
Key Points
-
Hepatocellular carcinoma (HCC) is one of the most lethal cancers, and affects many of the world's populations.
-
Various aetiologies have been linked to HCC development, the most prominent of which include chronic hepatitis B (HBV) and C (HCV) viral infection, chronic alcohol consumption and aflatoxin-B1-contaminated food. Virtually all cirrhosis-inducing conditions can cause HCC, pointing to important interactions with the host microenvironment.
-
HBV-induced hepatocarcinogenesis can involve an array of processes, including host–viral interactions, sustained cycles of necrosis–inflammation–regeneration, viral–endoplasmic-reticulum interactions (induction of oxidative stress), viral integration into the host genome (and associated host DNA deletions) and the targeted activation of oncogenic pathways by various viral proteins.
-
HCV-induced hepatocarcinogenesis also provokes similar biological processes, but is associated with a propensity of HCV to evade the host's immune responses and to promote cirrhosis.
-
Alcohol-induced hepatocarcinogenesis is associated with the induction of inflammation and, consequently, cycles of hepatocyte necrosis and regeneration, oxidative stress and cirrhosis. Aflatoxin-B1-induced hepatocarcinogenesis is mostly associated with carcinogenic mutations.
-
Various genetic events have been associated with the development of HCC, such as the inactivation of the tumour suppressor p53, mutations in β-catenin, overexpression of various ErbB receptor family members and overexpression of the Met receptor. In addition, various cancer-relevant genes seem to be targeted on the epigenetic level (methylation) in human HCC.
-
Genomic instability is a common feature of human HCC. Various mechanisms are thought to contribute, including telomere erosion, chromosome segregation defects and alterations in the DNA-damage-response pathways.
-
There are many genomic alterations in HCC. Comparative genomic hybridization studies so far have pointed to frequent chromosomal gains in 1q, 6p, 8q, 11q and 17q, and losses in 1p, 4q, 8p, 13q and 17p. Attempts have also been made to relate particular genomic alterations to aetiology and tumour-stage, albeit to a limited extent.
-
Gene-expression analyses of human HCCs have led to the successful molecular classification of HCCs on the basis of prognosis, aetiology and intrahepatic recurrence.
-
Many challenges and opportunities exist in this field, including the need for a more detailed and clinically grounded genomic characterization of human HCCs, deeper understanding of the mechanisms of genomic instability, host–viral interactions, microenvironmental processes (inflammation and cirrhosis), cell of origin in hepatocarcinogenesis and the identification of biomarkers to identify early stage disease as well as those at greatest risk of developing HCC.
Abstract
Hepatocellular carcinoma is among the most lethal and prevalent cancers in the human population. Despite its significance, there is only an elemental understanding of the molecular, cellular and environmental mechanisms that drive disease pathogenesis, and there are only limited therapeutic options, many with negligible clinical benefit. This Review summarizes the current state of knowledge of this, the most common and dreaded liver neoplasm, and highlights the principal challenges and scientific opportunities that are relevant to controlling this accelerating global health crisis.
Similar content being viewed by others

Main
The American Cancer Society estimates that, in 2005, there were over 667,000 new cases of liver cancer worldwide. The 5-year survival rate of individuals with liver cancer in the United States is only 8.9% despite aggressive conventional therapy, marking this malignancy as the second most lethal cancer after pancreatic ductal adenocarcinoma (4.4% survival at 5 years)1,2. The lethality of liver cancer stems in part from its resistance to existing anticancer agents, a lack of biomarkers that can detect surgically resectable incipient disease, and underlying liver disease that limits the use of chemotherapeutic drugs. In cases of limited disease, surgical tumour resection represents an effective therapeutic alternative (particularly in cases without underlying liver cirrhosis). Several local ablation treatment methods, such as ethanol injection or radiofrequency, are also used in cases of limited disease. Similarly, liver transplantation epitomizes a radical treatment option, but only when patients meet stringent specific criteria1.
Liver cancer comprises diverse, histologically distinct primary hepatic neoplasms, which include hepatocellular carcinoma (HCC), intrahepatic bile duct carcinoma (cholangiocarcinoma), hepatoblastoma, bile duct cystadenocarcinoma, haemangiosarcoma and epitheliod haemangioendothelioma3. Among these, HCC is the most common type of liver cancer, representing 83% of all cases2. This Review summarizes the magnitude of the HCC problem, current scientific knowledge of the pathobiology and genetics of HCC, and outstanding challenges and potential opportunities for the study and treatment of this prominent human disease. For information on the important topic of epidemiology of HCC, the reader is referred to an excellent recent review4.
Aetiologies of hepatocellular carcinoma
HCC affects all segments of the world population, although significant differences in HCC incidence in various countries reflect the regional differences in the prevalence of specific aetiological factors as well as ethnicity2. The most prominent factors associated with HCC include chronic hepatitis B and C viral infection, chronic alcohol consumption, aflatoxin-B1-contaminated food and virtually all cirrhosis-inducing conditions5. Other aetiological factors have also been proposed to lead to HCC, albeit at a lower frequency (Box 1). In addition, gender can also influence the risk and behaviour of HCC, with males accounting for a larger fraction of cases4.
Viral-induced hepatocarcinogenesis. There are two main hepatitis viruses associated with the development of HCC. Hepatitis B virus (HBV) infects approximately 2 billion individuals worldwide and causes an estimated 320,000 deaths annually. Approximately 30–50% of HBV-related deaths are attributable to HCC6. The impact of HBV infection on HCC development is reflected by the correlation between increased incidence of HCC in patients with increasing levels of HBV DNA in serum7. Hepatitis C virus (HCV) infects approximately 170 million individuals worldwide8. Approximately 20% of chronic HCV cases develop liver cirrhosis, and 2.5% develop HCC9. The viral-associated mechanisms driving hepatocarcinogenesis are complex and involve both host and viral factors.
HBV is a non-cytopathic, partially double-stranded hepatotropic DNA virus classified as a member of the hepadnaviridae family. The HBV genome encodes several viral proteins essential to its life cycle, including a reverse transcriptase/DNA polymerase (pol), the capsid protein known as hepatitis B core antigen (HBcAg), and the L, M and S envelope proteins that associate with the endoplasmic reticulum (ER) membrane as part of their replication process. HBV also encodes a number of proteins whose functions are not fully understood, such as protein x (HBx)10.
Several lines of evidence support the direct involvement of HBV in the transformation process. First, HBV genome integration has been associated with host DNA microdeletions11 that can target cancer-relevant genes including telomerase reverse transcriptase (TERT), platelet-derived-growth-factor receptor-β (PDGFR β), PDGF β and mitogen activated protein kinase 1 (MAPK1), among others12. Second, HBx transcriptional activation activity can alter the expression of growth-control genes, such as SRC tyrosine kinases, Ras, Raf, MAPK, ERK, JNK and others13,14,15,16. Finally, HBx can bind and inactivate the tumour suppressor p53 in vitro, therefore increasing cellular proliferation and survival and compromising DNA-damage checkpoints16,17. The hepatocarcinogenic potential of HBx has been genetically validated in HBx transgenic mice, of which 90% develop HCC18,19.
Host–viral interactions seem to contribute to hepatocarcinogenesis in several ways. A robust T-cell immune response is presumably elicited to combat viral infection, however, this response contributes to hepatocyte necrosis, inflammation and consequently regeneration, leading to carcinogenesis10,20,21,22. Although infection is clearly acute in most cases, 10% of adults experience inefficient HBV clearance and develop chronic active infection with associated sustained cycles of necrosis–inflammation–regeneration10,23. Such continuous replication of hepatocytes might enable the propagation of oncogenic lesions and telomere erosion with consequent genomic instability (see below) (Fig. 1). Another proposed mechanism of HBV-induced hepatocarcinogenesis might stem from viral–ER physical interactions24 that provoke ER stress and ultimately the induction of oxidative stress25, which can stimulate growth- and survival-signalling pathways, cause mutations through the generation of free radicals and activate stellate cells10,26. Therefore, HBV can engender a pro-carcinogenic state in the liver through many mechanisms10. HBV has also been proposed to increase liver disease and consequently hepatocarcinogenesis through HBV mutations that might enable the virus to escape the host's immune response and/or result in the retention of the virus within the cell, therefore damaging hepatocytes and leading to liver disease27.

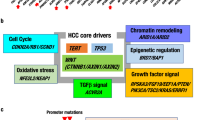
The suspected mechanisms of hepatocarcinogenesis for the various risk factors are shown. Commonalities are indicated using the same colour. In addition to these mechanisms, hepatitis B virus (HBV) and aflatoxin B1 share the characteristic of affecting the genome — HBV can integrate into the host genome and aflatoxin B1 is a mutagen. HCV, hepatitis C virus
HCV is a non-cytopathic virus of the flaviviridae family. The HCV positive-stranded RNA genome encodes non-structural proteins (NS2, NS3, NS4A, NS5A and NS5B), which associate with the ER membrane to form the viral replicase and viral envelope proteins (E1 and E2). An important recent advance has been the establishment of a cell-culture model supporting efficient HCV replication and infectious particle production22,28,29,30, enabling the molecular dissection of these processes for the first time.
HCV possesses three important clinico-biological distinctions from HBV that are relevant to hepatocarcinogenesis. First, HCV shows a higher propensity to yield chronic infection — 10% of HBV cases versus 60–80% of HCV22. This might relate to immune evasion by HCV quasi-species generated from high rates of replication errors22,31. The second key difference is the greater propensity of HCV to promote liver cirrhosis compared with HBV. 5–10% of HCV-infected patients develop liver cirrhosis after 10 years of infection, a frequency that is approximately 10–20-fold higher than HBV22, a highly relevant association as cirrhosis is a significant correlate of HCC development. Third, as HCV is an RNA virus without a DNA intermediate form, it cannot integrate into host genomes22.
Both viral and host factors are thought to contribute to HCC development in the setting of HCV infection10, analogous to HBV. One theory for HCV-induced hepatocarcinogenesis posits that the continuous cycles of hepatocyte death caused by the immune response to the virus and subsequent regeneration provide a context for the accumulation and propagation of mutations. In addition, it is possible that immune responses to viral infection promote hepatocarcinogenesis in a manner substantiated by a transgenic skin-tumour model in which deficiencies in pro-inflammatory T cells have been shown to correlate with decreased tumour incidence and impaired progression32. On the other hand, the relevance of this immune-mediated mechanism is less clear in light of the feeble immunological response in chronically HCV-infected livers10. In support of this notion, HCV RNA and/or core proteins have been suggested to impair dendritic cell functions that are important for T-cell activation33. Furthermore, the HCV core protein and the NS5A non-structural protein have been implicated in the evasion from immune-mediated cell killing by interacting with various factors involved in this process (such as tumour-necrosis factor-α (TNFα) receptor, interferon-α (IFNα) and others)34,35,36. In addition, the NS3 and NS4A HCV proteins use their protease function to cleave and activate components that are integral for signalling the immune response37,38. Various other immune-evasion mechanisms are used by HCV proteins, as detailed elsewhere39. Overall, the pathogenetic interactions between the immune system and HCV-induced HCC are extremely complex and not fully understood, and will therefore benefit from continued investigation.
