
Abstract
Small-molecule drug discovery can be viewed as a challenging multidimensional problem in which various characteristics of compounds — including efficacy, pharmacokinetics and safety — need to be optimized in parallel to provide drug candidates. Recent advances in areas such as microfluidics-assisted chemical synthesis and biological testing, as well as artificial intelligence systems that improve a design hypothesis through feedback analysis, are now providing a basis for the introduction of greater automation into aspects of this process. This could potentially accelerate time frames for compound discovery and optimization and enable more effective searches of chemical space. However, such approaches also raise considerable conceptual, technical and organizational challenges, as well as scepticism about the current hype around them. This article aims to identify the approaches and technologies that could be implemented robustly by medicinal chemists in the near future and to critically analyse the opportunities and challenges for their more widespread application.
Similar content being viewed by others


Main
'Automation of science' bears the promise of making better decisions faster1. In drug discovery, automated systems already have a long and fruitful history2 (Fig. 1). Medium-throughput to high-throughput robotic screening in specialized assays has become standard in the pharmaceutical industry (Fig. 2). The breadth of other applications of automated systems extends from decision-support systems, to computational molecular design to fully fledged robotic synthesis and hit finding3. Prominent examples include traditional rule-based and model-based approaches (for example, the archetypal DENDRAL system for analysing mass spectra4, LHASA5 software for synthesis planning and various in-house tools for accessing and analysing chemical and biological data similar to Amgen's AADAPT system6), various software tools for de novo molecular design7 and prototypical robotic systems such as ADAM and EVE for automated target and hit finding1,8.

Starting off from results obtained by high-throughput compound screening, fragment screening, computational modelling or data from the literature, this feedback-driven discovery process alternates between deduction and induction, eventually leading to optimized hit and lead compounds. Smart automation of the individual parts of the cycle can help to reduce randomness and error, thereby supporting less wasteful, more productive and efficient drug discovery. Miniaturization and advanced lab-on-a-chip technology, together with machine learning methods, represent enabling technologies. The whole design cycle can also be performed completely inside a software program. These adaptive de novo design methods are equipped with both chemical knowledge for in silico compound synthesis and meaningful virtual screening models as surrogates for biochemical and biological tests, while active learning algorithms enable chemical space navigation towards compounds with promising properties. Note that the terms 'deduction' and 'induction' in the context of drug discovery are not always used in a strictly logical sense. Induction refers to explanatory reasoning in generating hypotheses. Deductive inference necessarily results in a true statement if the underlying hypothesis is true. Because a hypothesis in drug design is based on incomplete, error-prone experimental data, the term 'abduction' may be formally better suited. (Q)SAR, (quantitative) structure–activity relationship.

a | Millions of compound samples are stored in compact high-capacity facilities and handled by robots. b | Robot systems perform both high-throughput and medium-throughput screening of up to ten thousand samples per day to determine the activity against the biological target of interest. Multiple arms and flexible workstations enable fully automated liquid dispensing, compound preparation and testing. These storage and screening systems have become cornerstones of contemporary drug discovery. c | A prototype of a novel miniaturized design–synthesize–test–analyse facility for rapid automated drug discovery at AstraZeneca is shown. Images a and b courtesy of Jan Kriegl, Boehringer–Ingelheim Pharma; image c courtesy of Michael Kossenjans, AstraZeneca.
Nevertheless, the full integration of all aspects of compound design, synthesis, testing and automated iteration throughout the molecular design cycle (Fig. 1) has not yet been productively applied on a broader scale, although there have been a few isolated proof-of-concept studies. For example, MacConnell et al.9 recently disclosed a microfluidics-based, miniaturized discovery platform for ultra-high-throughput hit deconvolution by sequencing. The device distributes DNA-encoded compound beads into picolitre-scale droplets, cleaves off the compounds from the beads by ultraviolet (UV) irradiation and performs a fluorescence-based binding assay, hit detection and subsequent hit identification by DNA barcode sequencing. By replicate analysis, the authors were able to reduce the false-positive hit rate to below 3%. This proof-of-concept study highlights the use of integrated microfluidics systems for large-scale screening within short, hour-scale time frames and with very low material consumption. Another example is provided by researchers at AbbVie, who have developed an integrated robotic platform for the automated parallel synthesis of small, focused compound libraries, built mainly from commercially available components10. Their system is able to perform liquid handling and evaporation for in-line analytics, purification and activity testing. Turnaround times of 24–36 hours were reported, which allow the project teams involved to obtain results from hypothesis testing within a day or two11. Similar robotic systems have been installed or are under construction in several pharmaceutical companies (for an example, see Fig. 2, right panel).
Now, advances in areas such as 'organ-on-a-chip' technologies and artificial intelligence are increasingly providing the basis for more widespread application of semi-autonomous or even fully autonomous processes to support project teams in identifying and optimizing tool and hit compounds in drug discovery. The benefits of automation include: diminished measurement errors and reduced material consumption by the application of standardized procedures with robotic support; shortened synthesize-and-test cycle times, enabling fast feedback loops and compound optimization; and 'objectified' molecular design towards multiple relevant biochemical and biological end points without personal bias. Furthermore, given the increased interest in the application of sophisticated cell-based assays12 — in an effort to more effectively recapitulate disease biology and thereby improve the likelihood of identifying compounds that show efficacy in humans — more rigorous compound prioritization aided by automated approaches could be particularly important because these assays are not always suitable for high-throughput compound testing13,14.
The potential value of more fully integrated automated systems in drug discovery is substantial. However, as with past technological advances that have raised hopes of revolutionizing drug discovery (but often not lived up to expectations), it is important to look beyond the hype, for example, around automated high-throughput combinatorial synthesis, 'big data' and artificial intelligence. This article aims to identify the key approaches and technologies that could be implemented robustly by medicinal chemists in the near future and to critically analyse the technological and conceptual challenges of doing so in the context of workflows in industry. It first summarizes the state of the art in the application of automated systems in separate aspects of the 'design–synthesize–test–analyse' cycle and then discusses progress in the integration of these aspects to fully harness the potential of automation in drug discovery.
Automation in molecule design
Medicinal chemists select, design and prioritize molecular structures on the basis of factors including the desired biological activity of the compounds, other characteristics important for drugs (such as absorption, distribution, metabolism, excretion and toxicity (ADMET) properties), the availability of compounds and retrosynthetic analysis (if the compounds are being synthesized rather than being sourced from existing libraries or commercial suppliers). Consequently, medicinal chemists routinely face complex multidimensional optimization problems, with the importance of different parameters changing as the drug discovery process progresses from the identification of initial screening hits (when identifying compounds with the relevant biological activity is crucial) via hit-to-lead expansion (which often requires massive synthetic effort to improve compound activity and developability) towards the selection of clinical candidates (when there may be a need to compromise to achieve the best possible mix between desirable biological activity and desirable ADMET properties). Given the vast size (cardinality) of the relevant 'chemical space', which is estimated to be in the range of 1030–1060 drug-like molecules, the key challenge for medicinal chemists could be summed up as 'what to make and test next?' Automated drug discovery platforms must be able to provide the right answers to this question.
Chemical design concepts. Traditionally, compound selection and/or design was the sole domain of medicinal chemists, drawing on their expert knowledge and providing a substantial role for intuitive decision making. Over the past two decades, various broad concepts have emerged to help guide compound library design, hit-to-lead expansion and the enrichment of compound collections with new chemical entities. For example, diversity-oriented synthesis (DOS) provides a rationale for generating collections of small molecules with diverse functional groups, stereochemistry and frameworks in a controlled fashion15,16. Following this concept, Maurya and Rana17 recently reported on the diversification of macrocycles by carbohydrate-derived building blocks. As a complement to DOS, biology-oriented synthesis (BIOS) takes natural products as templates for generating synthetically accessible derivatives and mimetics18,19, often relying on natural product-derived scaffolds20. Finally, so-called function-oriented synthesis (FOS)21 strategies take the BIOS concept to the next level by aiming to recapitulate or tune the function of a biologically active lead structure to obtain simpler scaffolds, increase their ease of synthesis and achieve synthetic innovation22. A recent example of the FOS approach is the successful design of oxazolidine derivatives with antibiotic activities as simplified analogues of the structurally intricate natural product caprazamycin from Streptomyces23.
A wide range of guidelines that aim to improve the lead-likeness or drug-likeness of compounds have also been introduced, beginning with Lipinski's recommendations (often referred to as the 'rule of 5')24,25 and combined ligand efficiency (LE) and lipophilic ligand efficiency (LLE) values, which can be applied automatically or semi-automatically as computational filters for existing compound libraries or candidates for synthesis (see Refs 26,27,28 for reviews). Early applications of artificial neural networks have contributed to rationalization of the drug-likeness concept in more sophisticated abstract terms and enabled on-the-fly computational compound profiling29,30. Importantly, it has been realized that compound quality can be controlled by appropriate lead selection and optimization based on informed decisions rather than by the naive application of empirical rules31. Today, fully fledged in silico decision-support systems that greatly extend and augment such concepts and guidelines can assist medicinal chemists in multi-objective compound design, selection and prioritization32,33. A consequent 'predict first' mindset has recently been advocated by researchers at Merck, drawing from positive experiences with their own integrated design–make–test activities34. The concepts and guidelines have been reviewed comprehensively in the articles cited above, and thus this article focuses on some selected illustrative examples, as well as the limitations and challenges of autonomous computational selection and design of compounds.
Automated de novo design. Importantly, the probabilities of the underlying research hypotheses are recorded as experimental metadata and stored in databases, which enables automated semantic analysis, generating both revised design hypotheses and deriving new examples (that is, chemical entities) for testing35,36. Numerous automated compound generators and selection operators have been conceived for this purpose, some of which use certain classes of 'deep' machine learning methods; for example, generative and recurrent neural networks37,38, inverse quantitative structure–relationship models39,40,41 and reaction-based compound assembly techniques42.
De novo molecular design methods in particular have matured enough to be applicable in prospective settings and are now receiving increasing attention. Figure 3 presents examples of recent compounds that were obtained by fully autonomous or semi-autonomous de novo computational design. In each of these cases, a computer-generated molecular design hypothesis guided the decision of which compound to make next. The first example (Fig. 3a) demonstrates how computational target prediction can prioritize combinatorial compound assays. A focused imidazopyridine (compound 1) library was obtained by linear microfluidic synthesis on a chip, with the building block selection performed by an ant colony algorithm and multi-target activity predictions43. Several active molecules, such as compound 2, were obtained within minutes. The results of this study provide support for the close integration of microfluidics-assisted synthesis with computer-based target prediction as a viable approach to rapidly generate bioactivity-focused combinatorial compound libraries with high success rates. We revisit this design concept in more detail in the subsequent sections of this article.

a | A focused library of compounds with an imidazopyridine scaffold (compound 1) was synthesized on a microfluidics chip, based on the Ugi three-component reaction43. Coupling building block prioritization to a computational method for predicting ligand–target association led to the rapid identification of several ligands for G protein-coupled receptors (GPCRs), such as the α1A and α1B adrenoceptor antagonist shown (compound 2). b | Integration of computational activity prediction at GPCRs with microfluidics-assisted synthesis based on a reductive amination reaction enabled the identification of ligands with various binding profiles (compounds 3–6)44,45,46. c | Automated scaffold hopping from the drug fasudil (known to be a moderate inhibitor of death-associated protein kinase 3 (DAPK3); compound 7) and structure determination enabled the identification of a novel DAPK3 inhibitor (compound 8). On the basis of its binding mode determined by crystallographic studies, the diuretic drug azosemide (compound 9) was identified as a DAPK3 inhibitor47. d | Compounds 10 and 11 are examples of ligand structures that were computationally optimized from weaker or less selective precursors by using design methods trained on publicly available activity data48,49. e | The natural product (−)-englerin A (compound 12) was computationally morphed52 into the synthetically accessible compound 13; both compounds inhibit the transient receptor potential cation channel subfamily M member 8 (TRPM8). IC50, half-maximal inhibitory concentration; Ki, inhibition constant; LE, ligand efficiency; VEGFR, vascular endothelial growth factor receptor.
