on a strict lactose-free diet throughout the trial. Patients in the transient galactose supplementation group also remained on a strict lactose-free diet except for the galactose supplementation (see below). Food diaries for 2 d were analyzed and lactose/galactose content of foods was calculated using standard literature (34). Calculated galactose intake at the start of the study was 10 ± 25 (range 0–80) mg/day in the study group (excluding galactose supplements), and 23 ± 60 (range 0–159) mg/day in the control group (NS).
Galactose Supplementation
We used average lactose values in milk from the literature (34) and confirmed galactose content in a series of whole cow’s milk samples in a commercial Food Research Laboratory using High performance Anion Exchange Chromatography (Leatherhead Food Research, Surrey, UK). In the study group, a daily amount of 13 ml of whole cow’s milk (Avonmore milk, Glanbia Consumer Foods, Dublin, Ireland) was given for 14 d to provide 300 mg of galactose per day (range 282–310 mg), and 22 ml to provide 500 mg of galactose per day (range 478–525).
Laboratory Methods
Soluble leptin receptor (sOb-R). The concentration of sOb-R was determined using a ligand immunofunctional assay as previously described (35,36). The lowest detectable sOb-R concentration was calculated to be less than 2.0 ng/ml. Intra- and interassay coefficients of variation for two control samples were lower than 11.7% (n = 10).
Leptin. Serum leptin was measured using a commercially available ELISA from Mediagnost (Tuebingen, Germany) as published earlier (35). Its intra- and interassay coefficients of variation were lower than 12.5% in the concentration range of 1–8 ng/ml leptin. The recovery of dilution experiments was 88–112% for the concentration range of 4–6 ng/ml.
Isolation of IgG from whole serum. Serum was prepared from freshly spun blood samples and immediately frozen at −20 °C. IgG isolation was performed on a robotized glycomics platform as described previously, using Protein G 96-well filter plates (Fisher Scientific, Pittsburgh, PA) (7,37). The purity of the isolated IgG was assessed using 10 % reducing SDS-PAGE in an Xcell SureLock Mini-Cell (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions (data not shown).
Removal of IgG N-linked glycans. N-glycans were released from IgG using the automated method and peptide N-glycanase F (1,000 U/ml) as described elsewhere (7,22). Released N-glycans were then labeled by reductive amination with the fluorophore 2-AB 41,42 using a Ludger Tag 2-AB glycan labeling kit as described by the manufacturer (Ludger, Oxford, UK). IgG N-glycan purification, fluorescence labeling with 2-AB, and sample clean-up was performed using an automated high-throughput glycoprofiling platform (37).
Hydrophilic interaction ultra-performance liquid chromatography (HILIC-UPLC). 2-AB labeled N-glycans were separated by HILIC-UPLC with fluorescence detection on a Waters Acquity UPLC H-Class instrument consisting of a binary solvent manager, sample manager and fluorescence detector under the control of Empower 3 chromatography workstation software (Waters, Milford, MA). The HILIC separations were performed using a Waters Ethylene Bridged Hybrid (BEH) Glycan column with 50 mmol/l ammonium formate as solvent A and acetonitrile as solvent B. The separation was performed using a linear gradient of 30–45% solvent A at 0.56 ml/min for 15 min for released undigested IgG N-glycan separation and a linear gradient of 30–39.8% solvent A at 0.56 ml/min for 15 min for released digested IgG N-glycan separation. We used an injection volume of 10 μl sample in 70% v/v acetonitrile. The fluorescence detection excitation/emission wavelengths were λex = 330 nm and λem = 420 nm, respectively. The system was calibrated using an external standard of hydrolyzed and 2-AB labeled glucose oligomers to create a dextran ladder (38).