HCV co-opts the ER as part of its replication process, and can cause ER stress with all the aforementioned pro-carcinogenic effects. In addition, HCV core proteins have been shown to interact with components of the MAPK signalling pathway (such as ERK, MEK and Raf) and therefore modulate cell proliferation40,41 (Fig. 1). NS5A has also been shown to interact with and inactivate p53 by sequestration to the perinuclear membrane, thereby affecting the p53-regulated pathways that control cell-cycle progression, cellular survival, response to hypoxic and genotypic stresses, and tumour angiogenesis42. The carcinogenic potential of HCV core proteins is also indicated by the development of hepatic steatosis, the induction of reactive oxygen species and the development of HCC in transgenic mice that harbour the entire HCV core gene under the control of HBV transcriptional regulatory elements, which raises the possibility that HCV-induced HCC involves an oxidative-stress-mediated mechanism43. Finally, core-E1-E2 transgenic mice develop HCCs whose robust tumour growth seems to be due to the inhibition of apoptosis by the E1/E2 HCV proteins44.
Alcohol-induced hepatocarcinogenesis. Alcohol is an important HCC risk factor. Chronic alcohol intake has been implicated in causing the production of pro-inflammatory cytokines through monocyte activation45 and provoking increased concentrations of circulating endotoxin, activating Küpffer cells which release many chemokines and cytokines (including TNFα, interleukin-1β (IL1β), IL6 and prostaglandin E2) with adverse effects on hepatocyte survival. In the setting of chronic ethanol exposure, hepatocytes show increased sensitivity to the cytotoxic effects of TNFα46, which sets the stage for chronic hepatocyte destruction–regeneration, stellate cell activation, cirrhosis and ultimately HCC.
Alcohol also damages the liver through oxidative-stress mechanisms. Alcoholic hepatitis shows increased isoprostane, a marker of lipid peroxidation45. Oxidative stress might contribute to hepatocarcinogenesis in several ways. First, oxidative stress promotes the development of fibrosis and cirrhosis, which are key features of a permissive HCC microenvironment. The pro-carcinogenic effect of the cirrhotic microenvironment has been shown in the mouse, where PDGF transgenic mice develop fibrosis that progresses to HCC47. As stellate cells are the main source of collagen deposition in the injured liver, it is notable that oxidative-stress induction of cultured stellate cells through treatment with isoprostanes can promote increased cell proliferation and collagen synthesis, which are characteristics of the fibrotic response48. Second, ethanol-induced oxidative stress might have an effect on HCC-relevant signalling pathways, such as the documented reduction in tyrosine phosphorylation of STAT1 (signal transducer and activator of transcription 1), decreased STAT1-directed activation of IFNγ signalling, and the loss of the protective effects of IFNγ with consequent hepatocyte damage49. Oxidative stress might also cause the accumulation of oncogenic mutations. For example, increased oxidative stress associated with iron overload (hereditary haemochromatosis) has been associated with p53 mutations in resultant HCCs50. Finally, although speculative, oxidative stress might accelerate telomere shortening, which would in turn fuel the development of liver cirrhosis, chromosomal instability and ultimately HCC (see below)51.
Aflatoxin-B1-induced hepatocarcinogenesis. Ingestion of the fungal toxin, aflatoxin B1, also poses an increased risk for the development of HCC. Aflatoxin B1 seems to function as a mutagen, and is associated with a specific p53 mutation16,52,53,54,55 and cooperating mutational activation of oncogenes such as HRAS56 (Fig. 1). Unlike HCV-induced and alcohol-induced hepatocarcinogenesis there is no clear connection between aflatoxin B1 exposure and the development of cirrhosis, indicating that the mutational actions of this toxin might be the primary driver of HCC development. It is worth noting that aflatoxin B1 exposure often coexists with HBV infection, and such individuals possess a 5–10-fold increased risk of developing HCC compared with exposure to only one of these factors57. The mechanistic basis for this synergy is not known, although it seems plausible that cooperation would derive from aflatoxin-B1-induced mutagenesis and continuous hepatocyte turnover and regeneration during chronic HBV infection.
Common molecular themes in hepatocarcinogenesis. A survey of the diverse hepatocarcinogenesis mechanisms points to common pathogenetic pathways and processes (Fig. 1). In particular, p53 inactivation or mutation seems to be a consistent event in HBV-, HCV- and aflatoxin-B1-induced HCC. Furthermore, inflammation, continuous rounds of necrosis and regeneration, and oxidative stress are characteristic of HBV-, HCV- and alcohol-induced hepatocarcinogenesis, suggesting that these processes contribute in fundamental ways to HCC development. Both HBV and HCV activate the MAPK pathway, which also indicates its pathogenetic relevance. The identification of additional common molecular changes among the different aetiological factors would be important in the drug-discovery industry, as it would hold the greatest return on investment and increase the economic probability of developing and deploying these agents, particularly in underserved populations.
Despite some common molecular changes, the cellular and molecular bases of hepatocarcinogenesis are likely to differ significantly across the various aetiological factors, and such differences might also hold mechanistic clues and preventive or therapeutic opportunities. One potential opportunity could relate to the propensity of HBV to integrate into host genomes (in contrast to HCV), causing alterations in cancer-relevant genes. Prevention of this integration process might provide new preventive approaches. The prominent changes in the liver microenvironment brought about by HCV and alcohol might also provide new therapeutic strategies for targeting host–tumour heterotypic interactions. Such wide-ranging differences across the various aetiologies also caution that limited pathogenetic and treatment insights will emerge from genomic profile data that have not been linked to a specific aetiological agent.
Genetic and epigenetic events in HCC
The next subsections describe the current knowledge of the genetic and genomic events associated with the development of HCC. The neoplastic evolution of HCC proceeds through a multi-step histological process that is less well defined than that of other cancer types (Fig. 2). As noted above, diverse HCC-inducing aetiologies provoke continuous rounds of hepatocyte damage and regeneration, culminating in chronic liver disease. Hyperplastic nodules of regenerating hepatocytes have normal cytological features, and represent a potential first step towards HCC. These lesions can progress to pre-malignant dysplastic nodules, which have abnormal cytological features including clear cell changes and nuclear crowding, and these lesions are associated with the increased thickening of the trabeculae, which indicates abnormal liver architecture. These dysplastic nodules can evolve to frank HCC which, in addition to all the aforementioned abnormal features, is endowed with the capacity to invade the surrounding fibrous stroma and vessels, and occasionally has metastatic potential58. The molecular analysis of human HCC has shown many genetic and epigenetic alterations that result in the deregulation of key oncogenes and tumour-suppressor genes including TP53, β-catenin, ErbB receptor family members, MET and its ligand hepatocyte growth factor (HGF), p16(INK4a), E-cadherin and cyclooxygenase 2 (COX2).

After hepatic injury incurred by any one of several factors (hepatitis B virus (HBV), hepatitis C virus (HCV), alcohol and aflatoxin B1), there is necrosis followed by hepatocyte proliferation. Continuous cycles of this destructive–regenerative process fosters a chronic liver disease condition that culminates in liver cirrhosis. Cirrhosis is characterized by abnormal liver nodule formation surrounded by collagen deposition and scarring of the liver. Subsequently, hyperplastic nodules are observed, followed by dysplastic nodules and ultimately hepatocellular carcinoma (HCC), which can be further classified into well differentiated, moderately differentiated and poorly differentiated tumours — the last of which represents the most malignant form of primary HCC. Telomere shortening is a feature of chronic liver disease and cirrhosis. Telomerase reactivation has been associated with hepatocarcinogenesis (its activation in the early versus late stages of disease is still a point of debate, and is discussed in the text). Loss and/or mutation of p53 and genomic instability also characterize hepatocarcinogenesis. p53 loss and/or mutation is shown to occur during progression to HCC, however, there is some evidence that loss and mutation of p53 might also occur in the initial stages of hepatocarcinogenesis (this is discussed in the text in more detail).
The p53 tumour suppressor. Although it is widely accepted that p53 deficiency participates in the development of HCC, whether p53 mutation contributes to cancer initiation, progression or both remains an area of active investigation. In an HBx transgenic mouse model16,18, the functional inactivation of p53 by HBx (through the sequestration of p53 to the cytoplasm) was documented in HCCs but not in altered foci (initiation foci), implicating p53 in constraining progression to HCC. Furthermore, mutations in p53 were only detected in larger HCCs, suggesting that full genetic inactivation of p53 is associated with progression to late-stage disease. In humans, analyses of HBV- and HCV-related HCCs have shown a greater frequency of p53 mutations in advanced malignancies (43%) than in regenerative nodules (∼7%)59. Although such patterns are indicative of a role in progression, they do not exclude the possibilities that p53 mutant regenerative nodules are those that have 'initiated' a productive carcinogenic process, or that rare p53 mutant cells are much more common in regenerative nodules but not detected by conventional sequencing approaches. In the context of aflatoxin B1, regions of high aflatoxin B1 exposure show frequent p53 mutations in early-stage HCC lesions52, whereas regions of low aflatoxin B1 exposure show p53 mutations in much later stages of HCC60,61,62. However, no correlation was found between p53 mutation and tumour stages in a study of HCCs from southern Africa, where aflatoxin B1 exposure is common16,53, although this could relate to the limited number of tumours that were examined in this study.
On the basis of the above data, it seems plausible that p53 mutation might operate in either HCC initiation or progression, depending on the context. In the setting of aflatoxin B1, this mutation might serve to drive initiation with other cooperating events (the need for cooperating events seems evident from the lack of an HCC-prone phenotype in p53 mutant mice). In the context of other aetiologies such as those that provoke regeneration, oxidative stress and telomere erosion, the loss of p53 might have a more prominent role in HCC progression by facilitating continued proliferative potential in the face of activated DNA-damage signalling, which could also contribute to genomic instability in HCC (see below). Along these lines, p53 heterozygosity through germline mutation enables HCC progression in mice with short telomeres in a chronic liver disease model of HCC, suggesting that p53 cooperates with telomere-induced chromosomal instability in liver tumour progression63.