The second example (Fig. 3b) showcases the benefits of using virtual library enumeration in concert with target-panel prediction for focused library design and building block selection. Compounds 3–6 originated from the same chemical space accessible by reductive amination reaction products but possess different target preferences, validating the computational selection strategies employed. Compounds 3 and 4 were identified as potent and target-subtype selective ligands and synthesized in flow on a microfluidics chip44. Compound 5 was obtained as a target-subtype selective serotonin receptor 5-HT2B antagonist based on computational prediction, with no activities towards a large panel of off-targets45. By contrast, compound 6 was deliberately designed as an 'ultimately promiscuous' ligand, without showing aggregation in solution or possessing undesired frequent-hitter properties46. Importantly, very few compounds had to be synthesized to reach the design objectives.
The example shown in Fig. 3c demonstrates the advantageous interplay between ligand-based and structure-based hypothesis generation for scaffold hopping. With the known drug fasudil (a vasodilator, potent Rho kinase inhibitor and moderate inhibitor of death-associated kinase 3 (DAPK3)) as a template, computational de novo design suggested several scaffold hops47. A target prediction method relying on self-organizing neural networks prioritized these frameworks to obtain a novel DAPK3 inhibitor, compound 8. Subsequent crystallographic studies confirmed the binding of inhibitor 8 in the ATP–substrate pocket of the kinase (Protein Data Bank identifier: 5a6n). On the basis of the known binding mode of the de novo generated ligand, the diuretic drug azosemide (compound 9) could be identified as a DAPK3 inhibitor. This particular study succeeded in lead identification through the combination of automated scaffold hopping and experimental structure determination.
Compounds 10 and 11 are examples of computationally optimized ligand structures, starting from weaker or less selective precursors48,49 (Fig. 3d). In both cases, the design–synthesize–test cycles were guided by computational design methods trained on publicly available activity data, epitomizing the aforementioned 'predict first' philosophy.
The last de novo design example shown in Fig. 3e highlights the concept of automated morphing of natural products into synthetically accessible, isofunctional compounds, and illustrates the FOS design concept introduced previously. The natural anticancer compound (−)-englerin A (compound 12)50, which is synthetically accessible in a 14-step process51, was computationally (and by subsequent manual refinement) converted into compound 13, which could be afforded in only three synthetic steps52. Both compounds potently block transient receptor potential cation channel subfamily M member 8 (TRPM8) calcium channels, as correctly predicted by the software.
These selected examples of computer-assisted molecular design illustrate some of the potential of contemporary in silico methods for hypothesis generation. There is no doubt that state-of-the-art computational de novo design delivers new synthesizable chemical entities with desired properties. Multi-objective compound selection strategies have shown their applicability to de novo design, which is not only useful for prioritizing chemically attractive lead-like and drug-like molecular structures but also relevant in light of ligand–target promiscuity (estimates range between up to 5 and 11 pharmacologically relevant targets per drug)53,54,55,56. The logical next step is to combine these and related techniques with automated synthesis and compound testing in an integrated discovery platform.
Automation in compound synthesis
The automation and parallelization of chemical synthesis offer benefits such as increased speed and throughput, greater reproducibility, lower consumption of materials and, consequently, the possibility to explore wider areas of chemical space within a given time frame compared with manual, serial compound synthesis57. Historically, the first automated synthetic processes and robots were conceived for peptides58,59 (Merrifield's method for amide bond formation), oligonucleotides60,61 (solid-phase phosphoramidite method for internucleotide linkage) and later for oligosaccharides62 (for example, the trichloroacetimidate method for glycosidic bond formation).
A key element in each of these processes is the use of a small set of building blocks (including larger fragments) and a well-defined, robust chemical reaction to afford large sets of diverse products in high yields by iterative building block assembly, orthogonal protection group chemistry and purification. Various methodological and technical improvements, including stereoselective synthesis, parallelization of subprocesses and preparatory steps, miniaturization (small volumes and compact synthesis arrays) and automated in-line purification, have resulted in highly reliable synthesis machines for increasingly complex oligomeric structures. Their underlying general design concept mimics the biosynthesis of most natural products. Furthermore, combinatorial thinking has led to methods for the massively parallelized scaffold-centric synthesis of structurally diverse compound libraries63. Many of these approaches are readily amenable to miniaturization and inclusion in automated design cycles64. Researchers at Eli Lilly have established a superb example of such a fully automated robotic synthesis laboratory that can be remotely controlled, which is a major step towards advancing the efficiency and effectiveness of chemical synthesis for drug discovery65,66.
Some reaction schemes have been shown to be more agreeable than others for straightforward automation and parallelization67,68. Typically, these reactions do not require exotic reaction conditions, can be standardized, are amenable to a wide variety of (readily available or obtainable) educts and can be optimized for maximum yield. Prominent examples include scaffold-forming reactions (for example, the Pictet–Spengler reaction and metathesis reactions)69,70. Other desirable linkage reactions (for example, palladium-free C–C bond forming reactions) have been scarcely used in medicinal chemistry or automated synthesis set-ups71,72.
However, automated discovery processes may be crucial for exploring new chemistry73. One of the most versatile automated synthesis platforms for drug-like small molecules to date was developed by Burke and co-workers74. The synthesis of Csp3-rich macrocyclic and polycyclic natural products, pharmaceuticals and natural product-like cores was achieved by iterative building block assembly via automated C–C bond formation and cyclization reactions75 (Fig. 4). Cartridged bifunctional N-methyliminodiacetic acid (MIDA) boronate building blocks were prepared for this purpose, complementing the commercially available samples. Importantly, a small set of building blocks was sufficient for generating remarkable structural core diversity in the final products. The authors developed an in-line catch-and-release purification protocol for realizing a seamless three-step reaction cycle. Similarly to the automated synthesis of oligomers, this important advancement in automated synthesis was enabled by standardizing the synthesis and purification processes involved.

The example demonstrates the concept of sequential boronate building block assembly. Four building blocks (coloured circles) are combined in a standardized deprotection, coupling and purification process. Synthesizers implementing this and other combinatorial reaction schemes can serve as chemistry modules in automated drug discovery platforms. Adapted with permission from Ref. 74, Science/AAAS.
Microfluidics-based synthesis. 'From batch to continuous' is a general trend in industry and not limited to chemical production processes76,77. Evidently, miniaturized microfluidic synthetic and analytical devices will play a central role in drug discovery automation. Microfluidic reactors integrated with real-time product detection and a command-and-control system can, in theory, perform and analyse thousands of reactions on timescales that are not possible with conventional macroscale technologies.
Embracing such advantages demands the substitution of widespread, but inefficient, one-parameter-at-a-time methods with more sophisticated and specialized algorithms. For example, trial-and-error scanning of the experimental parameter space can identify local optima but often fails to find global optima. In the field of medicinal chemistry, reagents and products are often expensive. Furthermore, many reagents and intermediates have unknown hazards and must be treated with extreme caution owing to their unknown pharmacology. Microfluidics can offer an advantage by decreasing opportunities for human exposure and minimizing material usage78.
There are also several other technologies that can be used for this purpose. For instance, acoustic liquid handling systems for precision droplet dispensing are well-accepted tools in chemical synthesis that increase the reproducibility of experiments and reduce the amount of consumables needed, thereby cutting costs79,80. Exceptionally high precision has been reported for transferring microlitre droplets into well plates81. Nevertheless, each automation process requires skilled chemists and solid chemical engineering, as the individual usage of acoustic droplet ejection and its applicability depend on the types of liquids and mixtures handled82.
As a distinct feature of microfluidics systems, converging streams of fluids flow in parallel without turbulence (that is, the conditions of laminar flow are fulfilled), with characteristically low Reynolds numbers (the ratio of inertial forces to viscous forces, a dimensionless parameter indicating whether a flow condition will be laminar or turbulent)83. In addition to allowing miniaturized bioassays in flow, this property of microfluidics systems enables fine-tuned, diffusion-controlled synthetic reactions84. The short distances in microfluidic channels guarantee the desired rapid and controlled transport of heat and mass. Complex channel geometries, pulsed flow conditions and the high surface-to-volume ratio of miniaturized reactors can result in a dramatic increase in throughput and yield in microreactors85.
Ley and colleagues pioneered the field of flow chemistry, which has numerous practical applications in drug discovery; for example, the synthesis of imatinib in flow86, the translation of four sequential steps into a continuous-flow system to generate (E/Z)-tamoxifen with 100% conversion and 84% yield87 and numerous natural product syntheses88. Their seminal work has introduced single-step and multistep microscale and mesoscale flow systems, which enable otherwise difficult reactions with low yields or reactions that require special safety measures to be performed, such as hydrogenation or ozonolysis89,90,91. Warrington and co-workers have explored numerous reactions and microreactor designs, which have paved the way for advanced applications92,93,94,95. The technical capability of multistep continuous-flow synthesis was demonstrated by the Ley group in the generation of key intermediates for the total synthesis of the polyketide spirangien A96. This high-yielding system consists of heterogeneous reactor coils and microfluidics components, requiring minimal downstream processing.
Some of these techniques are already being applied in the pharmaceutical industry. For example, researchers at the Novartis–Massachusetts Institute of Technology (MIT) Center for Continuous Manufacturing succeeded in assembling a compact system for the continuous end-to-end synthesis of diphenhydramine hydrochloride, lidocaine hydrochloride, diazepam and fluoxetine hydrochloride in qualities that meet US Pharmacopeia standards97. Continuous-flow syntheses have also been used early on to obtain drug-like combinatorial compound libraries with heterocyclic scaffolds98,99.
Nagaki and co-workers noted the specific advantage of flow microreactors to enable 'flash' chemistry reactions that cannot be performed in batch100. The high-resolution reaction time control possible in microreactors allows access to a multitude of otherwise difficult synthetic procedures101. One such prominent example is the sequential synthesis of the subtype-selective retinoic acid receptor-α (RARα) ligand TAC-101 with a total on-chip residence time of 13 seconds and a productivity of 100–200 mg min−1 (Ref. 102). Another example is the high-temperature, high-pressure continuous-flow synthesis of 1H-4-substituted imidazoles103. The use of microfluidics technology to simulate the cytochrome P450-catalysed oxidation of drug molecules bears the promise of substituting in vitro metabolite identification by on-chip chemotransformations of compounds in the near future (for example, aromatic hydroxylation, C–H oxidation, glutathione conjugation and sulfoxidation)104,105. For further instances of advanced continuous-flow applications in chemical synthesis, see the topical review by Britton and Raston106.
Automated optimization of reaction conditions. Single-step and multistep syntheses can be optimized by feedback control107. Jensen and co-workers108 pioneered self-optimizing microscale and mesoscale reactor systems, for example, for C–C bond forming reactions. A recent example of such reaction optimization by suitable algorithms to achieve the maximum product yield, highest throughput and lowest production cost is the palladium-catalysed Heck–Matsuda arylation reaction109. Our group used microfluidic synthesis with in-line analytics to determine the optimal flow rate, temperature range, catalyst loading and reagent concentrations for continuous imidazopyridine formation on a chip43. Comparable conversion rates were obtained in a microwave procedure, albeit with much longer reaction times (15 min in the microwave reactor versus 0.3 s in flow). In-line mass spectrometry has enabled the optimization of atropine synthesis in microdroplets obtained by preparative electrospray (ES), as recently demonstrated by researchers from Purdue University110. They devised several continuous-flow set-ups with multistep or telescoped preparative ES, yielding up to 47% conversion of the starting material to atropine in residence times of a few minutes. Microfluidics techniques have also simplified the set-up and improved the functions of ambient mass spectrometry by integrating probe sampling and ES on a single glass microchip111.
Nevertheless, there are limitations to continuous-flow systems including the (in)stability of the fluidic interfaces between microscopic and macroscopic fluid handling and the deposition of reactive by-products, and automated batch synthesis and fast parallel synthetic strategies have been suggested as alternatives112. For example, researchers at Merck recently presented their 'chemical high-throughput experimentation' (HTE) platform in 3,456-well microtitre plates, aiming to optimize a key synthetic step in a drug discovery programme. HTE successfully identified the preferred catalyst, reaction conditions, reagents and solvents for the given transformation. The authors conclude that hypothesis-driven HTE allows a scientist to 'go fast' and may be considered the logical extension of traditional chemical experimentation113. Chow and Nelson114 have argued that automated HTE discovery workflows may enable expansion of the synthetic chemistry toolkit and increase innovation in medicinal chemistry.