IgG N-glycan G-ratios. Galactosylation of IgG is one of the most comprehensively studied glycosylation features, and galactose ratios (G-ratios) have been identified as highly informative indicators of the level of galactose incorporation (8). We determined peak % areas of agalactosylated (G0), mono- (G1) and digalactosylated (G2) structures to provide a quantitative measurement of galactose incorporation into released undigested IgG N-glycans. We focused on the G0/G2 ratio because digalactosylated (G2) structures were found to vary more than (G1) structures which remain more constant in healthy individuals (6,23). The technical replicate coefficients of variations for IgG N-glycans was below 10%. The average biological replicate coefficients of variation for the IgG N-glycan individual peak percentage areas was 9% with a biological replicate coefficients of variation for G0/G1 and G0/G2 ratios below 5%.
Biochemistry. Plasma urea, creatinine, electrolytes (Na, K, Cl, P, and Ca), total protein, albumin, AST, ALT, bilirubin, and alkaline phosphatase were measured using standard procedures on a Beckman Coulter UNICEL DXc 600. Prothrombin time (PT), activated partial thromboplastin time (APTT), and plasma fibrinogen levels were measured using a STAGO COMACT coagulation analyser according to the manufacturer’s instructions.
Galactose-1-phosphate (Gal-1-P). Gal-1-P in blood was measured using a RIA as published earlier (39). This method involves the quantitation of Gal-1-P in washed red blood cell pellets through the addition of yeast UDP-glucose:α-D-galactose-1-phosphate uridyltransferase and UDP-[14C]glucose to samples. The reagents interact with Gal-1-P to generate [14C]glucose -1-phosphate, which is eluted from the sample using DEAE-cellulose mini-column chromatography. Formation of [14C]glucose -1-phosphate is proportional to the corresponding Gal-1-P concentration in the sample. The normal range is 0–0.3 mg/dl; for patients on a low-galactose diet Gal-1-P levels of 2–3 mg/dl are considered acceptable. In an intoxicated newborn with galactosemia, Gal-1-P levels can be extremely high (up to 70 mg/dl) and subsequently decrease with treatment (39).
Statistical Analysis
Data were expressed as mean ± SD or using descriptive statistics including medians, quartiles and range (i.e., box- and-whisker diagram) as appropriate. In the case of non-normally distributed data, e.g., for leptin and sOb-R, nonparametric statistical tests were used, including Spearman’s correlation, Mann-Whitney test, Kruskal-Wallis test with Dunn’s correction, and Wilcoxon matched-pairs signed rank test as appropriate. Otherwise, parametric tests were used. A significance level of P < 0.05 was chosen for all comparisons.
IQ Assessment
Wechsler Pre-school and Primary Scale of Intelligence was applied in patients at age 3–7 y and Wechsler Intelligence Scale for Children 4th Edition UK (WISC-IV, UK) in children of 6 y and above. The composite score (i.e., full-scale IQ, FSIQ) was used for further analysis.
Statement of Financial Support
We would like to thank the Medical Research Charities Group (MRCG, Dublin, Ireland) and the Health Research Board (HRB, Dublin, Ireland) for financial assistance to perform these studies.
Disclosure
The authors declare that they have no relevant or material financial interests that relate to the research described in this publication.
References
Coss KP, Doran PP, Owoeye C, et al. Classical Galactosaemia in Ireland: incidence, complications and outcomes of treatment. J Inherit Metab Dis 2013;36:21–7.
Hughes J, Ryan S, Lambert D, et al. Outcomes of siblings with classical galactosemia. J Pediatr 2009;154:721–6.
Rubio-Gozalbo ME, Gubbels CS, Bakker JA, Menheere PP, Wodzig WK, Land JA. Gonadal function in male and female patients with classic galactosemia. Hum Reprod Update 2010;16:177–88.
Spencer JB, Badik JR, Ryan EL, et al. Modifiers of ovarian function in girls and women with classic galactosemia. J Clin Endocrinol Metab 2013;98:E1257–65.
Coman DJ, Murray DW, Byrne JC, et al. Galactosemia, a single gene disorder with epigenetic consequences. Pediatr Res 2010;67:286–92.
Coss KP, Byrne JC, Coman DJ, et al. IgG N-glycans as potential biomarkers for determining galactose tolerance in Classical Galactosaemia. Mol Genet Metab 2012;105:212–20.