The role of p53 in hepatocarcinogenesis has also been shown in other mouse models. HBV large envelop protein (HBsAg) transgenic mice have an HCC-prone condition64,65,66 that is accelerated and made fully penetrant when combined with aflatoxin B1 treatment and the p53ser246 allele, which corresponds to the p53 codon 249 mutation of HCC patients exposed to aflatoxin B1 (Refs 64, 66). Similarly, increased progression to high-grade HCC is observed in aflatoxin-B1-treated HBsAg mice heterozygous for a p53 null allele (Refs 65, 66).
β-catenin. β-catenin is a crucial downstream component of the Wnt signalling pathway. When Wnt signalling is engaged, the adenomatosis polyposis coli (APC) and Axin proteins no longer bind β-catenin, with consequent β-catenin stabilization and translocation to the nucleus where it associates with the Tcf family of transcription factors. This transcription factor complex transactivates a host of target genes governing cancer-relevant processes, including MYC, cyclin D1, COX2, and matrix metalloproteinase 7 (MMP7)67.
β-catenin mutations and increased nuclear expression have been detected in human HCC68,69. In some reports, β-catenin overexpression and mutations have been related to early-stage HCCs70,71, and in others to HCC progression72. Overexpression and mutations of β-catenin occur more frequently in HCV-related HCCs compared with HBV-related HCCs70,73,74. However, β-catenin is stabilized by the HBx protein in hepatoma cells, and therefore implicated in HBV-related HCC75.
Some studies suggest that β-catenin mutations function independently of other cancer-relevant genes, such as TP53. For example, Torbenson et al. recently suggested that the overexpression of p53 and β-catenin in HCCs were not correlated76. However, Prange et al. found that the accumulation of nuclear β-catenin was correlated with increased nuclear p53 expression and the loss of E-cadherin77. It should be noted that Torbenson and colleagues did not examine the mutational status of p53, and therefore the possibillity cannot be excluded that the mutational inactivation of p53 might be present in the same tumours with β-catenin overexpression.
It has also been suggested that β-catenin mutations are typical in HCCs that are not characterized by genomic instability. Human HCCs with high rates of genomic instability, as detected by their increased loss of heterozygosity (LOH), show very low levels of β-catenin mutations78. Studies in the mouse have shown that HCCs have either a high rate of β-catenin mutations or high genomic instability. These provocative findings indicate that abrogation of the Wnt signalling pathway could represent an alternative route to hepatocarcinogenesis79.
ErbB receptor family. The ErbB family of receptor tyrosine kinases consists of four members (ERBB1–ERBB4), which have been implicated in the development of various types of human cancers. The examination of these receptor tyrosine kinases has documented the overexpression of ERBB1 (also known as epidermal growth factor receptor (EGFR)) in 68% of HCC cases, ERBB3 in 84%, ERBB2 (also known as HER2) in 21% and ERBB4 in 61% (but at a lower level)80. ERBB1 and ERBB3 expression correlated with a more aggressive presentation, such as a high proliferation index, intrahepatic metastasis, de-differentiation and tumour size80. Further support that ERBB1 could be integral to hepatocarcinogenesis comes from the inhibition of ERBB1 by gefitinib, which results in growth inhibition, cell-cycle arrest and apoptosis in human HCC cell lines81, and also shows activity in a rat model of HCC82. In a limited phase II clinical study of 38 patients with HCC, another ERBB1 inhibitor, erlotinib, showed clinical efficacy as evidenced by disease control in 59% of participating patients and 32% of patients remaining progression-free at 6 months after treatment83. Further evidence for the involvement of the ErbB family of receptors in hepatocarcinogenesis comes from studies in mice. Mice transgenic for transforming growth factor-α (TGFα) (a member of the EGF family and a ligand for ErbB receptors) develop HCCs84,85. Hepatocarcinogen treatment accelerates HCC development in the TGFα transgenic strain, whereas it generates smaller hepatic neoplasms in TGFα-deficient mice86,87. Compound TGFα and MYC transgenic mice show increased hepatocarcinogenesis that is associated with the disruption of TGFβ1 signalling16,88,89,90,91 and chromosomal losses, some of which are syntenic to those in human HCCs that include the retinoblastoma (RB) tumour-suppressor locus89. TGFα can also cooperate with HBV transgenic strains to effectively increase HCC incidence. Together, these studies underscore the prime importance of TGFα signalling in HCC development92.
MET and HGF. Overexpression of the MET receptor has been reported in advanced human HCCs93. The role of MET signalling in HCC development has been confirmed in mouse models, whereby mice transgenic for the MET ligand HGF, one of the most potent hepatocyte mitogens, develop HCCs by 1.5 years of age94. Correspondingly, hepatic-directed expression of an inducible activated MET transgene resulted in the development of HCC, and the extinction of transgene expression resulted in tumour regression by apoptosis and impaired cell proliferation95.
Methylation of cancer-relevant genes. Aberrant DNA methylation patterns have been reported in human HCC70,96,97,98,99. Methylation has been detected in the earliest stages of hepatocarcinogenesis, and to a greater extent in tumour progression100. Specific hypermethylation events in HCC have targeted p16(INK4a), E-cadherin, COX2, apoptosis-associated speck-like protein (ASC) and deleted in liver cancer 1 (DLC1), among others16,101,102,103,104,105,106,107. The biological significance of the hypermethylation of some of these genes in hepatocarcinogenesis has been evaluated in HCC cell lines. Specifically, treatment of hepatoma cells with a demethylating agent increases p16(INK4a) (Ref. 107) and COX2 expression104, both of which are associated with the inhibition of cell proliferation. Therefore, the epigenetic silencing of key cancer genes seems relevant to hepatocarcinogenesis, and might provide another route to the development of a new class of anti-oncological agents.
Genomic instability in HCC
Telomeres and telomerase. Telomere shortening has been described as a key feature of chronic hyperproliferative liver disease108,109,110,111, specifically occurring in the hepatocyte compartment112 (Fig. 2). These observations have fueled speculation that telomere shortening associated with chronic liver disease and hepatocyte turnover contribute to the induction of genomic instability that drives human HCC. In this regard, the analysis of human hepatoma has established a correlation between telomere shortening and increased chromosomal instability113,114. Correspondingly, the modeling of HCC in a telomerase knockout mouse model has shown that telomere dysfunction has a role in increasing liver cancer initiation115.
Another aspect of telomere biology common to HCC is the robust activation of telomerase (correlating with increased TERT mRNA levels) in nearly 90% of human HCCs116,117,118. Intriguingly, HBV has been shown to integrate in the TERT locus in human HCCs, indicating that the proximal placement of viral enhancers might increase TERT gene expression119. Amplification of the gene that encodes the telomerase RNA component (TERC)120 and the allelic loss of chromosome 10p (in a region that encodes a putative telomerase repressor)121 has been reported in HCCs.
Telomerase re-activation has been suggested to promote HCC progression (increased microvessel density122 and HCC recurrence after resection123). Some studies have implicated telomerase in the early stages of hepatocarcinogenesis124,125,126,127; however, these inconsistent findings might stem from telomerase-positive infiltrating lymphocytes and resident sinusoidal cells128. Moreover, the shorter telomeres of HCCs compared with normal livers point to the later stage activation of telomerase in HCC development. Collectively, the current data suggest a model wherein telomere shortening drives chromosomal instability and cancer-promoting lesions during early stages of hepatocarcinogenesis, whereas telomerase re-activation is necessary for malignant progression as it restores chromosomal stability to a level compatible with cancer-cell viability. This is supported by studies in the TERC null mouse model, where telomere dysfunction increased the initiation of hepatic neoplasms whereas the absence of telomerase activity impaired HCC progression115. It should be noted that telomerase might also possess oncogenic properties aside from its telomere maintenance function129.
It is interesting to note that telomere shortening might also contribute to HCC development through the creation of a cirrhotic microenvironment. Telomerase knockout mice with short telomeres show increased liver fibrosis and cirrhosis following injury compared with mice with intact telomeres110, possibly because of increased hepatocyte death resulting from the telomere checkpoint response.
Chromosome segregation defects. Defects in chromosome segregation during mitosis result in aneuploidy, a common cytogenetic feature of cancer cells including HCC. Many genes have been linked to the complex series of orchestrated events that insure proper and accurate chromosome segregation and, as expected, the mutation of these genes results in aneuploidy and increased cancer incidence130.
Among the factors involved in chromosome segregation, Aurora kinase A, along with its kinase target hepatoma upregulated protein (HURP), are overexpressed in HCC131,132. Although the biological function of HURP remains to be defined, HURP expression enables cells to grow in low serum conditions, which points to an oncogenic potential132. Aurora-kinase inhibitors, which are currently under development133, could be particularly useful in HCC because of the increased expression of Aurora kinase in this cancer.
The spindle-assembly checkpoint has been shown to be defective in human HCC cell lines, as evidenced by the lack of accumulation of cells in metaphase following treatment with nocodazole or colcemid (which normally arrest the cells at metaphase)134. For some cancers, the deactivation of this checkpoint has been ascribed to mutations in spindle-checkpoint genes; however, such mutations have yet to be identified in HCC. It is possible that spindle-checkpoint defects in HCC might stem from mutations in p53, p38, BRCA2 and cohesin, molecules that have also been linked to similar cytogenetic defects134.
DNA-damage-response pathways. Two of the key molecules that are involved in the DNA damage response, p53 and BRCA2, seem to have roles in destabilizing the HCC genome130. As noted above, the inactivation of p53 through mutation or viral oncoprotein sequestration is a common event in HCC, and p53 knock-in alleles containing dominant point mutations have been shown to cause genomic instability in mouse models135,136. Therefore, it is reasonable to speculate that p53 mutation promotes genomic instability in HCC. In addition, LOH of the BRCA2 locus has been documented in HCCs, providing another route to genomic instability in this disease137,138. It is worth noting that, despite documentation of deletions or mutations in these and other DNA-damage-network genes, their direct roles in the genomic instability of HCC have yet to be established in genetic model systems. In addition, there remain many other network components that merit in-depth study on the basis of their expression and functional roles in HCC genomic instability.