An advantage of batch approaches, namely the ability to collect data from many time points in a single experiment, and a limitation of one-at-a-time flow experiments, has been addressed by recording time-series reaction and interaction data in-flow for kinetic analysis115. Similarly, microfluidics systems are no longer restricted to single-step reactions. For all these applications, in-line spectroscopy and purification of intermediates are vital to ensure maximal yields. Various fluorescence-based and infrared-based detectors, as well as Raman, NMR and mass-spectrometric analytical devices, have been integrated into continuous mix and flow systems116,117,118. Steady progress in miniaturized manufacturing of analytical devices facilitates system integration. In particular, 3D printing provides opportunities for building versatile multifunctional microfluidics modules with embedded in-line reaction monitoring and analytical capability119.
Droplet reactors. Although there are several off-the-shelf instruments available (for example, for hydrogenation reactions), the majority of current microfluidics platforms require a custom set-up, and one should carefully weigh the pros and cons of microfluidic versus batch technologies before deciding on a particular technology.
Coupling the individual components is an engineering challenge. The majority of platforms currently being introduced in industry for the automated parallel synthesis of small, focused compound libraries seem to operate without making extensive use of microfluidics-assisted chemical synthesis, probably because for certain microfluidic reactors, clogging of the reactor channels and leakage due to back-pressure issues or incompatibility of the solvents and materials remain a major problem. Performing chemical flow reactions in droplet environments offers a potential solution to several of these problems. Droplets may be considered isolated mini-reactors with volumes reduced to the femtolitre scale120,121, facilitating sorting and process control122. DeMello and co-workers123,124 have demonstrated that droplet-based microfluidics systems are precise tools for studying and optimizing the synthetic parameters of chemical reactions, leading to the production of materials with superior characteristics (Fig. 5).

The image shows a microreactor channel with droplets containing multinary (Cs/FA)Pb(Br/I)3 perovskite nanocrystals123. Each droplet exhibits different, composition- dependent emission under ultraviolet excitation, revealing the compositional gradient along the reactor. The flow rates of the individual precursor streams provided control over reaction times as well as precursor concentration ratios. This example from the field of nanomaterials demonstrates the unique capabilities of droplet- based synthesis for the production of chemical matter. Image courtesy of Andrew J. deMello and Richard Maceiczyk, ETH Zürich.
A challenge for drug discovery is the slow reaction time of many chemical transformations. Furthermore, any realistic application of such high-throughput miniaturized synthetic devices in drug discovery requires rapid in-line analytics of the generated products. Belder and co-workers125 have recently presented a droplet-based microfluidics system with seamless coupling to ES–mass spectrometry. In a proof-of-concept study, they applied the device to an amino-catalysed domino reaction in nanolitre droplets (Knoevenagel condensation followed by an intramolecular hetero-Diels–Alder reaction), with only picomolar amounts of catalyst needed. The greatly increasing numbers of applications and technological advances in the field of continuous microfluidic synthesis showcase the potential of these platforms for the high-throughput generation of diverse chemical entities for subsequent testing. The concept of continuous microfluidic reactors, which were originally designed for the continuous production of single compounds, has been augmented by their suitability for producing many compounds within very short time frames.
Microfluidics technologies for screening
The use of miniaturized microfluidics devices not only supports chemistry but also enables the use of human cell lines, biopsy material and organ models for screening, thereby helping to address the well-known issues with species-specific variations and poorly predictive animal models126,127. For example, liver-on-a-chip technology based on human hepatocytes can be used to swiftly screen compounds for cytochrome P450 binding to substrates and inhibitors, as well as subsequent high-performance liquid chromatography (HPLC)–mass spectroscopy for metabolite identification128. Combined with computational predictive models, this technology is ready for prospective practical application129. Cancer-on-a-chip systems that use single cells or 3D cancer models bear the promise of replicating the pathophysiology of human tumours and tumour environments in vitro130,131. Again, as with the many other organ-on-a-chip models, this technology has the potential to produce relevant readouts within short time frames and to enable informed hit and lead prioritization and optimization.
Physiologically relevant microfluidic environments are stable over weeks and have a footprint of a few square millimetres. For example, Loskill et al.132 recently presented a white adipose tissue (WAT)-on-a-chip system, allowing drug–WAT interactions to be studied by convective transport. Cao et al.133 reported a microfluidics system for rapid epigenetic DNA scanning to monitor drug effects on stem cells, using as few as 100 cells. Microfluidics platforms have been developed for the high-throughput (thousands of samples) analysis of DNA methylation patterns in low volumes on a chip, greatly extending chemical base modification studies for epigenetics-related drug effects134. Dittrich and co-workers135 demonstrated the possibility of determining the concentration of intracellular cAMP in response to extracellular stimuli in single cells, thereby greatly extending the capabilities of continuous chip-based assay systems for measuring relevant biochemical parameters for drug discovery. In addition, 3D triple co-culture microfluidics devices have been established as functional surrogates for the blood–brain barrier136.
Advanced nanotechnology offers even farther-reaching opportunities such as micromachines (nanobots) for drug delivery137. In fact, the prospect of combining nanotechnological devices with on-chip testing of computationally designed compounds does not seem far-fetched. Advances in chemical imaging further augment the capabilities of on-chip monitoring, for example, by miniature electrode arrays for high-resolution peak analysis138. 'Plug-and-play' microfluidics modules are the next step towards fully integrated on-chip drug discovery. Miled and co-workers developed such a modular lab-on-a-chip device for automated monitoring and modulating of the concentrations of neurotransmitters such as dopamine and serotonin, thereby opening new possibilities for functional drug screening with feedback control139.
Integration for automated design cycles
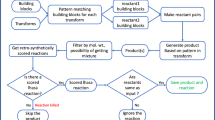
Coupling synthesis and testing. The Automated Lead Optimization Equipment (ALOE) platform is a prototypical example of an adaptive molecular design process140. Its software control contains an algorithm for building predictive bioactivity models and prioritizing the selection of starting materials for subsequent rounds of on-chip compound generation. The system can adapt to the underlying structure–activity relationship (SAR) and rapidly find optima in chemical space, with low reagent consumption.
Basic schematics of integrated microfluidics synthesize-and-test platforms are shown in Fig. 6, and a selection of applications is listed in Table 1. These methods operate on small volumes of fluids in geometrically well-controlled environments composed of different functional units, for example, dispensers, mixers, reactors and detectors. Solvent exchange may be required when transferring newly synthesized compounds to biochemical or biological testing, which is typically performed in aqueous media. Some of the integrated flow systems allow for slow solvent mixing and direct in-line testing. Fast evaporation and reformatting has also proved suitable and may represent an alternative working solution, especially in combination with batch synthesis. For example, researchers at Cyclofluidics developed a flow technology platform integrating the key elements of adaptive SAR modelling to the discovery of novel ABL1 kinase inhibitors141. Similarly, Tseng and co-workers142 devised a complex microfluidics chip for 'click' chemistry and subsequent hit identification. In their proof-of-concept study, throughput was limited by the employment of an eight-channel mass spectrometer for reaction monitoring, but the authors argue that substantially higher throughput could be achieved by expanding the instrumentation.

The classic linear layout shown in part a does not contain automated feedback from the assay to the reagent selection, whereas the cyclic layout shown in part b includes an adaptive computer model for reactant prioritization based on the assay readout. LC, liquid chromatography; MS, mass spectrometry; UV, ultraviolet light.
For biological experimentation and integration with chemical synthesis devices, droplet microfluidics systems and biological readouts from single cells seem to be reasonable choices143,144 (Fig. 7). These systems are suitable for creating concentration gradients and generating microdroplets of varying compositions for biochemical and cell-based screening applications. Similar to chemical microreactors, compared to single-layer microfluidics systems, 3D droplet-based systems have been shown to be more efficient and amenable to ultra-high-throughput analysis145. Droplets are especially suitable for performing enzyme-controlled processes146,147 and may contain cells for probing drug effects in continuous flow148. In this way, single cells may be addressed, thereby eliminating potential issues of readout interpretability caused by cell heterogeneity, for example, for studying cancer cells149. Often, a fluorescence-based readout of phenotypic drug effects is obtained for further analysis150. The rapidly developing and progressing field of microfluidics-assisted lab-on-a-chip platforms has recently been reviewed by Nakajima and co-workers151.

A microfluidics system for the continuous screening of compound effects on single cells is shown. It consists of a double-layer device containing an array of chambers. Each chamber has a central trap for capturing cells or vesicles (individual traps are visible in the enlarged illustration of a section of the device) and a round valve that can be opened and closed for fluid exchange. For analysis, the valve is usually closed. The volume of the chambers depends on the particular chip design and is typically 150–500 picolitres. Reproduced with permission from Lucas Armbrecht and Petra S. Dittrich, Bioanalytics Group, ETH Zürich.
The full automation of compound synthesis also requires reliable planning tools for synthesis and retrosynthesis. In fact, numerous such programmes have been conceived, dating back to Corey's pioneering work from the 1960s152, employing rigorous physical models (for example, reactivity prediction), rule-based approaches (for example, synthons and reaction schemes) or empirical models (for example, precedent-based database searching). Classic approaches have been reviewed elsewhere153,154,155. Their main drawbacks are their limited scope and often inaccurate results caused by insufficient chemical background knowledge captured by the software tools, paired with low execution speed.
Current computational tools are largely data driven. For example, ReactionExplorer is based on thousands of manually curated rules (electron-transfer steps) that represent basic chemical transformations to devise a mechanistic interpretation of a plausible reaction pathway156. More recently, machine learning models have been developed for automated synthesis planning, enabled by large curated reaction databases. ReactionPredictor is such a method and automatically identifies and ranks electron-transfer steps by use of a simplified molecular orbital description157. The number of prospective applications of these and other tools is still limited, and there is not much experience, if any, with integrating such tools in automated synthesis platforms. However, the continuously growing 'Network of Organic Chemistry' (NOC) contains approximately ten million reactions and reactants for synthesis planning158. One may consider such a collection of facts 'big data' in chemistry. Szymkuc et al.159 presented an innovative approach to reaction pathway construction based on NOC, using fast graph-analysis methods borrowed from bioinformatics. These algorithms are able to efficiently navigate through the entire breadth of chemical synthesis knowledge to identify optimal synthetic pathways. Alternative synthetic routes leading from the reactants to the products are compared using a function that includes the number of steps and the cost of synthesis. Finally, algorithmically identified optimal syntheses are obtained.
These and related data-driven machine learning approaches, with continuously increasing accuracy and chemical reaction space coverage, are no longer science fiction and will enable fully integrated drug discovery platforms to be built. One such straightforward approach implements a combination of forward reaction templates for generating a set of chemically plausible candidate products and a machine learning classifier for virtual product scoring160. This system is based on more than one million reactions compiled from United States patent literature. Importantly, the model does not predict quantitative yields but merely spots plausible true reaction products in the pool of potential solutions. Although this overall concept may not be entirely new, the availability of suitable reaction databases and advanced machine learning models has enabled the development of robust classifiers.
Artificial intelligence in molecular design. Aside from the required robotic hardware and synthesize-and-test machinery, the learning aspect probably represents the most crucial part of the automated design cycle. If the design hypothesis is wrong, then even the most advanced synthesize-and-test approach will fail to deliver, irrespective of the technology used. It is important to note that if we can achieve partial predictability of SAR models in this situation and build on iterative adjustments of our underlying molecular design hypothesis, we can gradually approximate the underlying function. This process is referred to as 'adaptive design' or 'active learning' (Refs 161,162). The key requirement for active learning is rapid feedback, and for hit and lead discovery, rapid feedback can be achieved by fast synthesize-and-test cycles.
Considering this situation from an information-theoretical viewpoint, the full-deck screening of hundreds of thousands of compounds by contemporary technology (for example, as shown in Fig. 2) may be not only cost intensive but also inefficient. Such an approach does not include feedback but relies on a single library design step before brute-force compound testing. The necessary continuous adjustment of the molecular design hypothesis is performed only in the later stages of hit optimization and lead expansion. This design concept is prone to fail when relying on noisy data, personal bias and poor intuitive choices ('gut feeling').
The active learning concept is central to automated drug discovery. This concept is based on iteratively adapting a design hypothesis — for example, a quantitative SAR model — by adjusting its free variables on the basis of newly acquired compound activity data. The modified design hypothesis is then used to select new compound sets for synthesis and testing. Dating back to the early 1990s, there have been several attempts to use adaptive de novo drug design guided by artificial neural networks and other machine learning techniques (see Refs 163,164,165 for reviews), although these attempts have been isolated. In a recent article, Hunter166 advanced the view that adopting and exploiting the full potential of artificial intelligence methods for pharmaceutical research might be essential to creating a sustainable drug discovery process.