Coss KP, Hawkes CP, Adamczyk B, et al. N-glycan abnormalities in children with galactosemia. J Proteome Res 2014;13:385–94.
Pucić M, Knezević A, Vidic J, et al. High throughput isolation and glycosylation analysis of IgG-variability and heritability of the IgG glycome in three isolated human populations. Mol Cell Proteomics 2011;10: M111.010090.
Knerr I, Coss KP, Doran PP, et al. Leptin levels in children and adults with classic galactosaemia. JIMD Rep 2013;9:125–31.
Meissner U, Hänisch C, Ostreicher I, et al. Differential regulation of leptin synthesis in rats during short-term hypoxia and short-term carbon monoxide inhalation. Endocrinology 2005;146:215–20.
Elias CF, Purohit D. Leptin signaling and circuits in puberty and fertility. Cell Mol Life Sci 2013;70:841–62.
Kratzsch J, Lammert A, Bottner A, et al. Circulating soluble leptin receptor and free leptin index during childhood, puberty, and adolescence. J Clin Endocrinol Metab 2002;87:4587–94.
Gordeladze JO, Drevon CA, Syversen U, Reseland JE. Leptin stimulates human osteoblastic cell proliferation, de novo collagen synthesis, and mineralization: Impact on differentiation markers, apoptosis, and osteoclastic signaling. J Cell Biochem 2002;85:825–36.
Kratzsch J, Knerr I, Galler A, et al. Metabolic decompensation in children with type 1 diabetes mellitus associated with increased serum levels of the soluble leptin receptor. Eur J Endocrinol 2006;155:609–14.
Knerr I, Herzog D, Rauh M, Rascher W, Horbach T. Leptin and ghrelin expression in adipose tissues and serum levels in gastric banding patients. Eur J Clin Invest 2006;36:389–94.
Nobili V, Manco M, Ciampalini P, et al. Leptin, free leptin index, insulin resistance and liver fibrosis in children with non-alcoholic fatty liver disease. Eur J Endocrinol 2006;155:735–43.
Trinko R, Gan G, Gao XB, Sears RM, Guarnieri DJ, DiLeone RJ. Erk1/2 mediates leptin receptor signaling in the ventral tegmental area. PLoS One 2011;6:e27180.
Lavens D, Piessevaux J, Tavernier J. Review: Negative regulation of leptin receptor signalling. Eur Cytokine Netw 2006;17:211–9.
Lee PJ, Lilburn M, Wendel U, Schadewaldt P. A woman with untreated galactosaemia. Lancet 2003;362:446.
Panis B, Bakker JA, Sels JP, Spaapen LJ, van Loon LJ, Rubio-Gozalbo ME. Untreated classical galactosemia patient with mild phenotype. Mol Genet Metab 2006;89:277–9.
Krabbi K, Uudelepp ML, Joost K, Zordania R, Õunap K. Long-term complications in Estonian galactosemia patients with a less strict lactose-free diet and metabolic control. Mol Genet Metab 2011;103:249–53.
Bosch AM, Bakker HD, Wenniger-Prick LJ, Wanders RJ, Wijburg FA. High tolerance for oral galactose in classical galactosaemia: dietary implications. Arch Dis Child 2004;89:1034–6.
Parekh R, Roitt I, Isenberg D, Dwek R, Rademacher T. Age-related galactosylation of the N-linked oligosaccharides of human serum IgG. J Exp Med 1988;167:1731–6.
Coss KP, Treacy EP, Cotter EJ, et al. Systemic gene dysregulation in classical Galactosaemia: Is there a central mechanism? Mol Genet Metab 2014;113:177–87.
Berry GT, Nissim I, Lin Z, Mazur AT, Gibson JB, Segal S. Endogenous synthesis of galactose in normal men and patients with hereditary galactosaemia. Lancet 1995;346:1073–4.
Schadewaldt P, Kamalanathan L, Hammen HW, Wendel U. Age dependence of endogenous galactose formation in Q188R homozygous galactosemic patients. Mol Genet Metab 2004;81:31–44.