Genomic alterations in HCC. Significant effort has been directed towards charting the genomic events in HCC, with the prime goals of understanding the genetic basis of the disease and identifying new therapeutic targets. To date, karyotypic analyses70,139,140, chromosomal comparative genomic hybridization (CGH)70,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155, and LOH mapping16,70,143,144,145,147,148,149 have identified recurrent regions of copy number change and allelic imbalances (Table 1). For some loci, the resident cancer gene has been identified and validated, as for example: RB (13q); BRCA2 (13q); FLT2 (13q); TP53 (17p); HBVS2 (hepatitis B virus integration site 2) (4q); hepatoma-derived growth factor (HDGF) (1q); ERBB2 (17q); PIM1 (6p); and MYC (8q). However, it should be emphasized that the large regional gains and losses leave open the possibility that additional oncogenes and tumour-suppressor genes reside in these cancer hotspots. Notably, for most amplified and deleted loci, the cancer gene targets are not certain.
Several studies have attempted to categorize genomic changes in relation to tumour stage. In general, high levels of chromosomal instability seem to correlate with the de-differentiation and progression of HCC156. Several studies have suggested certain chromosomal changes to be specific to dysplastic lesions70,151, early-stage and late-stage HCCs16,70,141,143,147,150,155,157, and metastases149 (Table 1), with several oncogenes and tumour-suppressor genes residing at these loci: MYC (8q24); EMS1 (11q13), RB (13q13-14), liver related putative tumour suppressor (LTPS) (8p23.3); TP53 (17p13.1); downregulated in liver malignancy (DRLM) (4q21-22); and cyclin A (4q32-q-ter). It is important to note that the studies that have attempted to compare genomic profiles and tumour stage are few in number, often did not classify HCCs on the basis of aetiology, and used relatively low-resolution genome-scanning platforms.
Several studies have examined whether genomic changes seem to track with specific aetiological factors. In HBV-associated HCCs, gains in 8q and 20q and the loss of 4q are observed more frequently in HCCs with no underlying cirrhosis16,157, suggesting that these genetic aberrations might facilitate malignant transformation in the absence of pro-tumorigenic signals derived from a fibrotic matrix. Several loci have been specifically identified in HBV-, HCV- or non-viral-related HCCs16,70,141,142,143,144,150,151,152,153,154 (Table 1). It should be emphasized that, although these genome–aetiology correlates are intriguing, several studies have failed to uncover significant differences in genomic changes between the different aetiological groups70,145,155, although this outcome might relate to small sample sizes and the low-resolution genome-scanning platform used. All together, when these genomic studies are viewed in aggregate, we reiterate that expanded efforts are warranted, in which greater emphasis is placed on the use of precisely annotated HCCs, sufficiently powered sample size and high-resolution microarray platforms.
Gene-expression analysis and HCC molecular sub-classification. Microarray analyses have been used to investigate gene-expression changes in HCC. Results from such studies have attempted to classify HCCs based on their gene-expression patterns, and have successfully led to the identification of gene-expression profiles that distinguish HBV- from HCV-associated HCCs158, early from late intrahepatic recurrence of HCC159, and patients with different prognoses160. The molecular classification of HCC on the basis of prognosis in the third study was further compared with gene-expression profiles of HCCs from seven different mouse models161. Interestingly, HCCs from some of these mice (Myc, E2F1 and Myc–E2F1 transgenics) showed similar gene-expression patterns to the ones of HCCs from patients with better survival. Coincidentally, in the aforementioned CGH studies, 8q24 gain (encompassing MYC) was found in early-stage HCCs. Interestingly, murine HCCs derived from the Myc–TGFα transgenic model or diethylnitrosamine-treated mice show similar gene-expression patterns to HCCs from patients with poor survival. As previously mentioned, ERBB1 and ERBB3 expression correlate with a more aggressive presentation in human HCCs. The application of comparative oncogenomic analyses has also led to the identification and validation of new oncogenes in HCC. Specifically, the search for common recurrent amplifications that are present in both human and mouse HCC has identified the cell-death inhibitor cIAP1 and the transcription factor YAP1. This approach promises to facilitate human cancer genome discovery efforts and provide animal models for testing drugs directed against such validated HCC oncogenes162. Together, these results point to the promise of genomic classification and comparative genomic studies in understanding tumour characteristics and in providing molecular markers for patient classification and, potentially, treatment163,164.
Challenges and opportunities
HCC is an aggressive and enigmatic disease driven by diverse aetiologies, ranging from metabolic disorders to hepatotoxins to viruses. An important question is whether HCCs represent very heterogeneous collections of tumour molecular subtypes, and whether specific aetiologies drive distinct or common (epi)genetic and genomic events during tumour development. As seems to be the case for many other solid tumours, the available genomic profiles of HCC, with its many amplifications and deletions, strongly suggests that there is currently limited knowledge of the compendium of genes that drive HCC pathogenesis. A comprehensive genomic analysis of HCC samples, using a battery of high-resolution approaches, is urgently needed. We believe that such an atlas would transform the field, as this is particularly relevant to the development and application of effective agents to target specific genetic mutations and specific aetiological factors.
As comparative array-CGH analysis of various murine cancers has shown that such aberrations often target syntenic loci in the analogous human cancer type165, we further suggest that a concerted comparative genomic analysis of available mouse models of HCC might be particularly helpful in filtering through the complex human cancer genome. At the same time, comparative oncogenomic studies would serve to validate murine models by assessing the extent to which acquired genomic alterations are shared in human and mouse HCCs. Concurrently, emphasis should be placed on the design of new mouse models that recapitulate key features of the human disease, including hepatocyte regeneration, genomic instability and the fibrotic tumour microenvironment (Fig. 3). Ultimately, mouse models that share features with human HCCs could serve as valuable tools for gene identification and drug development. However, one needs to keep in mind key differences between mice and humans. For example, as noted in certain human HCC cases, telomere shortening might drive the genomic instability that enables the accumulation of cancer-relevant changes for hepatocarcinogenesis. As mice have long telomeres, this aspect of hepatocarcinogenesis might be fundamentally different between the species and provide additional opportunities for model refinement and testing of this mechanism through the use of a telomerase-deficient mouse model. These and other cross-species differences, and limitations in the use of human cell-culture systems, must be considered in any interpretation of data from the various model systems.

At present there is a need for new mouse models of hepatocellular carcinoma (HCC) that recapitulate human disease. The integration of information from genomic analyses (and the consequent identification of new oncogenes and tumour-suppressor genes involved in hepatocarcinogenesis) would help to build better animal models of HCC that share many features with the human disease. This should enable the identification of the cell of origin in HCC, increase understanding of the role of microenvironmental factors in HCC development (inflammation and cirrhosis) and improve the design of studies that address chemoprevention, biomarker discovery and drug testing. These efforts would aid in targeting HCC at its earliest stages before progression to incurable disease. CGH, comparative genomic hybridization.
Another outstanding and important question in HCC pathogenesis involves the cellular origin of this cancer. The resolution of the HCC cell of origin issue could affect the development of useful preventive strategies to target nascent neoplasms, foster an understanding of how HCC-relevant genetic lesions function in that specific cell-developmental context and increase our ability to develop more accurate mouse models in which key genetic events are targeted to the appropriate cellular compartment. Oval cells have been described as multipotent progenitor cells that can give rise to both hepatocytes and bile duct epithelial cells (cholangiocytes)166,167. The precursors of oval cells (that is, adult stem cells) reside in the Canal of Hering, and oval cells represent the replicating cellular compartment after liver injury167. At present, analyses of human HCCs for oval-cell markers, comparison of their gene-expression patterns with rat fetal hepatoblasts and the cellular characteristics of HCCs from various animal models have provided contrasting results about the cellular origin of HCC166,168,169,170,171,172. Although these results imply dual origins from either oval cells or mature hepatocytes, it seems that the conclusive answers will require definitive lineage-tracing approaches in refined model systems. The liver-stem-cell niche might have a crucial role in promoting or suppressing the effect of cancer-relevant genetic alterations, and virtually nothing is known about these microenvironmental factors and the effect of fibrosis or cirrhosis on the liver stem cell niche173. Finally, the failure to identify a clear cell of origin for HCC might stem from the fact that there are multiple cells of origin, perhaps reflecting the developmental plasticity of the hepatocyte lineage. Therefore, the comparison of hepatocarcinogenesis in genetically modified mice that express oncogenes (or lack the expression of tumour-suppressor genes) in the hepatocyte or stem cell compartment would aid in identifying the elusive cell of origin in this cancer (Fig. 3).
A special effort must be directed towards understanding the nature of HBV and HCV viral and host interactions, particularly the viral and host factors that lead to viral persistence and chronic liver destruction. Identifying the signalling pathways in the host cells that enable viral propagation would aid in developing strategies to control viral infection, therefore minimizing the risk of the development of chronic liver disease, cirrhosis and ultimately HCC. In addition, viral-associated inflammation and the recent links between inflammation and cancer might highlight an opportunity to pharmacologically quell the pro-carcinogenic effects of immunocytes during the formative stages of cancer. Along these lines, intra-tumoral immunocytes have been shown to enable the angiogenic switch174, and inflammation seems to activate pathways that support tissue stem cell growth175. Interestingly, as discussed previously, some early-stage HCCs show proliferating oval cells in the centre of the lesions, which indicates that tissue stem cell growth might be favoured in some cases176. Considering that inflammation is a common theme across many of the HCC aetiologies, it is conceivable that it could contribute to such oval-cell proliferation177. These examples make obvious how mechanistic insights could lead to the effective application of preventive therapies targeting the specific cancer-promoting mediators of tumour immune cells.
Beyond viral-associated inflammation, there exists a poor understanding of the role of the cirrhotic microenvironment and its constituents in the development of liver cancer. Further motivation for an in-depth analysis of host–tumour heterotypic interactions is provided by anti-angiogenesis studies showing that bevacizumab shows promising results (in combination with gemcitabine and oxaliplatin) in phase II trials of patients with HCC178. Model systems are needed that would enable the systematic analysis of various extracellular matrix components and stromal cells. Particularly important would be investigations of how the disruption of liver tissue architecture might disturb the liver stem cell niche and possibly promote an expansion of those cells at greatest risk of malignant transformation. Using a mouse model of HCC in which hepatocarcinogenesis is preceded by cirrhosis would seem to be a necessary starting point. In this regard, the carbon tetrachloride model of hepatocarcinogenesis would be useful for such studies, given its propensity to develop a classical fibrotic liver microenvironment and HCC115.