A specific advantage of machine-driven hypothesis generation is that new compounds may be designed according to numerous criteria in parallel, for example, activity, synthesizability, predicted off-target effects and so on. Importantly, these models are able to capture essential non-additive (nonlinear) feature contributions to the design objectives, which cannot be appropriately considered by linear substituent contribution models (for example, Free−Wilson analysis and matched-molecular-pair analysis)167,168. Non-additive models of protein−ligand binding are a basic prerequisite for rational drug design169.
While explorative selection by active learning aims to add new information to the model with each iteration through the design cycle, exploitive selection maximizes compound quality with regard to certain design criteria, such as activity and selectivity. Balanced selection strategies compromising between these two extremes seem to be particularly suitable for both finding potent compounds (exploitive selection) with novel scaffolds (explorative selection) and optimal SAR model building170,171. This principle of model adaptation by active learning offers the additional advantage of limiting both the number of iterations that are required to find compounds with the desired properties and the number of compounds to be synthesized and tested in each iteration of the design cycle172. Visualization of the fitness landscape ('activity landscape') modelled during each iteration can additionally help to navigate the chemical space173 (Fig. 8). Compound 14 is a new subtype-selective antagonist of the dopamine D4 receptor found by active learning with an ant colony algorithm (MAntA, Molecular Ant Algorithm)174 for compound selection44. Similarly, new CXC-chemokine receptor 4 (CXCR4) antagonists have been identified by active learning with a random forest model175.

Knowledge of the underlying structure–activity relationship (SAR) captured by a machine learning model is very limited in the beginning of a discovery project but grows over time with each active learning step. The 'fitness landscapes' visualize the areas of chemical space that are associated with low (transparent) and high (strong colour intensity) predictive confidence (part a). In the example, d1 and d2 denote meaningful coordinates of chemical space, which can be obtained, for example, by projection or dimensionality-reduction techniques245. The distributions shown in part b illustrate four stages of a SAR model during active learning. The average predictive confidence increases (and the margin of error decreases) with each iteration (models 1–4). The initial model 1 was trained on literature data (in this case, CXC-chemokine receptor 4 (CXCR4) ligands)175. Models 2 and 3 were obtained after testing 30 additional compounds per learning step. Model 4 was trained with all tested compounds taken together. The small discrepancy of predictive confidence between models 3 and 4 demonstrates the efficiency of the active learning process. D4R, dopamine D4 receptor; KD, dissociation constant; Ki, inhibition constant; P, pseudo- probability density function.
'Deep learning' from 'big data'. The possibilities of computational molecular structure generation and property–activity prediction seem virtually unlimited. A particular appeal of automated structure generators lies in their trainability on complex chemical data, extreme speed and consideration of several design objectives in parallel. The young research field of constructive machine learning offers innovative methods for learning multidimensional SARs and iteratively navigating in very large chemical spaces to suggest chemical entities for testing that optimally fit the design hypothesis.
Based on the body of assay data stored in public and proprietary databases, it is now possible to train learning machines on arbitrary target−target, ligand−target and ligand−effect associations. Algorithms are able to recognize hidden patterns in molecules that escape medicinal chemical rationales and intuition because of the large set of variables and drug design objectives that should be considered in parallel. Suitable molecular structures that fit these patterns can then be computationally generated and forwarded to chemical synthesis and analytics and subsequent biophysical, biochemical and biological testing. A new design hypothesis is formed after updating the machine learning model with the newly obtained assay data (feedback loop), and swift compound optimization can take place. With such a set-up, one can expect to make informed choices of starting points for lead optimization.
Drug design can be regarded as a pattern recognition process. Medicinal chemists are skilled in visual chemical structure recognition and their association with retrosynthetic routes and pharmacological properties. In this context, various 'deep-learning' concepts are currently being evaluated as potentially enabling technology for drug discovery and automation because these systems aim to mimic the chemist's pattern recognition process and to take it to the next level by considering all available domain-specific data and associations during model development. While acknowledging their usefulness, we should not fool ourselves with the term 'deep learning' or consider these methods 'magic wands'. These systems are reincarnations of artificial neural network prototypes for automated molecular design from the 1990s176,177,178,179 that, in augmented and expanded form, can now be trained and optimized on complex pattern recognition tasks, largely owing to substantial improvements in available hardware and software180,181. One of the prominent machine learning toolkits harnessing the computational power of specifically developed tensor processing units (TPUs; application-specific integrated circuits developed by Google)182 is the TensorFlow open-source software library for numerical computation183,184. This software library provides access to contemporary machine learning methods and has found widespread use for cheminformatics and bioinformatics modelling and medicinal informatics185,186,187,188. For a review on toolkits and software libraries for deep learning, see Ref. 189.
To date, most machine learning applications in the field have been 'shallow' — that is, using a single layer of feature transformation to achieve their goals. This class of algorithms includes various clustering and regression methods (for example, nearest neighbour approaches, support vector machines, standard neural networks and decision trees). The successes of these methods in activity prediction and lead suggestion are, in part, due to the development of useful, often domain-specific, molecular representations, which enable comparably simple machine learning architectures to make reasonable predictions. In the process of engineering and applying these descriptor systems, we include a measure of our chemical knowledge and understanding in the depiction of the actuality of these molecules. Now, 'deep' methods based on learning directly from molecular graphs and other physically oriented models of complex molecular objects have been proposed that remove some of this input-level abstraction190,191,192. This more general approach, however, benefits from a more sophisticated machine learning methodology for pattern recognition, as the input data are much less amenable to producing useful output with 'shallow' transformation methods.
Essentially, deep-learning models are hypothesis generators. Their secret lies in a cascaded feature extraction and transformation process from the training data representation and in nonlinear function estimation based on these features (Fig. 9). While passing information from the input to the output layer, increasingly intricate features are formed in the subsequent layers of such models. Each network layer may contain heterogeneous processing units that select and refine features in different ways. Such a learning process often results in models that elude our immediate interpretation in chemical terms193,194. Nonetheless, such models can be extremely useful195,196.

Deep neural networks transform the input data (for example, molecular structures or microscopic images) by cascaded feature extraction and compute a nonlinear function of the input, f(x). They essentially represent universal function estimators. Each network layer can vary in size and architecture, can have alternating functionality and can contain different types of processing units. When trained on compound activity data, the overall network function adapts to the underlying structure–activity relationship and, after successful training, can be used for automated compound design. Essentially, such learning systems are able to incorporate new data (for example, new compound–target activities or chemogenomics data) and continuously adjust their internal model of the input–output relationship. The depicted network architecture highlights only one of several related deep-learning concepts.
From a chemogenomics viewpoint197,198, deep-learning methods for model building may indeed represent a breakthrough199,200,201. Currently, there are approximately 70 million SAR data points stored in public databases, not accounting for the very large volumes of proprietary data from deep sequencing and other massively parallel and ultra-high-throughput assays. Deep-learning networks provide appropriate technology for analysing such large amounts of data to find meaningful relationships between ligands, proteins, genotypes and phenotypes202,203,204,205. Several heterogeneous deep-learning systems with high prediction accuracies have been developed for drug–target association, drug repurposing opportunities and target identification, among other tasks202,206,207,208. Deep network models have also been shown to improve conventional virtual screening methods, such as automated ligand docking209, and to accelerate otherwise computationally costly chemical computing tasks210. Various applications of deep learning in biomedicine have been comprehensively reviewed211.
Curated consistent data are a prerequisite for improved model building. A consortium of industrial and academic partners has recently published a new comprehensive database of standardized chemical and biological data for chemogenomics data analysis (ExCAPE-DB212, Exascale Compound Activity Prediction Engine)213. Although the number of compound structures and activity values stored in these databases may appear impressive from a chemistry-oriented viewpoint, they are vanishingly small in comparison with other fields, such as computer vision214. With the exception of virtual chemical space, one may indeed wonder if big experimental data exist in chemistry215. In this context, Tetko et al.216 suggested the definition of big data as “out of the scale of traditional applications, which require efforts beyond the traditional analysis”. Data sharing and open software between research organizations will further expedite successful model building for automated drug discovery217. Importantly, big data as such are not a prerequisite or guarantee for obtaining good predictive models. Similarly, it is advisable not to simply try and apply deep models to any given classification or regression task in drug discovery, but to carefully evaluate the required model complexity and its applicability domain beforehand210,218,219.
Conceptual and practical challenges
Judging from successful proof-of-concept studies and pilot applications, potentially major benefits for drug design from the integration of automated discovery processes can be anticipated. These include low error rates (for example, reduced risk of false positives), high speed of execution (for example, faster hit and lead identification), low consumption of materials (advancing green chemistry), straightforward synthetic schemes for ease of compound production, potentially patentable compound structures (in combination with scaffold hopping), ease of instrument handling (low maintenance) and, ultimately, improved decision making for hit and lead candidate selection.
Nevertheless, molecular design is governed by nonlinear relationships between the chemical structures and their biological activities, random events (serendipity), measurement and judgement errors and the incompleteness of available drug discovery data. In addition, erroneous assay readouts hamper accurate model building, and poor data curation can easily be a limiting factor for machine learning. Reducing errors in data annotation and relying on suitable assays will therefore be mandatory for future success. Progress in automatically detecting and recovering false negatives (that is, active compounds misidentified as inactive by the test) points to new means of hit selection besides relying on primary activity alone220. Automated retesting of suspicious compounds could be performed by autonomous robots. Researchers at Pfizer recently disclosed success rates of 13–51% of true false negatives from HTS that were rescued based on computational prediction221.
Although the required flexibility and adaptability of the design hypothesis have long been adopted in software solutions for de novo molecular design and model building, real-life applications have only recently been demonstrated. Minimizing the time gap between synthesis and testing may be the vital factor for increased productivity of drug discovery projects. A high program speed increases the number of design loops that can be made and limits the risk of generating new compounds agnostically, without full integration of the test results into the design hypothesis. There is no learning without reflection and feedback.
Lab-on-a-chip and other miniaturized and/or mobile platforms with a small footprint seem to be suited to address this bottleneck in hit expansion. As appealing as this technology may be, however, seamless integration of the heterogeneous instrumentation faces technical challenges. New continuous-flow platforms may provide a complement or even an alternative to these mixed-method systems. Similar to conventional robot-assisted systems, in continuous-flow devices, the lack of direct in-line methods for compound profiling in dose–response format has prevented the emergence of fully automated hit discovery and optimization in the past.
Another limiting factor is the currently restricted versatility of automated synthesis platforms. Each chemical reaction requires optimization and often hardware modifications (for example, seals, reactors and piping); the reagents must be prepared for handling, detection and purification protocols must be adjusted and so on. On-the-fly switching from one chemical transformation scheme to another and sequentially performing multiple steps automatically may be straightforward in silico, but remains challenging in real life. Although one-step syntheses of individual compounds or focused libraries can be robustly performed in parallel batches or in flow, we still need to identify the sweet spots of such platforms for seamless integration in drug discovery. The elegant automated synthetic strategy devised by Burke and co-workers74, which enabled the generation of structurally diverse compounds from a limited set of simple building blocks (Fig. 4), points to a direction of future research to address this issue.
With all the current excitement about sophisticated artificial intelligence systems and the maturation of rapid automation, it is crucial to identify approaches and technologies that could be implemented robustly by medicinal chemists in the near future and to discuss the challenges of doing so in the context of industrial workflows. Computational molecular design has always raised hopes that some computer wizardry might come to the rescue of stalled discovery projects. The prospect of process automation in the age of 'big data' further stimulates a drug designer's fantasies. What will the laboratory of the future look like? Are we facing the automation of drug discovery with autonomous molecular design robots replacing medicinal chemists?
There is no doubt that the automation of science has already begun. The use of robotic devices is not limited to improving the reproducibility of experiments; a particular feature of 'robot scientists' is their explicit foundation of scientific reasoning, which contrasts with the more polymorphic, generalized human mind222. The key technology drivers are hardware and software improvements and data availability. However, there may be limitations to the applicability of machine learning in chemistry, as recently noted by Gambin and co-workers223. According to their study, fundamental mathematical theorems impose upper bounds on the accuracy with which reaction yields and times can be predicted, which in turn will limit the scope of autonomous drug discovery platforms. Furthermore, the hundreds of thousands (or more) data points required for deep learning will be unavailable in many drug discovery projects. Alternative methods for equally robust feature extraction and hypothesis generation from 'small data' sets need to be identified. Pande and co-workers recently suggested 'one-shot' learning for such instances224.