Berry GT, Palmieri M, Gross KC, et al. The effect of dietary fruits and vegetables on urinary galactitol excretion in galactose-1-phosphate uridyltransferase deficiency. J Inherit Metab Dis 1993;16:91–100.
Haniu M, Arakawa T, Bures EJ, et al. Human leptin receptor. Determination of disulfide structure and N-glycosylation sites of the extracellular domain. J Biol Chem 1998;273:28691–9.
Kamikubo Y, Dellas C, Loskutoff DJ, Quigley JP, Ruggeri ZM. Contribution of leptin receptor N-linked glycans to leptin binding. Biochem J 2008;410:595–604.
Schaab M, Kausch H, Klammt J, et al. Novel regulatory mechanisms for generation of the soluble leptin receptor: implications for leptin action. PLoS One 2012;7:e34787.
Lammert A, Kiess W, Bottner A, Glasow A, Kratzsch J. Soluble leptin receptor represents the main leptin binding activity in human blood. Biochem Biophys Res Commun 2001;283:982–8.
Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, Canbay A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol 2012;56:952–64.
McGregor GI, Malekizadeh Y, Harvey J. Food for thought: Regulation of synaptic function by metabolic hormones. Mol Endocrinol 2015;29:3–13.
McCance and Widdowson’s. The Composition of Foods, Royal Society of Chemistry. 7th edn. London Public Health England, 2015. pp. 107–47.
Kratzsch J, Berthold A, Lammert A, Reuter W, Keller E, Kiess W. A rapid, quantitative immunofunctional assay for measuring human leptin. Horm Res 2002;57:127–32.
Kratzsch J, Schubring C, Stitzel B, et al. Inverse changes in the serum levels of the soluble leptin receptor and leptin in neonates: relations to anthropometric data. J Clin Endocrinol Metab 2005;90:2212–7.
Stöckmann H, Adamczyk B, Hayes J, Rudd PM. Automated, high-throughput IgG-antibody glycoprofiling platform. Anal Chem 2013;85:8841–9.
Royle L, Radcliffe CM, Dwek RA, Rudd PM. Detailed structural analysis of N-glycans released from glycoproteins in SDS-PAGE gel bands using HPLC combined with exoglycosidase array digestions. Methods Mol Biol 2006;347:125–43.
Shin YS. Galactose metabolites and disorders of galactose metabolism. New York: Wiley-Liss, 1990. pp. 267–84.
Acknowledgements
We would like to thank all our patients who volunteered for the study. We would like to acknowledge the expert help of our clinical Psychologists, Aoife Brinkley and Yvonne Rogers. Frances Gibbons, Colm Carolan, and the staff in the laboratory at Temple Street Children’s University Hospital are thanked for their help with the patient samples. Ahmad A. Monavari and Joanne Hughes are thanked for their stimulating discussion.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Knerr, I., Coss, K., Kratzsch, J. et al. Effects of temporary low-dose galactose supplements in children aged 5–12 y with classical galactosemia: a pilot study. Pediatr Res 78, 272–279 (2015). https://doi.org/10.1038/pr.2015.107
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2015.107
This article is cited by
-
The natural history of classic galactosemia: lessons from the GalNet registry
Orphanet Journal of Rare Diseases (2019)
-
Fertility in classical galactosaemia, a study of N-glycan, hormonal and inflammatory gene interactions
Orphanet Journal of Rare Diseases (2018)
-
Profiling of intracellular metabolites produced from galactose and its potential for galactosemia research
Orphanet Journal of Rare Diseases (2018)
-
Rigor of non‐dairy galactose restriction in early childhood, measured by retrospective survey, does not associate with severity of five long‐term outcomes quantified in 231 children and adults with classic galactosemia
Journal of Inherited Metabolic Disease (2017)
-
Classical galactosaemia: novel insights in IgG N-glycosylation and N-glycan biosynthesis
European Journal of Human Genetics (2016)