As genomic instability is a prevalent feature of HCC, and probably contributes to the transformation process, the elucidation of the mechanisms driving genomic instability might provide new approaches designed to detect, reduce or eliminate this process. Whether telomere shortening, chromosome segregation defects, inactivation of DNA-damage-response components or other yet-to-be identified mechanisms function alone or together to drive genomic instability remains an open question, although accumulating experimental evidence favours a role for telomere dysfunction in promoting both cirrhosis and carcinogenesis, as supported by the telomerase knockout mouse model110,115 and shorter telomeres in chronic liver disease and HCC in humans108,109,110,111. If telomere erosion does prove to drive cirrhosis and genomic instability in humans, then a serial assessment of telomere reserves might provide a useful biomarker to gauge both the cirrhotic potential and risk for chromosomal instability in the liver. Along these lines, it is plausible that the most rapid advances might come not from new therapies for advanced HCC, but rather the prevention of disease by viral elimination and/or the detection of early neoplasms that are surgically resectable. Here again, technological advances in protein science and comparative-serum profiling of humans and mice with HCC could prove to be highly productive, with the potential for immediate clinical impact. Prevention would also be aided by the identification of those individuals who are at greatest risk of HCC following exposure to an inciting agent. This effort would require large cohort studies involving serial follow-up, such as the Health Professional Study, which would enable the identification of risk loci in the broader population.
Concluding remarks
At present, the liver cancer field faces many challenges but equally compelling opportunities to address fundamental questions relating to the genetic, cell biological and environmental mechanisms of hepatocarcinogenesis, especially those that relate to aetiology. As our understanding of human hepatocarcinogenic mechanisms increases, the field will gain the foundation needed to build refined animal models that recapitulate the full range of human disease. The lethal nature of this cancer and the high levels of genomic instability in advanced disease make treatment at an earlier stage imperative. Better animal models will enable the identification of biomarkers of the earliest disease stages, and will aid in the development of successful therapeutic interventions. Such efforts are of vital interest given the large number of HCV-infected individuals and the looming epidemic of associated HCC. In the United States alone, there are an estimated 2.7 million individuals with chronic HCV infection (1988–1994 study)2,179, from which 68,000 HCC cases would be predicted in the coming years. Worldwide, there could be ∼120 million individuals with chronic HCV infection from which about 3 million HCC cases are expected to occur. In the absence of an HCV vaccine, this pool of HCV-infected individuals would be maintained for the foreseeable future. At the same time, the availability of an effective HBV vaccine brings to light the challenges of distributing preventive medicines to underserved populations worldwide. A concerted international effort involving several research approaches, from molecular analysis to vaccine development to population studies to distribution infrastructure, is urgently needed.
References
Hertl, M. & Cosimi, A. B. Liver transplantation for malignancy. Oncologist 10, 269–281 (2005).
American Cancer Society. Cancer Facts and FIGS 2005. American Cancer Society [online], (2005).
Anthony, P. in Pathology of the Liver (eds MacSween, R., Burt, A., Portmann, B., Ishak, K., Scheuer P. & Anthony, P.) 711–775 (Churchill Livingstone, London, New York, Sydney, Toronto, 2002).
Sherman, M. Hepatocellular carcinoma: epidemiology, risk factors, and screening. Semin. Liver Dis. 25, 143–154 (2005).
Badvie, S. Hepatocellular carcinoma. Postgrad. Med. J. 76, 4–11 (2000).
Lavanchy, D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral. Hepat. 11, 97–107 (2004).
Chen, C. J. et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA 295, 65–73 (2006).
Chisari, F. V. Unscrambling hepatitis C virus-host interactions. Nature 436, 930–932 (2005).
Bowen, D. G. & Walker, C. M. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature 436, 946–952 (2005).
Block, T. M., Mehta, A. S., Fimmel, C. J. & Jordan, R. Molecular viral oncology of hepatocellular carcinoma. Oncogene 22, 5093–5107 (2003).
Tokino, T., Tamura, H., Hori, N. & Matsubara, K. Chromosome deletions associated with hepatitis B virus integration. Virology 185, 879–882 (1991).
Murakami, Y. et al. Large scaled analysis of hepatitis B virus (HBV) DNA integration in HBV related hepatocellular carcinomas. Gut 54, 1162–1168 (2005).
Tarn, C., Lee, S., Hu, Y., Ashendel, C. & Andrisani, O. M. Hepatitis B virus X protein differentially activates RAS-RAF-MAPK and JNK pathways in X-transforming versus non-transforming AML12 hepatocytes. J. Biol. Chem. 276, 34671–34680 (2001).
Nijhara, R. et al. Sustained activation of mitogen-activated protein kinases and activator protein 1 by the hepatitis B virus X protein in mouse hepatocytes in vivo. J. Virol. 75, 10348–10358 (2001).
Nijhara, R., Jana, S. S., Goswami, S. K., Kumar, V. & Sarkar, D. P. An internal segment (residues 58–119) of the hepatitis B virus X protein is sufficient to activate MAP kinase pathways in mouse liver. FEBS Lett. 504, 59–64 (2001).
Feitelson, M. A. et al. Genetic mechanisms of hepatocarcinogenesis. Oncogene 21, 2593–2604 (2002).
Ueda, H. et al. Functional inactivation but not structural mutation of p53 causes liver cancer. Nature Genet. 9, 41–47 (1995).
Kim, C. M., Koike, K., Saito, I., Miyamura, T. & Jay, G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 351, 317–320 (1991). This article shows the carcinogenic potential of hepatitis B viral factors (more specifically, the HBx gene), as HBx transgenic animals develop HCC.
Yu, D. Y. et al. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitis B virus X-protein. J. Hepatol. 31, 123–132 (1999).
Nowak, M. A. et al. Viral dynamics in hepatitis B virus infection. Proc. Natl Acad. Sci. USA 93, 4398–4402 (1996).
Wieland, S., Thimme, R., Purcell, R. H. & Chisari, F. V. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl Acad. Sci. USA 101, 6669–6674 (2004).
Rehermann, B. & Nascimbeni, M. Immunology of hepatitis B virus and hepatitis C virus infection. Nature Rev. Immunol. 5, 215–229 (2005).
Lok, A. S., Heathcote, E. J. & Hoofnagle, J. H. Management of hepatitis B: 2000 —summary of a workshop. Gastroenterology 120, 1828–1853 (2001).
Kojima, T. Immune electron microscopic study of hepatitis B virus associated antigens in hepatocytes. Gastroenterol. Jpn 17, 559–575 (1982).
Shimoda, R. et al. Increased formation of oxidative DNA damage, 8-hydroxydeoxyguanosine, in human livers with chronic hepatitis. Cancer Res. 54, 3171–3172 (1994).
Galli, A. et al. Oxidative stress stimulates proliferation and invasiveness of hepatic stellate cells via a MMP2-mediated mechanism. Hepatology 41, 1074–1084 (2005).
Chen, B. F. et al. High prevalence and mapping of pre-S deletion in hepatitis B virus carriers with progressive liver diseases. Gastroenterology 130, 1153–1168 (2006).
Lindenbach, B. D. & Rice, C. M. Unravelling hepatitis C virus replication from genome to function. Nature 436, 933–938 (2005).
Lindenbach, B. D. et al. Complete replication of hepatitis C virus in cell culture. Science 309, 623–626 (2005). This article reports a significant advance for research on HCV, as it shows successful HCV replication and viral particle production in cell culture. These culture systems will now provide a better opportunity to study viral replication and propagation, and provide clues for antiviral therapies.
Wakita, T. et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nature Med. 11, 791–796 (2005).
Weiner, A. et al. Persistent hepatitis C virus infection in a chimpanzee is associated with emergence of a cytotoxic T lymphocyte escape variant. Proc. Natl Acad. Sci. USA 92, 2755–2759 (1995).
Daniel, D. et al. Immune enhancement of skin carcinogenesis by CD4+ T cells. J. Exp. Med. 197, 1017–1028 (2003).
Pachiadakis, I., Pollara, G., Chain, B. M. & Naoumov, N. V. Is hepatitis C virus infection of dendritic cells a mechanism facilitating viral persistence? Lancet Infect. Dis. 5, 296–304 (2005).
Park, K. J. et al. 1Hepatitis C virus NS5A protein modulates c-Jun N-terminal kinase through interaction with tumor necrosis factor receptor-associated factor 2. J. Biol. Chem. 278, 30711–8 (2003).
Majumder, M. et al. Hepatitis C virus NS5A protein impairs TNF-mediated hepatic apoptosis, but not by an anti-FAS antibody, in transgenic mice. Virology 294, 94–105 (2002).
Melen, K., Fagerlund, R., Nyqvist, M., Keskinen, P. & Julkunen, I. Expression of hepatitis C virus core protein inhibits interferon-induced nuclear import of STATs. J. Med. Virol. 73, 536–547 (2004).
Foy, E. et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl Acad. Sci. USA 102, 2986–2991 (2005).
Li, K. et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl Acad. Sci. USA 102, 2992–2997 (2005).
Gale, M., Jr. & Foy, E. M. Evasion of intracellular host defence by hepatitis C virus. Nature 436, 939–945 (2005).
Hino, O., Kajino, K., Umeda, T. & Arakawa, Y. Understanding the hypercarcinogenic state in chronic hepatitis: a clue to the prevention of human hepatocellular carcinoma. J. Gastroenterol. 37, 883–887 (2002).
Macdonald, A. et al. The hepatitis C virus non-structural NS5A protein inhibits activating protein-1 function by perturbing ras- pathway signaling. J. Biol. Chem. 278, 17775–17784 (2003).
Majumder, M., Ghosh, A. K., Steele, R., Ray, R. & Ray, R. B. Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J. Virol. 75, 1401–1407 (2001). This report shows a potential carcinogenic mechanism of one of the HCV viral proteins (NS5A) through the interaction and consequent functional inactivation of p53.
Moriya, K. et al. Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 61, 4365–4370 (2001).
Kamegaya, Y. et al. Hepatitis C virus acts as a tumor accelerator by blocking apoptosis in a mouse model of hepatocarcinogenesis. Hepatology 41, 660–667 (2005).
McClain, C. J., Hill, D. B., Song, Z., Deaciuc, I. & Barve, S. Monocyte activation in alcoholic liver disease. Alcohol 27, 53–61 (2002).
Hoek, J. B. & Pastorino, J. G. Ethanol, oxidative stress, and cytokine-induced liver cell injury. Alcohol 27, 63–68 (2002).
Campbell, J. S. et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc. Natl Acad. Sci. USA 102, 3389–3394 (2005). This article provides evidence for the hepatocarcinogenic potential of the cirrhotic microenvironment, and provides a useful mouse model to investigate the molecular mechanisms underlying cirrhosis-induced HCC.
Comporti, M. et al. F(2)-isoprostanes stimulate collagen synthesis in activated hepatic stellate cells: a link with liver fibrosis? Lab. Invest. 85, 1381–1391 (2005).