More conventional modelling techniques are not expected to become outdated. The combination of 'big data' and 'deep learning' per se does not solve problems; it is the ability of the researchers involved who devise appropriate representations of chemistry and biology for computational analysis. Their scientific skills will be needed even more in future drug discovery settings. This notion becomes especially relevant when contemplating the fragility of autonomous discovery platforms. Although there have been reports about robots that can adapt to damage and show outwardly 'intelligent' behaviour225,226, at least in the foreseeable future, it will remain the task of the skilled scientists, technicians and engineers who design, run and maintain these discovery platforms.
Irrespective of the success or failure of individual technologies, this fresh view on drug discovery goes far beyond traditional approaches and will deliver innovative methodologies and potentially ground-breaking solutions that may have a substantial impact on future discovery concepts. One could envisage the future development of benchtop instruments equipped with building block cartridges for chemical synthesis and cassette-like bespoke assay panels for in-line screening, opening up great opportunities for small and medium-sized technology companies; for example, such a mobile instrument could be made available for project teams in many laboratories. Certainly, this concept does not make medicinal chemistry obsolete, as one might mistakenly deduce from some published comments on this topic227,228; in reality, the opposite expectation is probably closer to the truth. However, medicinal chemistry training needs to adapt to this new situation and to prepare chemists accordingly229,230,231.
The well-controlled conditions possible using microfluidic synthesis technology enable otherwise strongly exothermic, dangerous or difficult reactions to be performed safely, potentially making novel molecular scaffolds more accessible. However, chemists will still have to design these experiments to be performed by a machine, and the tool compounds obtained will not represent perfect lead compounds for immediate expansion and development. Furthermore, because the design machine will be able to produce chemical starting points very quickly, future hit-to-lead optimization and scaffold morphing will require strong chemical expertise and will probably generate demand for increased conventional synthesis capacity.
The possibilities of bioinspired molecular machines allow for even farther-reaching goals: for example, in the performance of diverse operations in response to chemical triggers. A recent example is provided by a DNA nanomachine that uses DNA origami command tracks to control a microfluidics device232. One may also envisage automated drug discovery platforms that include modules for dynamic combinatorial chemistry with biocompatible reactions; that is, the in situ generation of drugs binding to a protein target233,234. In light of the rather limited compound library sizes used in such projects to date, automated adaptive feedback control offers opportunities for the optimal exploration of chemical space for dynamic combinatorial chemistry.
There is no doubt that drug discovery demands the right mix of human mind, automation and machine intelligence. In the future, the 'intranet/internet of things' may enable fully autonomous cross-platform drug discovery. In combination with the appropriate test systems and metrics of success, such integrated environments bear the promise not only of stable system performance but also of increasing the competitiveness and efficiency of drug discovery processes by sharing resources and data intramurally and extramurally235,236.
Conclusions and future perspectives
The drug discovery process has characteristics of chaotic systems, including nonlinear behaviour, error, incompleteness, random serendipitous events and partial predictability237. Not surprisingly, good compounds may be overlooked for various reasons. Clearly, drug discovery is a challenging endeavour that requires skilful navigation in a multidimensional, multimodal search space. For example, 'activity cliffs' may affect lead optimization238, and unexpected biochemical and pharmacological effects can derail lead compound expansion and development.
The three challenges for automated drug design are the assembly of synthetically accessible structures, scoring and property prediction, and the systematic optimization of promising molecules in adaptive learning cycles. Over the past three decades, numerous guidelines, methods, algorithms and heuristics have been proposed to address each of these problems. Although the generation of new chemical entities with attractive chemical scaffolds has become feasible and although the algorithmic optimization problem can also be considered largely solved, the persisting issue of compound scoring — that is, picking the best compounds from a large pool of accessible possibilities — remains difficult. While compound elimination by appropriate scoring models discards the bulk of the designs ('negative design') with acceptable accuracy, the selection of the best or most promising ('positive design') remains prone to error. More accurate activity prediction models that extend the capabilities of existing approaches could originate from advanced machine learning methods.
Prognoses of the sustainability of customary pharmaceutical discovery and development practices imply the need for adjusted strategies for the future239,240,241,242. In such a situation, one can and must be creative. Given the prospects of labs-on-a-chip, human organoid assay systems, automated synthesis and intelligent learning software, we are currently witnessing a new wave of excitement about the changes in pharmaceutical research and development243,244. The concept of automated drug discovery could help to considerably reduce the number of compounds to be tested in a medicinal chemistry project and, at the same time, establish a rational unbiased foundation of adaptive molecular design. Recent advances in both lab-on-a-chip and computer technology, as well as the development of self-teaching artificial intelligence systems, could allow bottlenecks in the molecular design cycle to be addressed, thereby enabling better decision making in the future. Automation will play a central role in this process.
The envisaged drug discovery engine imitates human decision making by transferring responsibility to an objective machine learning system as a core aspect of the discovery process. If successful in the long run, the approach will amalgamate a continuously learning machine intelligence with the synthesis of pharmacologically relevant chemical matter. Thus, the medicinal chemist will gain the freedom to draw inspiration from potentially surprising solutions delivered by computational models, have fast access to initial tool compounds for a given discovery project and save precious material.
Rapid feedback cycles require the customization of instrumentation and the adjustment of work processes. Establishing this concept in pharmaceutical discovery may require considerable investment in terms of money and the reorganization of laboratory structures and processes. It will be necessary to evaluate the feasibility of fully autonomous molecular design with the aid of computers and robotic devices and, at the same time, to analyse which aspects of compound generation are best left to a chemically savvy artificial intelligence or a skilled human mind. The answers to these questions may vary depending on the particular discovery context, and keeping an open mind to many different viewpoints is advisable. Medicinal chemistry has always borrowed methodological thinking from engineering and experimental design so that tailored solutions could be implemented to meet challenges in chemistry, and continuing to do so would be wise. While keeping a healthy scepticism of automation for its own sake, embracing new technologies for planning and performing compound design, synthesis and testing, without fearing a loss of control, could enable substantial improvements in the effectiveness of drug discovery.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
King, R. D. et al. The automation of science. Science 324, 85–89 (2009).
Chapman, T. Lab automation and robotics: automation on the move. Nature 421, 661–666 (2003).
Sanderson, K. March of the synthesis machines. Nat. Rev. Drug Discov. 14, 299–300 (2015).
Lindsay, R. K., Buchanan, B. G., Feigenbaum, E. A. & Lederberg, J. DENDRAL: a case study of the first expert system for scientific hypothesis formation. Artif. Intell. 61, 209–261 (1993).
Johnson, A. P. & Marshall, C. Starting material oriented retrosynthetic analysis in the LHASA program. 3. Heuristic estimation of synthetic proximity. J. Chem. Inf. Comput. Sci. 32, 426–429 (1992).
Cho, S. J., Sun, Y. & Harte, W. ADAAPT: Amgen's data access, analysis, and prediction tools. J. Comput. Aided Mol. Des. 20, 249–261 (2006).
Schneider, G. De novo Molecular Design (Wiley–VCH, 2013).
Sparkes, A. et al. Towards robot scientists for autonomous scientific discovery. Autom. Exp. 4, 1 (2010).
MacConnell, A. B., Price, A. K. & Paegel, B. M. An integrated microfluidic processor for DNA-encoded combinatorial library functional screening. ACS Comb. Sci. 19, 181–192 (2017).
Baranczak, A. et al. Integrated platform for expedited synthesis-purification-testing of small molecule libraries. ACS Med. Chem. Lett. 8, 461–465 (2017).
Vasudevan, A., Bogdan, A. R., Koolman, H. F., Wang, Y. & Djuric, S. W. Enabling chemistry technologies and parallel synthesis-accelerators of drug discovery programmes. Prog. Med. Chem. 56, 1–35 (2017).
Esch, E. W., Bahinski, A. & Huh, D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Discov. 14, 248–260 (2015).
Eglen, R. M. & Randle, D. H. Drug discovery goes three-dimensional: goodbye to flat high-throughput screening? Assay Drug Dev. Technol. 13, 262–265 (2015).
Jones, L. H. & Bunnage, M. E. Applications of chemogenomic library screening in drug discovery. Nat. Rev. Drug Discov. 16, 285–296 (2017).
Schreiber, S. L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 287, 1964–1969 (2000).
O' Connor, C. J., Beckmann, H. S. & Spring, D. R. Diversity-oriented synthesis: producing chemical tools for dissecting biology. Chem. Soc. Rev. 41, 4444–4456 (2012).
Maurya, S. K. & Rana, R. An eco-compatible strategy for the diversity-oriented synthesis of macrocycles exploiting carbohydrate-derived building blocks. Beilstein J. Org. Chem. 13, 1106–1118 (2017).
Maier, M. E. Design and synthesis of analogues of natural products. Org. Biomol. Chem. 13, 5302–5343 (2015).
Wetzel, S., Bon, R. S., Kumar, K. & Waldmann, H. Biology-oriented synthesis. Angew. Chem. Int. Ed. 50, 10800–10826 (2011).
Wilk, W., Zimmermann, T. J., Kaiser, M. & Waldmann, H. Principles, implementation, and application of biology-oriented synthesis (BIOS). Biol. Chem. 391, 491–497 (2010).
Wender, P. A., Verma, V. A., Paxton, T. J. & Pillow, T. H. Function-oriented synthesis, step economy, and drug design. Acc. Chem. Res. 41, 40–49 (2008).
Wender, P. A., Quiroz, R. V. & Stevens, M. C. Function through synthesis-informed design. Acc. Chem. Res. 48, 752–760 (2015).
Ichikawa, S. Function-oriented synthesis: how to design simplified analogues of antibacterial nucleoside natural products? Chem. Rec. 16, 1106–1115 (2016).
Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Del. Rev. 46, 3–26 (2001).
Walters, W. P., Ajay & Murcko, M. A. Recognizing molecules with drug-like properties. Curr. Opin. Chem. Biol. 3, 384–387 (1999).
Leeson, P. D. & Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 6, 881–890 (2007).
Bickerton, G. R., Paolini, G. V., Besnard, J., Muresan, S. & Hopkins, A. L. Quantifying the chemical beauty of drugs. Nat. Chem. 4, 90–98 (2012).
Yusof, I. & Segall, M. D. Considering the impact drug-like properties have on the chance of success. Drug Discov. Today 18, 659–666 (2013).
Ajay, A., Walters, W. P. & Murcko, M. A. Can we learn to distinguish between “drug-like” and “nondrug-like” molecules? J. Med. Chem. 41, 3314–3324 (1998).
Sadowski, J. & Kubinyi, H. A scoring scheme for discriminating between drugs and nondrugs. J. Med. Chem. 41, 3325–3329 (1998).
Leeson, P. D. Molecular inflation, attrition and the rule of five. Adv. Drug Deliv. Rev. 101, 22–33 (2016).
Leahy, D. E. & Sykora, V. Automation of decision making in drug design. Drug Discov. Today Technol. 10, e437–e441 (2013).
Nicolaou, C. A. & Brown, N. Multi-objective optimization methods in drug design. Drug Discov. Today Technol. 10, e427–e435 (2013).
Harrison, S. et al. Extending 'predict first' to the design-make-test cycle in small-molecule drug discovery. Future Med. Chem. 9, 533–536 (2017).
Soldatova, L. N., Rzhetsky, A., De Grave, K. & King, R. D. Representation of probabilistic scientific knowledge. J. Biomed. Semantics 4 (Suppl. 1), S7 (2013).
Zhu, Q. et al. Semantic inference using chemogenomics data for drug discovery. BMC Bioinformatics 12, 256 (2011).
White, D. & Wilson, R. C. Generative models for chemical structures. J. Chem. Inf. Model. 50, 1257–1274 (2010).
Gupta, A. et al. Generative recurrent networks for de novo design. Mol. Inf. 36, 1700111 (2017).
Miyao, T., Arakawa, M. & Funatsu, K. Exhaustive structure generation for inverse-QSPR/QSAR. Mol. Inf. 29, 111–125 (2010).
Miyao, T., Kaneko, H. & Funatsu, K. Inverse QSPR/QSAR analysis for chemical structure generation (from y to x). J. Chem. Inf. Model. 56, 286–299 (2016).
Gaspar, H. A., Baskin, I. I., Marcou, G., Horvath, D. & Varnek, A. Stargate GTM: bridging descriptor and activity spaces. J. Chem. Inf. Model. 55, 2403–2410 (2015).