Osna, N. A., Clemens, D. L. & Donohue, T. M., Jr. Ethanol metabolism alters interferon-γ signaling in recombinant HepG2 cells. Hepatology 42, 1109–1117 (2005).
Marrogi, A. J. et al. Oxidative stress and p53 mutations in the carcinogenesis of iron overload-associated hepatocellular carcinoma. J. Natl Cancer Inst. 93, 1652–1655 (2001).
Kurz, D. J. et al. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J. Cell Sci. 117, 2417–2426 (2004).
Aguilar, F., Harris, C. C., Sun, T., Hollstein, M. & Cerutti, P. Geographic variation of p53 mutational profile in nonmalignant human liver. Science 264, 1317–1319 (1994).
Bressac, B., Kew, M., Wands, J. & Ozturk, M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature 350, 429–431 (1991). This article shows the occurrence of p53 mutations in human HCCs induced after aflatoxin B1 exposure. It points to the mutagenic properties of aflatoxin, as well as the key role that p53 inactivation might have in hepatocarcinogenesis.
Hsu, I. C. et al. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature 350, 427–428 (1991).
Ozturk, M. p53 mutation in hepatocellular carcinoma after aflatoxin exposure. Lancet 338, 1356–1359 (1991).
Riley, J., Mandel, H. G., Sinha, S., Judah, D. J. & Neal, G. E. In vitro activation of the human Harvey-ras proto-oncogene by aflatoxin B1. Carcinogenesis 18, 905–910 (1997).
Kew, M. C. Synergistic interaction between aflatoxin B1 and hepatitis B virus in hepatocarcinogenesis. Liver Int. 23, 405–409 (2003).
Okuda, K. Hepatocellular carcinoma. J. Hepatol. 32, 225–237 (2000).
Minouchi, K., Kaneko, S. & Kobayashi, K. Mutation of p53 gene in regenerative nodules in cirrhotic liver. J. Hepatol. 37, 231–239 (2002).
Nose, H., Imazeki, F., Ohto, M. & Omata, M. p53 gene mutations and 17p allelic deletions in hepatocellular carcinoma from Japan. Cancer 72, 355–360 (1993).
Nishida, N. et al. Role and mutational heterogeneity of the p53 gene in hepatocellular carcinoma. Cancer Res. 53, 368–372 (1993).
Hosono, S., Chou, M. J., Lee, C. S. & Shih, C. Infrequent mutation of p53 gene in hepatitis B virus positive primary hepatocellular carcinomas. Oncogene 8, 491–496 (1993).
Farazi, P. A., Glickman, J., Horner, J. & Depinho, R. A. Cooperative Interactions of p53 Mutation, Telomere Dysfunction, and Chronic Liver Damage in Hepatocellular Carcinoma Progression. Cancer Res. 66, 4766–4773 (2006).
Huang, S. N. & Chisari, F. V. Strong, sustained hepatocellular proliferation precedes hepatocarcinogenesis in hepatitis B surface antigen transgenic mice. Hepatology 21, 620–626 (1995).
Ghebranious, N. & Sell, S. Hepatitis B injury, male gender, aflatoxin, and p53 expression each contribute to hepatocarcinogenesis in transgenic mice. Hepatology 27, 383–391 (1998).
Ghebranious, N. & Sell, S. The mouse equivalent of the human p53ser249 mutation p53ser246 enhances aflatoxin hepatocarcinogenesis in hepatitis B surface antigen transgenic and p53 heterozygous null mice. Hepatology 27, 967–973 (1998).
Gregorieff, A. & Clevers, H. Wnt signaling in the intestinal epithelium: from endoderm to cancer. Genes Dev. 19, 877–890 (2005).
Ishizaki, Y. et al. Immunohistochemical analysis and mutational analyses of β-catenin, Axin family and APC genes in hepatocellular carcinomas. Int. J. Oncol. 24, 1077–1083 (2004). This article shows alterations of the Wnt signalling pathway in human HCC. It shows increased nuclear expression of β-catenin, as well as mutations in β-catenin, axin1 and axin2.
Edamoto, Y. et al. Alterations of RB1, p53 and Wnt pathways in hepatocellular carcinomas associated with hepatitis C, hepatitis B and alcoholic liver cirrhosis. Int. J. Cancer 106, 334–341 (2003).
Thorgeirsson, S. S. & Grisham, J. W. Molecular pathogenesis of human hepatocellular carcinoma. Nature Genet. 31, 339–346 (2002).
Peng, S. Y. et al. High α-fetoprotein level correlates with high stage, early recurrence and poor prognosis of hepatocellular carcinoma: significance of hepatitis virus infection, age, p53 and β-catenin mutations. Int. J. Cancer 112, 44–50 (2004).
An, F. Q. et al. Tumor heterogeneity in small hepatocellular carcinoma: analysis of tumor cell proliferation, expression and mutation of p53 AND β-catenin. Int. J. Cancer 93, 468–474 (2001).
Hsu, H. C. et al. β-catenin mutations are associated with a subset of low-stage hepatocellular carcinoma negative for hepatitis B virus and with favorable prognosis. Am. J. Pathol. 157, 763–770 (2000).
Huang, H. et al. Beta-catenin mutations are frequent in human hepatocellular carcinomas associated with hepatitis C virus infection. Am. J. Pathol. 155, 1795–1801 (1999).
Cha, M. Y., Kim, C. M., Park, Y. M. & Ryu, W. S. Hepatitis B virus X protein is essential for the activation of Wnt/β-catenin signaling in hepatoma cells. Hepatology 39, 1683–1693 (2004).
Torbenson, M. et al. Concurrent evaluation of p53, β-catenin, and α-fetoprotein expression in human hepatocellular carcinoma. Am. J. Clin. Pathol. 122, 377–382 (2004).
Prange, W. et al. β-catenin accumulation in the progression of human hepatocarcinogenesis correlates with loss of E-cadherin and accumulation of p53, but not with expression of conventional WNT-1 target genes. J. Pathol. 201, 250–259 (2003).
Gross-Goupil, M. et al. Analysis of chromosomal instability in pulmonary or liver metastases and matched primary hepatocellular carcinoma after orthotopic liver transplantation. Int. J. Cancer 104, 745–751 (2003).
Calvisi, D. F., Factor, V. M., Ladu, S., Conner, E. A. & Thorgeirsson, S. S. Disruption of β-catenin pathway or genomic instability define two distinct categories of liver cancer in transgenic mice. Gastroenterology 126, 1374–1386 (2004).
Ito, Y. et al. Expression and clinical significance of erb-B receptor family in hepatocellular carcinoma. Br. J. Cancer 84, 1377–1383 (2001).
Hopfner, M. et al. Targeting the epidermal growth factor receptor by gefitinib for treatment of hepatocellular carcinoma. J. Hepatol. 41, 1008–1016 (2004).
Schiffer, E. et al. Gefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosis. Hepatology 41, 307–314 (2005).
Philip, P. A. et al. Phase II study of Erlotinib (OSI-774) in patients with advanced hepatocellular cancer. J. Clin. Oncol. 23, 6657–6663 (2005).
Jhappan, C. et al. TGF alpha overexpression in transgenic mice induces liver neoplasia and abnormal development of the mammary gland and pancreas. Cell 61, 1137–1146 (1990). This work points to the hepatocarcinogenic potential of TGFα signalling, and complements findings of ErbB receptor overexpression in human HCCs.
Sandgren, E. P., Luetteke, N. C., Palmiter, R. D., Brinster, R. L. & Lee, D. C. Overexpression of TGF α in transgenic mice: induction of epithelial hyperplasia, pancreatic metaplasia, and carcinoma of the breast. Cell 61, 1121–1135 (1990).
Webber, E. M., Wu, J. C., Wang, L., Merlino, G. & Fausto, N. Overexpression of transforming growth factor-α causes liver enlargement and increased hepatocyte proliferation in transgenic mice. Am. J. Pathol. 145, 398–408 (1994).
Russell, W. E., Kaufmann, W. K., Sitaric, S., Luetteke, N. C. & Lee, D. C. Liver regeneration and hepatocarcinogenesis in transforming growth factor-alpha-targeted mice. Mol. Carcinog. 15, 183–189 (1996).
Sandgren, E. P. et al. Transforming growth factor-α dramatically enhances oncogene-induced carcinogenesis in transgenic mouse pancreas and liver. Mol. Cell Biol. 13, 320–330 (1993).
Sargent, L. M. et al. Nonrandom cytogenetic alterations in hepatocellular carcinoma from transgenic mice overexpressing c-Myc and transforming growth factor-α in the liver. Am. J. Pathol. 154, 1047–1055 (1999).
Murakami, H. et al. Transgenic mouse model for synergistic effects of nuclear oncogenes and growth factors in tumorigenesis: interaction of c-myc and transforming growth factor alpha in hepatic oncogenesis. Cancer Res. 53, 1719–1723 (1993).
Santoni-Rugiu, E., Nagy, P., Jensen, M. R., Factor, V. M. & Thorgeirsson, S. S. Evolution of neoplastic development in the liver of transgenic mice co-expressing c-myc and transforming growth factor-α. Am. J. Pathol. 149, 407–428 (1996).
Jakubczak, J. L., Chisari, F. V. & Merlino, G. Synergy between transforming growth factor α and hepatitis B virus surface antigen in hepatocellular proliferation and carcinogenesis. Cancer Res. 57, 3606–3611 (1997).
Daveau, M. et al. Hepatocyte growth factor, transforming growth factor-α, and their receptors as combined markers of prognosis in hepatocellular carcinoma. Mol. Carcinog. 36, 130–141 (2003).
Sakata, H. et al. Hepatocyte growth factor/scatter factor overexpression induces growth, abnormal development, and tumor formation in transgenic mouse livers. Cell Growth Differ. 7, 1513–1523 (1996).
Wang, R., Ferrell, L. D., Faouzi, S., Maher, J. J. & Bishop, J. M. Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J. Cell Biol. 153, 1023–1034 (2001).
Kanai, Y., Ushijima, S., Tsuda, H., Sakamoto, M. & Hirohashi, S. Aberrant DNA methylation precedes loss of heterozygosity on chromosome 16 in chronic hepatitis and liver cirrhosis. Cancer Lett. 148, 73–80 (2000).
Kanai, Y. et al. DNA hypermethylation at the D17S5 locus and reduced HIC-1 mRNA expression are associated with hepatocarcinogenesis. Hepatology 29, 703–709 (1999).