Schneider, G., Funatsu, K., Okuno, J. & Winkler, D. De novo drug design — ye olde scoring problem revisited. Mol. Inf. 36, 1681031 (2017).
Reutlinger, M., Rodrigues, T., Schneider, P. & Schneider, G. Combining on-chip synthesis of a focused combinatorial library with computational target prediction reveals imidazopyridine GPCR ligands. Angew. Chem. Int. Ed. 53, 582–585 (2014).
Reutlinger, M., Rodrigues, T., Schneider, P. & Schneider, G. Multi-objective molecular de novo design by adaptive fragment prioritization. Angew. Chem. Int. Ed. 53, 4244–4248 (2014).
Rodrigues, T. et al. Multidimensional de novo design reveals 5-HT2B receptor-selective ligands. Angew. Chem. Int. Ed. 54, 1551–1555 (2015).
Schneider, P., Röthlisberger, M., Reker, D. & Schneider, G. Spotting and designing promiscuous ligands for drug discovery. Chem. Commun. 52, 1135–1138 (2016).
Rodrigues, T. et al. De novo fragment design for drug discovery and chemical biology. Angew. Chem. Int. Ed. 54, 15079–15083 (2015).
Rodrigues, T. et al. Steering target selectivity and potency by fragment-based de novo drug design. Angew. Chem. Int. Ed. 52, 10006–10009 (2013).
Besnard, J. et al. Automated design of ligands to polypharmacological profiles. Nature 492, 215–220 (2012).
Willot, M. et al. Total synthesis and absolute configuration of the guaiane sesquiterpene Englerin A. Angew. Chem. Int. Ed. 48, 9105–9108 (2009).
Kusama, H., Tazawa, A., Ishida, K. & Iwasawa, N. Total synthesis of (±)-Englerin A using an intermolecular [3 + 2] cycloaddition reaction of platinum-containing carbonyl ylide. Chem. Asian J. 11, 64–67 (2016).
Friedrich, L., Rodrigues, T., Neuhaus, C. S., Schneider, P. & Schneider, G. From complex natural products to simple synthetic mimetics by computational de novo design. Angew. Chem. Int. Ed. 55, 6789–6792 (2016).
Antolín, A. A. & Mestres, J. Distant polypharmacology among MLP chemical probes. ACS Chem. Biol. 10, 395–400 (2015).
Reker, D., Rodrigues, T., Schneider, P. & Schneider, G. Identifying the macromolecular targets of de novo-designed chemical entities through self-organizing map consensus. Proc. Natl Acad. Sci. USA 111, 4067–4072 (2014).
Schneider, P. & Schneider, G. Privileged structures revisited. Angew. Chem. Int. Ed. 56, 7971–7974 (2017).
Schneider, P. & Schneider, G. A computational method for unveiling the target promiscuity of pharmacologically active compounds. Angew. Chem. Int. Ed. 56, 11520–11524 (2017).
Ley, S. V., Fitzpatrick, D. E., Ingham, R. J. & Myers, R. M. Organic synthesis: march of the machines. Angew. Chem. Int. Ed. 54, 3449–3464 (2015).
Merrifield, R. B. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 85, 2149–2154 (1963).
Palomo, J. M. Solid–phase peptide synthesis: an overview focused on the preparation of biologically relevant peptides. RSC Adv. 4, 32658–32672 (2014).
Kosuri, S. & Church, G. M. Large-scale de novo DNA synthesis: technologies and applications. Nat. Methods 11, 499–507 (2014).
Wan, W. B. & Seth, P. P. The medicinal chemistry of therapeutic oligonucleotides. J. Med. Chem. 59, 9645–9667 (2016).
Seeberger, P. H. & Werz, D. B. Synthesis and medical applications of oligosaccharides. Nature 446, 1046–1051 (2007).
Koppitz, M. & Eis, K. Automated medicinal chemistry. Drug Discov. Today 11, 561–568 (2006).
Liu, R., Li, X. & Lam, K. S. Combinatorial chemistry in drug discovery. Curr. Opin. Chem. Biol. 38, 117–126 (2017).
Godfrey, A. G., Masquelin, T. & Hemmerle, H. A remote-controlled adaptive medchem lab: an innovative approach to enable drug discovery in the 21st Century. Drug Discov. Today 18, 795–802 (2013).
Nicolaou, C. A., Watson, I. A., Hu, H. & Wang, J. The Proximal Lilly Collection: mapping, exploring and exploiting feasible chemical space. J. Chem. Inf. Model. 56, 1253–1266 (2016).
Crooks, S. L. & Charles, L. J. Overview of combinatorial chemistry. Curr. Protoc. Pharmacol. 9, Unit 9.3 (2001).
Long, A. Parallel chemistry in the 21st century. Curr. Protoc. Pharmacol. 9, Unit9.16 (2012).
Ingallina, C. et al. The Pictet-Spengler reaction still on stage. Curr. Pharm. Des. 22, 1808–1850 (2016).
Pirrung, M. C. Molecular Diversity and Combinatorial Chemistry (Elsevier, 2004).
Roughley, S. D. & Jordan, A. M. The medicinal chemist's toolbox: an analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 54, 3451–3479 (2011).
Brown, D. G. & Boström, J. Analysis of past and present synthetic methodologies on medicinal chemistry: where have all the new reactions gone? J. Med. Chem. 59, 4443–4458 (2016).
Collins, K. D., Gensch, T. & Glorius, F. Contemporary screening approaches to reaction discovery and development. Nat. Chem. 6, 859–871 (2014).
Li, J. et al. Synthesis of many different types of organic small molecules using one automated process. Science 347, 1221–1226 (2015).
Li, J., Grillo, A. S. & Burke, M. D. From synthesis to function via iterative assembly of N-methyliminodiacetic acid boronate building blocks. Acc. Chem. Res. 48, 2297–2307 (2015).
LaPorte, T. L. & Wang, C. Continuous processes for the production of pharmaceutical intermediates and active pharmaceutical ingredients. Curr. Opin. Drug Discov. Devel. 10, 738–745 (2007).
Chin, P., Barney, W. S. & Pindzola, B. A. Microstructured reactors as tools for the intensification of pharmaceutical reactions and processes. Curr. Opin. Drug Discov. Devel. 12, 848–861 (2009).
Dressler, O. J., Maceiczyk, R. M., Chang, S. I. & deMello, A. J. Droplet-based microfluidics: enabling impact on drug discovery. J. Biomol. Screen. 19, 483–496 (2014).
Shultz, S. et al. Miniaturized GPCR signaling studies in 1536-well format. J. Biomol. Tech. 19, 267–274 (2008).
Kanigowska, P., Shen, Y., Zheng, Y., Rosser, S. & Cai, Y. Smart DNA fabrication using sound waves: applying acoustic dispensing technologies to synthetic biology. J. Lab. Autom. 21, 49–56 (2016).
Sackmann, E. K. et al. Technologies that enable accurate and precise nano- to milliliter-scale liquid dispensing of aqueous reagents using acoustic droplet ejection. J. Lab. Autom. 21, 166–177 (2016).
Hadimioglu, B., Stearns, R. & Ellson, R. Moving liquids with sound: the physics of acoustic droplet ejection for robust laboratory automation in life sciences. J. Lab. Autom. 21, 4–18 (2016).
Squires, T. M. & Quake, S. R. Microfluidics: fluid physics at the nanoliter scale. Rev. Mod. Phys. 77, 977–1026 (2005).
Yoshida, J., Nagaki, A. & Yamada, D. Continuous flow synthesis. Drug Discov. Today Technol. 10, e53–e59 (2013).
Rodrigues, T., Schneider, P. & Schneider, G. Accessing new chemical entities through microfluidic systems. Angew. Chem. Ind. Ed. 53, 5750–5758 (2014).
Hopkin, M. D., Baxendale, I. R. & Ley, S. V. A flow-based synthesis of imatinib: the API of Gleevec. Chem. Commun. 46, 2450–2452 (2010).
Murray, P. R. D. et al. Continuous flow-processing of organometallic reagents using an advanced peristaltic pumping system and the telescoped flow synthesis of (E/Z)-tamoxifen. Org. Process Res. Dev. 17, 1192–1208 (2013).
Pastre, J. C., Browne, D. L. & Ley, S. V. Flow chemistry syntheses of natural products. Chem. Soc. Rev. 42, 8849–8869 (2013).
Saaby, S., Knudsen, K. R., Ladlow, M. & Ley, S. V. The use of a continuous flow-reactor employing a mixed hydrogen-liquid flow stream for the efficient reduction of imines to amines. Chem. Commun. 23, 2909–2911 (2005).
Baxendale, I. R., Hayward, J. J. & Ley, S. V. Microwave reactions under continuous flow conditions. Comb. Chem. High Throughput Screen. 10, 802–836 (2007).
Brzozowski, M., O'Brien, M., Ley, S. V. & Polyzos, A. Flow chemistry: intelligent processing of gas-liquid transformations using a tube-in-tube reactor. Acc. Chem. Res. 48, 349–362 (2015).
Wong-Hawkes, S. Y., Matteo, J. C., Warrington, B. H. & White, J. D. in New Avenues to Efficient Chemical Synthesis Vol. 2006 (eds Seeberger, P. H. & Blume, T.) 39–55 (2007).
Fernandez-Suarez, M., Wong, S. Y. & Warrington, B. H. Synthesis of a three-member array of cycloadducts in a glass microchip under pressure driven flow. Lab Chip 2, 170–174 (2002).
Jönsson, D., Warrington, B. H. & Ladlow, M. Automated flow-through synthesis of heterocyclic thioethers. J. Comb. Chem. 6, 584–595 (2004).
Garcia-Egido, E., Spikmans, V., Wong, S. Y. & Warrington, B. H. Synthesis and analysis of combinatorial libraries performed in an automated micro reactor system. Lab Chip 3, 73–76 (2003).
Newton, S. et al. Accelerating spirocyclic polyketide synthesis using flow chemistry. Angew. Chem. Int. Ed. 53, 4915–4920 (2014).
Adamo, A. et al. On-demand continuous-flow production of pharmaceuticals in a compact, reconfigurable system. Science 352, 61–67 (2016).
Hochlowski, J. E. et al. An integrated synthesis-purification system to accelerate the generation of compounds in pharmaceutical discovery. J. Flow Chem. 2, 56–61 (2011).
Lange, P. P. & James, K. Rapid access to compound libraries through flow technology: fully automated synthesis of a 3-aminoindolizine library via orthogonal diversification. ACS Comb. Sci. 14, 570–578 (2012).
Yoshida, J., Nagaki, A. & Yamada, T. Flash chemistry: fast chemical synthesis by using microreactors. Chemistry 14, 7450–7459 (2008).
Yoshida, J., Takahashi, Y. & Nagaki, A. Flash chemistry: flow chemistry that cannot be done in batch. Chem. Commun. 49, 9896–9904 (2013).
Nagaki, A., Imai, K., Kim, H. & Yoshida, J. Flash synthesis of TAC-101 and its analogues from 1,3,5-tribromobenzene using integrated flow microreactor systems. RSC Adv. 1, 758–760 (2011).
Carneiro, P. F., Gutmann, B., de Souza, R. O. M. A. & Kappe, O. Process intensified flow synthesis of 1H-4-substituted imidazoles: toward the continuous production of Daclatasvir. ACS Sustain. Chem. Eng. 3, 3445–3453 (2015).
Stalder, R. & Roth, G. P. Preparative microfluidic electrosynthesis of drug metabolites. ACS Med. Chem. Lett. 4, 1119–1123 (2013).
Genovino, J., Sames, D., Hamann, L. G. & Touré, B. B. Accessing drug metabolites via transition-metal catalyzed C-H oxidation: the liver as synthetic inspiration. Angew. Chem. Int. Ed. 55, 14218–14238 (2016).
Britton, J. & Raston, C. L. Multi-step continuous-flow synthesis. Chem. Soc. Rev. 46, 1250–1271 (2017).
Reizman, B. J. & Jensen, K. F. Feedback in flow for accelerated reaction development. Acc. Chem. Res. 49, 1786–1796 (2016).
McMullen, J. P., Stone, M. T., Buchwald, S. L. & Jensen, K. F. An integrated microreactor system for self-optimization of a Heck reaction: from micro- to mesoscale flow systems. Angew. Chem. Int. Ed. 49, 7076–7080 (2010).