Kanai, Y. et al. Aberrant DNA methylation on chromosome 16 is an early event in hepatocarcinogenesis. Jpn J. Cancer Res. 87, 1210–1217 (1996).
Yu, J. et al. Methylation profiling of twenty four genes and the concordant methylation behaviours of nineteen genes that may contribute to hepatocellular carcinogenesis. Cell Res. 13, 319–333 (2003).
Lee, S. et al. Aberrant CpG island hypermethylation along multistep hepatocarcinogenesis. Am. J. Pathol. 163, 1371–1378 (2003). This article shows the presence of epigenetic alterations (hypermethylation) in human HCCs. This work shows hypermethylation in the earliest stages of hepatocarcinogenesis, and to a greater extent in tumour progression.
Wong, I. H. et al. Detection of aberrant p16 methylation in the plasma and serum of liver cancer patients. Cancer Res. 59, 71–73 (1999).
Matsuda, Y., Ichida, T., Matsuzawa, J., Sugimura, K. & Asakura, H. p16(INK4) is inactivated by extensive CpG methylation in human hepatocellular carcinoma. Gastroenterology 116, 394–400 (1999).
Liew, C. T. et al. High frequency of p16INK4A gene alterations in hepatocellular carcinoma. Oncogene 18, 789–795 (1999).
Murata, H. et al. Promoter hypermethylation silences cyclooxygenase-2 (Cox-2) and regulates growth of human hepatocellular carcinoma cells. Lab. Invest. 84, 1050–1059 (2004).
Kubo, T. et al. Apoptotic speck protein-like, a highly homologous protein to apoptotic speck protein in the pyrin domain, is silenced by DNA methylation and induces apoptosis in human hepatocellular carcinoma. Cancer Res. 64, 5172–5177 (2004).
Wong, C. M., Lee, J. M., Ching, Y. P., Jin, D. Y. & Ng, I. O. Genetic and epigenetic alterations of DLC-1 gene in hepatocellular carcinoma. Cancer Res. 63, 7646–7651 (2003).
Maeta, Y., Shiota, G., Okano, J. & Murawaki, Y. Effect of promoter methylation of the p16 gene on phosphorylation of retinoblastoma gene product and growth of hepatocellular carcinoma cells. Tumour Biol. 26, 300–305 (2005).
Urabe, Y. et al. Telomere length in human liver diseases. Liver 16, 293–297 (1996). This article shows telomere shortening during human chronic liver disease and the induction of liver cirrhosis. It also suggests that telomerase might be re-activated during the late stages of hepatocarcinogenesis as the telomere length of advanced HCCs is slightly longer than that of early-stage HCCs, suggestive of telomerase activation.
Miura, N. et al. Progressive telomere shortening and telomerase reactivation during hepatocellular carcinogenesis. Cancer Genet. Cytogenet. 93, 56–62 (1997).
Rudolph, K. L., DrPinho, R. A. in The Liver Biology and Pathobiology (eds Arias, I., Fausto, N., Boyer, J., Chisari, F. & Shafritz, D.) 1001–1011 (Lippincott Williams and Wilkins, Philadelphia, 2001).
Kitada, T., Seki, S., Kawakita, N., Kuroki, T. & Monna, T. Telomere shortening in chronic liver diseases. Biochem. Biophys. Res. Commun. 211, 33–39 (1995).
Wiemann, S. U. et al. Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. Faseb J. 16, 935–942 (2002).
Plentz, R. R. et al. Hepatocellular telomere shortening correlates with chromosomal instability and the development of human hepatoma. Hepatology 40, 80–86 (2004).
Plentz, R. R. et al. Telomere shortening correlates with increasing aneuploidy of chromosome 8 in human hepatocellular carcinoma. Hepatology 42, 522–526 (2005).
Farazi, P. A. et al. Differential impact of telomere dysfunction on initiation and progression of hepatocellular carcinoma. Cancer Res. 63, 5021–5027 (2003). The role of telomere dysfunction in HCC initiation and progression is addressed in this work. Whereas telomere dysfunction promotes liver cancer initiation in mice, it suppresses progression, therefore pointing to the importance of telomerase re-activation in HCC progression.
Nagao, K., Tomimatsu, M., Endo, H., Hisatomi, H. & Hikiji, K. Telomerase reverse transcriptase mRNA expression and telomerase activity in hepatocellular carcinoma. J. Gastroenterol. 34, 83–87. (1999).
Shimojima, M. et al. Detection of telomerase activity, telomerase RNA component, and telomerase reverse transcriptase in human hepatocellular carcinoma. Hepatol. Res. 29, 31–38 (2004).
Lee, C. M. et al. Telomerase activity and telomerase catalytic subunit in hepatocellular carcinoma. Hepatogastroenterology 51, 796–800 (2004).
Ferber, M. J. et al. Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene 22, 3813–3820 (2003).
Takeo, S. et al. Examination of oncogene amplification by genomic DNA microarray in hepatocellular carcinomas: comparison with comparative genomic hybridization analysis. Cancer Genet. Cytogenet. 130, 127–132 (2001).
Nishimoto, A. et al. Functional evidence for a telomerase repressor gene on human chromosome 10p15. 1. Oncogene 20, 828–835 (2001).
Piao, Y. F., He, M., Shi, Y. & Tang, T. Y. Relationship between microvessel density and telomerase activity in hepatocellular carcinoma. World J. Gastroenterol. 10, 2147–2149 (2004).
Kobayashi, T., Kubota, K., Takayama, T. & Makuuchi, M. Telomerase activity as a predictive marker for recurrence of hepatocellular carcinoma after hepatectomy. Am. J. Surg. 181, 284–288 (2001).
Hytiroglou, P. et al. Telomerase activity in precancerous hepatic nodules. Cancer 82, 1831–1838 (1998).
Tahara, H. et al. Telomerase activity in human liver tissues: comparison between chronic liver disease and hepatocellular carcinomas. Cancer Res. 55, 2734–2736 (1995).
Youssef, N., Paradis, V., Ferlicot, S. & Bedossa, P. In situ detection of telomerase enzymatic activity in human hepatocellular carcinogenesis. J. Pathol. 194, 459–465 (2001).
Oh, B. K. et al. Telomere shortening and telomerase reactivation in dysplastic nodules of human hepatocarcinogenesis. J. Hepatol. 39, 786–792 (2003).
Ogami, M. et al. Quantitative analysis and in situ localization of human telomerase RNA in chronic liver disease and hepatocellular carcinoma. Lab. Invest. 79, 15–26 (1999).
Artandi, S. E. et al. Constitutive telomerase expression promotes mammary carcinomas in aging mice. Proc. Natl Acad. Sci. USA 99, 8191–8196 (2002).
Gollin, S. M. Mechanisms leading to chromosomal instability. Semin. Cancer Biol. 15, 33–42 (2005).
Smith, M. W. et al. Identification of novel tumor markers in hepatitis C virus-associated hepatocellular carcinoma. Cancer Res. 63, 859–864 (2003).
Yu, C. T. et al. Phosphorylation and stabilization of HURP by Aurora-A: implication of HURP as a transforming target of Aurora-A. Mol. Cell Biol. 25, 5789–5800 (2005).
Andrews, P. D. Aurora kinases: shining lights on the therapeutic horizon? Oncogene 24, 5005–5015 (2005).
Saeki, A. et al. Frequent impairment of the spindle assembly checkpoint in hepatocellular carcinoma. Cancer 94, 2047–2054 (2002).
Olive, K. P. et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119, 847–860 (2004).
Lang, G. A. et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119, 861–872 (2004).
Kuroki, T. et al. Evidence for the presence of two tumour-suppressor genes for hepatocellular carcinoma on chromosome 13q. Br. J. Cancer 72, 383–385 (1995).
Martins, C., Kedda, M. A. & Kew, M. C. Characterization of six tumor suppressor genes and microsatellite instability in hepatocellular carcinoma in southern African blacks. World J. Gastroenterol. 5, 470–476 (1999).
Wong, N. et al. A comprehensive karyotypic study on human hepatocellular carcinoma by spectral karyotyping. Hepatology 32, 1060–1068 (2000).
Hwang, H. J. et al. A comprehensive karyotypic analysis on Korean hepatocellular carcinoma cell lines by cross-species color banding and comparative genomic hybridization. Cancer Genet. Cytogenet. 141, 128–137 (2003).
Okabe, H. et al. Comprehensive allelotype study of hepatocellular carcinoma: potential differences in pathways to hepatocellular carcinoma between hepatitis B virus-positive and-negative tumors. Hepatology 31, 1073–1079 (2000).
Wong, N. et al. Genomic aberrations in human hepatocellular carcinomas of differing etiologies. Clin. Cancer Res. 6, 4000–4009 (2000).
Kusano, N. et al. Chromosomal imbalances detected by comparative genomic hybridization are associated with outcome of patients with hepatocellular carcinoma. Cancer 94, 746–751 (2002).
Kitay-Cohen, Y. et al. Analysis of chromosomal aberrations in large hepatocellular carcinomas by comparative genomic hybridization. Cancer Genet. Cytogenet. 131, 60–64 (2001).
Koo, S. H. et al. Genetic alterations in hepatocellular carcinoma and intrahepatic cholangiocarcinoma. Cancer Genet. Cytogenet. 130, 22–28 (2001).
Marchio, A. et al. Recurrent chromosomal abnormalities in hepatocellular carcinoma detected by comparative genomic hybridization. Genes Chromosomes Cancer 18, 59–65 (1997).
Hashimoto, K. et al. Analysis of DNA copy number aberrations in hepatitis C virus-associated hepatocellular carcinomas by conventional CGH and array CGH. Mod. Pathol. 17, 617–622 (2004).
Sakakura, C. et al. Chromosomal aberrations in human hepatocellular carcinomas associated with hepatitis C virus infection detected by comparative genomic hybridization. Br. J. Cancer 80, 2034–2039 (1999).
Zhang, L. H. et al. Allelic imbalance regions on chromosomes 8p, 17p and 19p related to metastasis of hepatocellular carcinoma: comparison between matched primary and metastatic lesions in 22 patients by genome-wide microsatellite analysis. J. Cancer Res. Clin. Oncol. 129, 279–286 (2003).
Kusano, N. et al. Genetic aberrations detected by comparative genomic hybridization in hepatocellular carcinomas: their relationship to clinicopathological features. Hepatology 29, 1858–1862 (1999).
Zondervan, P. E. et al. Molecular cytogenetic evaluation of virus-associated and non-viral hepatocellular carcinoma: analysis of 26 carcinomas and 12 concurrent dysplasias. J. Pathol. 192, 207–215 (2000).