Cortés–Borda, D. et al. Optimizing the Heck-Matsuda reaction in flow with a constraint-adapted direct search algorithm. Org. Process Res. Dev. 20, 1979–1987 (2016).
Falcone, C. E. et al. Reaction screening and optimization of continuous-flow atropine synthesis by preparative electrospray mass spectrometry. Analyst 142, 2836–2845 (2017).
Huang, C. M., Zhu, Y., Jin, D. Q., Kelly, R. T. & Fang, Q. Direct surface and droplet microsampling for electrospray ionization mass spectrometry analysis with an integrated dual-probe microfluidic chip. Anal. Chem. 89, 9009–9016 (2017).
Hartman, R. L., McMullen, J. P. & Jensen, K. F. Deciding whether to go with the flow: evaluating the merits of flow reactors for synthesis. Angew. Chem. Int. Ed. 50, 7502–7519 (2011).
Shevlin, M. Practical high-throughput experimentation for chemists. ACS Med. Chem. Lett. 8, 601–607 (2017).
Chow, S. Y. & Nelson, A. Embarking on a chemical space odyssey. J. Med. Chem. 60, 3591–3593 (2017).
Moore, J. S. & Jensen, K. F. “Batch” kinetics in flow: online IR analysis and continuous control. Angew. Chem. Int. Ed. 53, 470–473 (2014).
Haeberle, S. & Zengerle, R. Microfluidic platforms for lab-on-a-chip applications. Lab Chip 7, 1094–10110 (2007).
Jeong, G. S., Chung, S., Kim, C. B. & Lee, S. H. Applications of micromixing technology. Analyst 135, 460–473 (2010).
Fratila, R. M. & Velders, A. H. Small-volume nuclear magnetic resonance spectroscopy. Annu. Rev. Anal. Chem. 4, 227–249 (2011).
Capel, A. J. et al. 3D printed fluidics with embedded analytic functionality for automated reaction optimisation. Beilstein J. Org. Chem. 13, 111–119 (2017).
Chiu, D. T. & Lorenz, R. M. Chemistry and biology in femtoliter and picoliter volume droplets. Acc. Chem. Res. 42, 649–658 (2009).
He, M. et al. Selective encapsulation of single cells and subcellular organelles into picoliter- and femtoliter-volume droplets. Anal. Chem. 77, 1539–1544 (2005).
Theberge, A. B. et al. Microdroplets in microfluidics: an evolving platform for discoveries in chemistry and biology. Angew. Chem. Int. Ed. 49, 5846–5868 (2010).
Lignos, I. et al. Synthesis of Cesium lead halide Perovskite nanocrystals in a droplet-based microfluidic platform: fast parametric space mapping. Nano Lett. 16, 1869–1877 (2016).
Krishnadasan, S., Brown, R. J., deMello, A. J. & deMello, J. C. Intelligent routes to the controlled synthesis of nanoparticles. Lab Chip 7, 1434–1441 (2007).
Beulig, R. J. et al. A droplet-chip/mass spectrometry approach to study organic synthesis at nanoliter scale. Lab Chip 17, 1996–2002 (2017).
Dittrich, P. S. & Manz, A. Lab-on-a-chip: microfluidics in drug discovery. Nat. Rev. Drug Discov. 5, 210–218 (2006).
Skardal, A., Shupe, T. & Atala, A. Organoid-on-a-chip and body-on-a-chip systems for drug screening and disease modeling. Drug Discov. Today 21, 1399–1411 (2016).
Zakhariants, A. A., Burmistrova, O. A., Shkurnikov, M. Y., Poloznikov, A. A. & Sakharov, D. A. Development of a specific substrate-inhibitor panel (liver-on-a-chip) for evaluation of cytochrome P450 activity. Bull. Exp. Biol. Med. 162, 170–174 (2016).
Kirchmair, J. et al. Predicting drug metabolism: experiment and/or computation? Nat. Rev. Drug Discov. 14, 387–404 (2015).
Zhang, Y. S., Zhang, Y. N. & Zhang, W. Cancer-on-a-chip systems at the frontier of nanomedicine. Drug Discov. Today 22, 1392–1399 (2017).
Galler, K., Bräutigam, K., Große, C., Popp, J. & Neugebauer, U. Making a big thing of a small cell — recent advances in single cell analysis. Analyst 139, 1237–1273 (2014).
Loskill, P. et al. WAT-on-a-chip: a physiologically relevant microfluidic system incorporating white adipose tissue. Lab Chip. 17, 1645–1654 (2017).
Cao, Z., Chen, C., He, B., Tan, K. & Lu, C. A microfluidic device for epigenomic profiling using 100 cells. Nat. Methods 12, 959–962 (2015).
Kurita, R. & Niwa, O. Microfluidic platforms for DNA methylation analysis. Lab Chip 16, 3631–3644 (2016).
Eyer, K., Stratz, S., Kuhn, P., Küster, S. K. & Dittrich, P. S. Implementing enzyme-linked imunosorbent assays on a microfluidic chip to quantify intracellular molecules in single cells. Anal. Chem. 85, 3280–3287 (2013).
Adriani, G., Ma, D., Pavesi, A., Gohm, E. L. & Kamm, R. D. Modeling the blood-brain barrier in a 3D triple co-culture microfluidic system. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2015, 338–341 (2015).
Huang, T. Y. et al. 3D printed microtransporters: compound micromachines for spatiotemporally controlled delivery of therapeutic agents. Adv. Mater. 42, 6644–6650 (2015).
Kara, A. et al. Electrochemical imaging for microfluidics: a full-system approach. Lab Chip 16, 1081–1087 (2016).
Kara, A. et al. Towards a multifunctional electrochemical sensing and niosome generation lab-on-chip platform based on a plug-and-play concept. Sensors 16, 778 (2016).
Hartmann, D. M. et al. Microfluidic chip apparatuses, systems and methods having fluidic and fiber optic interconnections. US Patent 20090147253 A1 (2007).
Desai, B. et al. Rapid discovery of a novel series of Abl kinase inhibitors by application of an integrated microfluidic synthesis and screening platform. J. Med. Chem. 56, 3033–3047 (2013).
Wang, Y. et al. An integrated microfluidic device for large-scale in situ click chemistry screening. Lab. Chip 9, 2281–2285 (2009).
Lombardi, D. & Dittrich, P. S. Advances in microfluidics for drug discovery. Expert Opin. Drug Discov. 5, 1081–1094 (2010).
Wen, N. et al. Development of droplet microfluidics enabling high-throughput single-cell analysis. Molecules 21, 881 (2016).
Kang, D. K. et al. 3D droplet microfluidic systems for high-throughput biological experimentation. Anal. Chem. 87, 10770–10778 (2015).
Agresti, J. J. et al. Ultrahigh-throughput screening in drop-based microfluidics for directed evolution. Proc. Natl Acad. Sci. USA 107, 4004–4009 (2010).
Obexer, R. et al. Emergence of a catalytic tetrad during evolution of a highly active artificial aldolase. Nat. Chem. 9, 50–56 (2017).
Du, G., Fang, Q. & den Toonder, J. M. Microfluidics for cell-based high throughput screening platforms — a review. Anal. Chim. Acta 903, 36–50 (2016).
Zhu, Z. & Yang, C. J. Hydrogel droplet microfluidics for high-throughput single molecule/cell analysis. Acc. Chem. Res. 50, 22–31 (2017).
Fenneteau, J., Chauvin, D., Griffiths, A. D., Nizak, C. & Cossy, J. Synthesis of new hydrophilic rhodamine based enzymatic substrates compatible with droplet-based microfluidic assays. Chem. Commun. 53, 5437–5440 (2017).
Khalid, N., Kobayashi, I. & Nakajima, M. Recent lab-on-chip developments for novel drug discovery. Wiley Interdiscip. Rev. Syst. Biol. Med. 6, e1381 (2017).
Corey, E. J. General methods for the construction of complex molecules. Pure Appl. Chem. 14, 19–38 (1967).
Ihlenfeldt, W. D. & Gasteiger, J. Computer-assisted planning of organic syntheses: the second generation of programs. Angew. Chem. Int. Ed. 34, 2613–2633 (1996).
Cook, A. et al. Computer-aided synthesis design: 40 years on. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2, 79–107 (2011).
Ravitz, O. Data-driven computer aided synthesis design. Drug Discov. Today Technol. 10, e443–e449 (2013).
Chen, J. H. & Baldi, P. No electron left behind: a rule-based expert system to predict chemical reactions and reaction mechanisms. J. Chem. Inf. Model. 49, 2034–2043 (2009).
Kayala, M. A. et al. Learning to predict chemical reactions. J. Chem. Inf. Model. 51, 2209–2222 (2011).
Kowalik, M. et al. Parallel optimization of synthetic pathways within the Network of Organic Chemistry. Angew. Chem. Int. Ed. 51, 7928–7932 (2012).
Szymkuc, S. et al. Computer-assisted synthetic planning: the end of the beginning. Angew. Chem. Int. Ed. 55, 5904–5937 (2016).
Coley, C. W., Barzilay, R., Jaakkola, T. S., Green, W. H. & Jensen, K. F. Prediction of organic reaction outcomes using machine learning. ACS Cent. Sci. 3, 434–443 (2017).
Whelan, K. E. & King, R. D. Intelligent software for laboratory automation. Trends Biotechnol. 22, 440–445 (2004).
Reker, D. & Schneider, G. Active learning strategies in computer-assisted drug discovery. Drug Discov. Today 20, 458–465 (2015).
Schneider, G. & Fechner, U. Computer-based de novo design of drug-like molecules. Nat. Rev. Drug Discov. 4, 649–663 (2005).
Hartenfeller, M. & Schneider, G. Enabling future drug discovery by de novo design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 1, 742–759 (2011).
Rodrigues, T. & Schneider, G. Flashback forward: reaction-driven de novo design of bioactive compounds. Synlett 25, 170–178 (2014).
Hunter, J. Adopting AI is essential for a sustainable pharma industry. Drug Discov. World Winter 2016/2017, 69–71 (2017).
Kramer, C., Fuchs, J. E. & Liedl, K. R. Strong nonadditivity as a key structure-activity relationship feature: distinguishing structural changes from assay artifacts. J. Chem. Inf. Model. 55, 483–494 (2015).
Scho¨nherr, H. & Cernak, T. Profound methyl effects in drug discovery and a call for new C-H methylation meactions. Angew. Chem. Int. Ed. 52, 12256–12267 (2013).
Kuhn, B., Fuchs, J. E., Reutlinger, M., Stahl, M. & Taylor, N. R. Rationalizing tight ligand binding through cooperative interaction networks. J. Chem. Inf. Model. 51, 3180–3198 (2011).
Reker, D., Schneider, P., Schneider, G. & Brown, J. B. Active learning for computational chemogenomics. Future Med. Chem. 9, 381–402 (2017).
Lang, T., Flachsenberg, F., von Luxburg, U. & Rarey, M. Feasibility of active machine learning for multiclass compound classification. J. Chem. Inf. Model. 56, 12–20 (2016).
Schüller, A. & Schneider, G. Identification of hits and lead structure candidates with limited resources by adaptive optimization. J. Chem. Inf. Model. 48, 1473–1491 (2008).
Reutlinger, M. et al. Neighborhood–preserving visualization of adaptive structure-activity landscapes: application to drug discovery. Angew. Chem. Int. Ed. 50, 11633–11636 (2011).
Hiss, J. A. et al. Combinatorial chemistry by ant colony optimization. Future Med. Chem. 6, 267–280 (2014).
Reker, D., Schneider, P. & Schneider, G. Multi-objective active machine learning rapidly improves structure-activity models and reveals new protein-protein interaction inhibitors. Chem. Sci. 7, 3919–3927 (2016).
Schneider, G., Schuchhardt, J. & Wrede, P. Artificial neural networks and simulated molecular evolution are potential tools for sequence-oriented protein design. Comput. Appl. Biosci. 10, 635–645 (1994).
Schneider, G. et al. Peptide design by artificial neural networks and computer-based evolutionary search. Proc. Natl Acad. Sci. USA 95, 12179–12184 (1998).
Schneider, G. & Wrede, P. Artificial neural networks for computer-based molecular design. Prog. Biophys. Mol. Biol. 70, 175–222 (1998).
Zupan, J. & Gasteiger, J. Neural networks: a new method for solving chemical problems or just a passing phase? Anal. Chim. Acta 248, 1–30 (1991).
LeCun, Y., Bengio, Y. & Hinton, G. Deep learning. Nature 521, 436–444 (2015).