Marchio, A. et al. Distinct chromosomal abnormality pattern in primary liver cancer of non-B, non-C patients. Oncogene 19, 3733–3738 (2000).
Balsara, B. R. et al. Human hepatocellular carcinoma is characterized by a highly consistent pattern of genomic imbalances, including frequent loss of 16q23. 1–24. 1. Genes Chromosomes Cancer 30, 245–253 (2001).
Kawai, H. et al. Quantitative evaluation of genomic instability as a possible predictor for development of hepatocellular carcinoma: comparison of loss of heterozygosity and replication error. Hepatology 31, 1246–1250 (2000).
Guan, X. Y. et al. Recurrent chromosome alterations in hepatocellular carcinoma detected by comparative genomic hybridization. Genes Chromosomes Cancer 29, 110–116 (2000).
Wilkens, L. et al. Induction of aneuploidy by increasing chromosomal instability during dedifferentiation of hepatocellular carcinoma. Proc. Natl Acad. Sci. USA 101, 1309–1314 (2004). This study shows a correlation between increasing levels of chromosomal instability and progression of HCC, suggesting that marked genome instability characterizes the more advanced stages of the disease.
Wong, N. et al. Assessment of genetic changes in hepatocellular carcinoma by comparative genomic hybridization analysis: relationship to disease stage, tumor size, and cirrhosis. Am. J. Pathol. 154, 37–43 (1999).
Okabe, H. et al. Genome-wide analysis of gene expression in human hepatocellular carcinomas using cDNA microarray: identification of genes involved in viral carcinogenesis and tumor progression. Cancer Res. 61, 2129–2137 (2001).
Iizuka, N. et al. Oligonucleotide microarray for prediction of early intrahepatic recurrence of hepatocellular carcinoma after curative resection. Lancet 361, 923–929 (2003).
Lee, J. S. et al. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 40, 667–676 (2004). This work shows the successful use of gene-expression profiling to classify human HCCs. Gene-expression patterns could distinguish between HCCs with different prognoses.
Lee, J. S. et al. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nature Genet. 36, 1306–1311 (2004). This paper compared gene-expression profiles of human HCCs with HCCs from various mouse models. It shows commonalities in gene-expression patterns in tumours from both species, and points to the promising use of comparative functional genomics to understand the molecular basis of human cancer.
Zender, L. et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 125, 1253–1267 (2006). This paper shows the successful use of comparative genomics in the identification and validation of new oncogenes important in hepatocarcinogenesis.
Lee, J. S. & Thorgeirsson, S. S. Genetic profiling of human hepatocellular carcinoma. Semin. Liver Dis. 25, 125–132 (2005).
Lee, J. S., Grisham, J. W. & Thorgeirsson, S. S. Comparative functional genomics for identifying models of human cancer. Carcinogenesis 26, 1013–1020 (2005).
O'Hagan, R. C. et al. Telomere dysfunction provokes regional amplification and deletion in cancer genomes. Cancer Cell 2, 149–155 (2002).
Roskams, T., Libbrecht, L. in Malignant Liver Tumours: Basic Concepts and Clinical Management (eds Berr, F., B. J., Hauss, J., Wands, J. & Wittekind, C.) 44–56 (Kluwer Academic Publishers, Dordrecht, 2003).
Fausto, N. & Campbell, J. S. The role of hepatocytes and oval cells in liver regeneration and repopulation. Mech. Dev. 120, 117–130 (2003).
Shachaf, C. M. et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 431, 1112–1117 (2004). This study shows the ability of tumour cells to differentiate into hepatocytes on oncogene inactivation, and their ability to restore their cancerous phenotype on oncogene re-activation. Therefore, this work suggests a possible stem cell origin of HCC.
Dumble, M. L., Croager, E. J., Yeoh, G. C. & Quail, E. A. Generation and characterization of p53 null transformed hepatic progenitor cells: oval cells give rise to hepatocellular carcinoma. Carcinogenesis 23, 435–445 (2002).
Lee, J. S. et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nature Med. 12, 410–416 (2006).
Sell, S. Cellular origin of hepatocellular carcinomas. Semin. Cell Dev. Biol. 13, 419–424 (2002).
Braun, L., Mikumo, R. & Fausto, N. Production of hepatocellular carcinoma by oval cells: cell cycle expression of c-myc and p53 at different stages of oval cell transformation. Cancer Res. 49, 1554–1561 (1989).
Fotiadu, A. et al. Progenitor cell activation in chronic viralhepatitis. Liver Int. 24, 268–274 (2004).
Coussens, L. M. et al. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 13, 1382–1397 (1999).
Beachy, P. A., Karhadkar, S. S. & Berman, D. M. Tissue repair and stem cell renewal in carcinogenesis. Nature 432, 324–331 (2004).
Wu, P. C. et al. Hepatocellular carcinoma expressing both hepatocellular and biliary markers also expresses cytokeratin 14, a marker of bipotential progenitor cells. J. Hepatol. 31, 965–966 (1999).
Huang, T., Chesnokov, V., Yokoyama, K. K., Carr, B. I. & Itakura, K. Expression of the Hoxa-13 gene correlates to hepatitis B and C virus associated HCC. Biochem. Biophys. Res. Commun. 281, 1041–1044 (2001).
Zhu, A. X. et al. Phase II study of gemcitabine and oxaliplatin in combination with bevacizumab in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 24, 1898–1903 (2006).
Sy, T. & Jamal, M. M. Epidemiology of hepatitis C virus (HCV) infection. Int. J. Med. Sci. 3, 41–46 (2006).
Limdi, J. K. & Crampton, J. R. Hereditary haemochromatosis. Qjm 97, 315–324 (2004).
Sarkany, R. P. The management of porphyria cutanea tarda. Clin. Exp. Dermatol. 26, 225–232 (2001).
Parfrey, H., Mahadeva, R. & Lomas, D. A. α(1)-antitrypsin deficiency, liver disease and emphysema. Int. J. Biochem. Cell Biol. 35, 1009–1014 (2003).
Tanguay, R. M., Jorquera, R., Poudrier, J. & St-Louis, M. Tyrosine and its catabolites: from disease to cancer. Acta Biochim. Pol. 43, 209–216 (1996).
El-Serag, H. B., Tran, T. & Everhart, J. E. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology 126, 460–468 (2004).
Farrell, G. C. & Larter, C. Z. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology 43, S99–S112 (2006).
Adams, L. A. & Angulo, P. Recent concepts in non-alcoholic fatty liver disease. Diabet. Med. 22, 1129–1133 (2005).
Acknowledgements
We would like to thank N. Fausto, G. Merlino, L. Rudolph, E. Sahin, and S. Thorgeirsson for critical reading of this manuscript and helpful suggestions. R.A.D. is an American Cancer Society Research Professor and an Ellison Medical Foundation Senior Scholar. This work is supported by an NIH grant and by the Robert A. and Renee E. Belfer Foundation Institute for Innovative Cancer Science. We apologize to those whose work could not be included owing to space limitations.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Related links
DATABASES
National Cancer Institute
FURTHER INFORMATION
R. DePinho's laboratory webpage
Glossary
- Liver cirrhosis
-
A pathological condition characterized by fibrotic scarring of the liver caused by excessive collagen deposition after chronic liver disease or damage.
- Aflatoxin B1
-
A toxin with mutagenic properties that is produced as a secondary metabolite by the fungus Aspergillus flavus, which is found on many food products such as nuts, spices and oilseeds.
- DNA microdeletions
-
In the case of hepatitis B virus, host DNA sequences are lost when the virus integrates into the genome.
- p53
-
A tumour suppressor that functions to promote apoptosis and induce cell-cycle arrest upon DNA damage or oncogene activation.
- Stellate cells
-
Liver stellate cells store retinol (vitamin A), and are in an otherwise quiescent state. Liver injury and exposure to various cytokines in the context of chronic liver disease provoke stellate cell activation, which is associated with cellular proliferation, the acquisition of myofibroblast morphology and the robust synthesis of extracellular matrix components such as collagen, therefore contributing to liver fibrosis.
- Hepatic steatosis
-
Also known as fatty liver, hepatic steatosis is a process that involves the accumulation of fatty acids in hepatocytes. One feature of steatohepatitis is an increase in reactive oxygen species generation, which results in lipid peroxidation.
- Küpffer cells
-
These are specialized macrophages located in the liver. The activation of these cells by various insults (as for example, exposure to bacterial endotoxin) results in the release of various cytokines in the liver that might lead to hepatocyte death or damage.
- Clear cell changes
-
The cytoplasm of hepatocytes seems to be clear due to the accumulation of glycogen or lipids.
- Nuclear crowding
-
The number of nuclei per unit area in a lesion is increased compared with normal hepatic tissue.
- Trabeculae
-
Fenestrated plates that are formed by hepatocytes in the liver. These rows of hepatocytes are separated by sinusoids (which function as bloodstreams within the liver and are lined by endothelial cells to prevent contact of hepatocytes with the sinusoidal blood). The normal thickness of the trabeculae is 1–2 cells.
- Sinusoidal cells
-
Cells such as endothelial cells, Küpffer cells and stellate cells that line the sinusoids in the liver.
- Canal of Hering
-
A channel lined by biliary epithelial cells, and partially by hepatocytes, located adjacent to the portal tract in the liver.
- Bevacizumab
-
A recombinant humanized monoclonal antibody targeting vascular endothelial growth factor.
Rights and permissions
About this article
Cite this article
Farazi, P., DePinho, R. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer 6, 674–687 (2006). https://doi.org/10.1038/nrc1934
Issue Date:
DOI: https://doi.org/10.1038/nrc1934
This article is cited by
-
Targeting DDX11 promotes PARP inhibitor sensitivity in hepatocellular carcinoma by attenuating BRCA2-RAD51 mediated homologous recombination
Oncogene (2024)
-
Outer dynein arm docking complex subunit 2 polymorphism rs7893462 modulates hepatocellular carcinoma susceptibility and can serve as an overall survival biomarker for hepatitis B virus-related hepatocellular carcinoma after hepatectomy: a cohort study with a long-term follow-up
World Journal of Surgical Oncology (2023)
-
The renin-angiotensin-aldosterone system (RAAS) signaling pathways and cancer: foes versus allies
Cancer Cell International (2023)
-
Anti-cancer effect of nano-encapsulated boswellic acids, curcumin and naringenin against HepG-2 cell line
BMC Complementary Medicine and Therapies (2023)
-
Loss of TP53 cooperates with c-MET overexpression to drive hepatocarcinogenesis
Cell Death & Disease (2023)