Baskin, I. I., Winkler, D. & Tetko, I. V. A renaissance of neural networks in drug discovery. Expert Opin. Drug Discov. 11, 785–795 (2016).
Jouppi, N. P. et al. in Proceedings of the 44th International Symposium on Computer Architecture (ISCA) http://dx.doi.org/10.1145/3079856.3080246 (Toronto, 2017).
Sato, K., Young, C. & Patterson, D. An in-depth look at Google's first Tensor Processing Unit (TPU). Google Cloud Platform https://cloud.google.com/blog/bigdata/2017/05/an-in-depth-look-at-googles-first-tensorprocessing-unit-tpu (2017).
Google. TensorFlow™www.tensorflow.org (2017)
Rampasek, L. & Goldenberg, A. TensorFlow: biology's gateway to deep learning? Cell Syst. 2, 12–14 (2016).
Litjens, G. et al. A survey on deep learning in medical image analysis. Med. Image Anal. 42, 60–88 (2017).
Holder, L. B., Haque, M. M. & Skinner, M. K. Machine learning for epigenetics and future medical applications. Epigenetics 19, 1–10 (2017).
Li, Y., Chen, C. Y. & Wasserman, W. W. Deep feature selection: theory and application to identify enhancers and promoters. J. Comput. Biol. 23, 322–336 (2016).
Erickson, B. J., Korfiatis, P., Akkus, Z., Kline, T. & Philbrick, K. Toolkits and libraries for deep learning. J. Digit. Imag. 30, 400–405 (2017).
Gasteiger, J. Physicochemical effects in the representation of molecular structures for drug designing. Mini Rev. Med. Chem. 3, 789–796 (2003).
Sawada, R., Kotera, M. & Yamanishi, Y. Benchmarking a wide range of chemical descriptors for drug–target interaction prediction using a chemogenomic approach. Mol. Inf. 33, 719–731 (2014).
Goh, G. B., Siegel, C., Vishnu, A., Hodas, N. O. & Baker, N. Chemception: a deep neural network with minimal chemistry knowledge matches the performance of expert-developed QSAR/QSPR models. arXiv, 1706.06689 (2017).
Castelvecchi, D. Can we open the black box of AI? Nature 538, 20–23 (2016).
Albrecht, T., Slabaugh, G., Alonso, E. & Al-Arif, M. R. Deep learning for single-molecule science. Nanotechnology 28, 423001 (2017).
Schneider, G. Neural networks are useful tools for drug design. Neural Netw. 13, 15–16 (2000).
Winkler, D. A. & Le, T. C. Performance of deep and shallow neural networks, the universal approximation theorem, activity cliffs, and QSAR. Mol. Inf. 36, 1600118 (2017).
Xie, L., Draizen, E. J. & Bourne, P. E. Harnessing big data for systems pharmacology. Annu. Rev. Pharmacol. Toxicol. 57, 157–160 (2017).
Del Sol, A., Thiesen, H. J., Imitola, J. & Carazo Salas, R. E. Big-data-driven stem cell science and tissue engineering: vision and unique opportunities. Cell Stem Cell 20, 157–160 (2017).
Schmid, M. & Lipson, H. Distilling free-form natural laws from experimental data. Science 324, 81–85 (2009).
Ekins, S. The next era: deep learning in pharmaceutical research. Pharm. Res. 33, 2594–2603 (2016).
Gawehn, E., Hiss, J. A. & Schneider, G. Deep learning in drug discovery. Mol. Inf. 35, 3–14 (2016).
Aliper, A. et al. Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data. Mol. Pharm. 13, 2524–2530 (2016).
Tian, K., Shao, M., Wang, Y., Guan, J. & Zhou, S. Boosting compound-protein interaction prediction by deep learning. Methods 110, 64–72 (2016).
Schneider, G. & Schneider, P. Macromolecular target prediction by self-organizing feature maps. Expert Opin. Drug Discov. 12, 271–277 (2017).
Filzen, T. M., Kutchukian, P. S., Hermes, J. D., Li, J. & Tudor, M. Representing high throughput expression profiles via perturbation barcodes reveals compound targets. PLoS Comput. Biol. 13, e1005335 (2017).
Zhang, L., Tan, J., Han, D. & Zhu, H. From machine learning to deep learning: progress in machine intelligence for rational drug discovery. Drug Discov. Today. http://dx.doi.org/10.1016/j.drudis.2017.08.010 (2017).
Zong, N., Kim, H., Ngo, V. & Harismendy, O. Deep mining heterogeneous networks of biomedical linked data to predict novel drug-target associations. Bioinformatics 33, 2337–2344 (2017).
Ragoza, M., Hochuli, J., Idrobo, E., Sunseri, J. & Koes, D. R. Protein-ligand scoring with convolutional neural networks. J. Chem. Inf. Model. 57, 942–957 (2017).
Pereira, J. C., Caffarena, E. R. & Dos Santos, C. N. Boosting docking-based virtual screening with deep learning. J. Chem. Inf. Model. 56, 2495–2506 (2016).
Goh, G. B., Hodas, N. O. & Vishnu, A. Deep learning for computational chemistry. J. Comput. Chem. 38, 1291–1307 (2017).
Mamoshina, P., Vieira, A., Putin, E. & Zhavoronkov, A. Applications of deep learning in biomedicine. Mol. Pharm. 13, 1445–1454 (2016).
ExCAPE-DB: ExCAPE chemogenomics database. https://solr.ideaconsult.net/search/excape/ (2017).
Sun, J. et al. ExCAPE–DB: an integrated large scale dataset facilitating Big Data analysis in chemogenomics. J. Cheminform. 9, 17 (2017).
Mondal, K. Design issues of Big Data parallelisms. Adv. Intell. Syst. Comput. 434, 209–217 (2016).
Tetko, I. V., Engkvist, O. & Chen, H. Does 'Big Data' exist in medicinal chemistry, and if so, how can it be harnessed? Future Med. Chem. 8, 1801–1806 (2016).
Tetko, I. V., Engkvist, O., Koch, U., Reymond, J. L. & Chen, H. BIGCHEM: challenges and opportunities for big data analysis in chemistry. Mol. Inf. 35, 615–621 (2016).
Ramsundar, B. et al. Is multitask deep learning practical for pharma? J. Chem. Inf. Model. 57, 2068–2076 (2017).
Mathea, M., Klingspohn, W. & Baumann, K. Chemoinformatic classification methods and their applicability domain. Mol. Inf. 35, 160–180 (2016).
Ochi, S., Miyao, T. & Funatsu, K. Structure modification toward applicability domain of a QSAR/QSPR model considering activity/property. Mol. Inf. http://dx.doi.org/10.1002/minf.201700076 (2017).
Posner, B. A., Xi, H. & Mills, J. E. Enhanced HTS hit selection via a local hit rate analysis. J. Chem. Inf. Model. 49, 2202–2210 (2009).
Zhang, L., Boehm, M. & Lovering, F. in ACS National Meeting & Exposition CINF82 (San Francisco, 2017).
Sparkes, A. et al. Towards robot scientists for autonomous scientific discovery. Autom. Exp. 2, 1 (2010).
Skoraczynski, G. et al. Predicting the outcomes of organic reactions via machine learning: are current descriptors sufficient? Sci. Rep. 7, 3582 (2017).
Altae-Tran, H., Ramsundar, B., Pappu, A. S. & Pande, V. Low data drug discovery with one-shot learning. ACS Cent. Sci. 3, 283–293 (2017).
Cully, A., Clune, J., Tarapore, D. & Mouret, J. B. Robots that can adapt like animals. Nature 521, 503–507 (2015).
Adami, C. Artificial intelligence: robots with instincts. Nature 521, 426–427 (2015).
[No authors listed.] Blogroll: Robot wars. Nat. Chem. 1, 173 (2009).
Peplow, M. Organic synthesis: the robo-chemist. Nature 512, 20–22 (2014).
Satyanarayanajois, S. D. & Hill, R. A. Medicinal chemistry for 2020. Future Med. Chem. 3, 1765–1786 (2011).
Rafferty, M. F. No denying it: medicinal chemistry training is in big trouble. J. Med. Chem. 59, 10859–10864 (2016).
Allen, D. Where will we get the next generation of medicinal chemists? Drug Discov. Today 21, 704–706 (2016).
Tomov, T. E. et al. DNA bipedal motor achieves a large number of steps due to operation using microfluidics–based interface. ACS Nano 11, 4002–4008 (2017).
Lehn, J. M. & Eliseev, A. V. Dynamic combinatorial chemistry: evolutionary formation and screening of molecular libraries. Science 291, 2331–2332 (2001).
Mondal, M. & Hirsch, A. K. Dynamic combinatorial chemistry. Chem. Soc. Rev. 44, 2455–2488 (2015).
Vermesan, O. & Friess, P. Internet of Things — Converging Technologies for Smart Environments and Integrated Ecosystems (River Publishers, 2013).
Carroll, G. P., Srivastava, S., Volini, A. S., Piñeiro-Núñez, M. M. & Vetman, T. Measuring the effectiveness and impact of an open innovation platform. Drug Discov. Today 22, 776–785 (2017).
Schneider, P. & Schneider, G. De novo design at the edge of chaos. J. Med. Chem. 59, 4077–4086 (2016).
Dimova, D., Heikamp, K., Stumpfe, D. & Bajorath, J. Do medicinal chemists learn from activity cliffs? A systematic evaluation of cliff progression in evolving compound data sets. J. Med. Chem. 56, 3339–3345 (2013).
Munos, B. Lessons from 60 years of pharmaceutical innovation. Nat. Rev. Drug Discov. 8, 959–968 (2009).
Sneddon, H. Embedding sustainable practices into pharmaceutical R&D: what are the challenges? Future Med. Chem. 6, 1373–1376 (2014).
Djuric, S. W., Hutchins, C. W. & Talaty, N. N. Current status and future prospects for enabling chemistry technology in the drug discovery process. F1000Res 5, 2426 (2016).
Scannell, J. W., Blanckley, A., Boldon, H. & Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 11, 191–200 (2012).
Mignani, S., Huber, S., Tomás, H., Rodrigues, J. & Majoral, J. P. Why and how have drug discovery strategies in pharma changed? What are the new mindsets? Drug Discov. Today 21, 239–249 (2016).
Gautam, A. & Pan, X. The changing model of big pharma: impact of key trends. Drug Discov. Today 21, 379–384 (2016).
Reutlinger, M. & Schneider, G. Nonlinear dimensionality reduction and mapping of compound libraries for drug discovery. J. Mol. Graph. Model. 34, 108–117 (2012).
Hawkes, S. Y. F. W., Chapela, M. J. V. & Montembault, M. Leveraging the advantages offered by microfluidics to enhance the drug discovery process. QSAR Comb. Sci. 24, 712–721 (2005).
Werner, M. et al. Seamless integration of dose–response screening and flow chemistry: efficient generation of structure–activity relationship data of β-secretase (BACE1) inhibitors. Angew. Chem. Int. Ed. 53, 1704–1708 (2014).
Czechtizky, W. et al. Integrated synthesis and testing of substituted xanthine based DPP4 inhibitors: application to drug discovery. ACS Med. Chem. Lett. 4, 768–772 (2013).
Pagano, N. et al. An integrated chemical biology approach reveals the mechanism of action of HIV replication inhibitors. Bioorg. Med. Chem. http://dx.doi.org/10.1016/j.bmc.2017.03.061 (2017).
Acknowledgements
P. Dittrich, A. deMello, Boehringer-Ingelheim Pharma and AstraZeneca contributed photographs of automated discovery devices. The author thanks M. Kossenjans, J. Hiss, P. Schneider, J. B. Brown, J. Kriegl and R. King for stimulating discussions on the future of drug discovery and process automation. The author was financially supported by the Swiss Federal Institute of Technology (ETH) Zurich, the Swiss National Science Foundation (grant numbers: 200021_157190, CR32I2_159737), the European Union Framework Programme for Research and Innovation (Horizon 2020, Marie Skłodowska–Curie ITN grant numbers: 676434 'BIGCHEM', 675555 'AEGIS') and the OPO-Foundation Zurich.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
G.S. is a life science industry consultant and a co-founder of inSili.com LLC, Zurich.
Rights and permissions
About this article

Cite this article
Schneider, G. Automating drug discovery. Nat Rev Drug Discov 17, 97–113 (2018). https://doi.org/10.1038/nrd.2017.232
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrd.2017.232
