
Abstract
A new understanding of the genetic basis of coronary artery disease (CAD) has recently emerged from genome-wide association (GWA) studies of common single-nucleotide polymorphisms (SNPs), thus far performed mostly in European-descent populations. To identify novel susceptibility gene variants for CAD and confirm those previously identified mostly in populations of European descent, a multistage GWA study was performed in the Japanese. In the discovery phase, we first genotyped 806 cases and 1337 controls with 451 382 SNP markers and subsequently assessed 34 selected SNPs with direct genotyping (541 additional cases) and in silico comparison (964 healthy controls). In the replication phase, involving 3052 cases and 6335 controls, 12 SNPs were tested; CAD association was replicated and/or verified for 4 (of 12) SNPs from 3 loci: near BRAP and ALDH2 on 12q24 (P=1.6 × 10−34), HLA-DQB1 on 6p21 (P=4.7 × 10−7), and CDKN2A/B on 9p21 (P=6.1 × 10−16). On 12q24, we identified the strongest association signal with the strength of association substantially pronounced for a subgroup of myocardial infarction cases (P=1.4 × 10−40). On 6p21, an HLA allele, DQB1*0604, could show one of the most prominent association signals in an ∼8-Mb interval that encompasses the LTA gene, where an association with myocardial infarction had been reported in another Japanese study. CAD association was also identified at CDKN2A/B, as previously reported in different populations of European descent and Asians. Thus, three loci confirmed in the Japanese GWA study highlight the likely presence of risk alleles with two types of genetic effects – population specific and common – on susceptibility to CAD.
Similar content being viewed by others

Introduction
Coronary artery disease (CAD) and its main complication, myocardial infarction (MI), are the leading causes of death and disability worldwide, with genetic component substantially contributing to the pathogenesis.1 Genome-wide association (GWA) studies of common single-nucleotide polymorphisms (SNPs) have recently been reported to identify susceptibility loci for CAD and/or MI mostly in populations of European descent.2, 3, 4, 5, 6, 7, 8, 9 Although reproducible evidence of disease association has been obtained at a few candidate loci that were originally detected through GWA scan, for example, near the CDKN2A/B gene on chromosome 9p21, in different populations of European descent and Asians,1, 10, 11 the extent to which disease association differs or overlaps between populations remains unknown. Besides comparing the genetic associations between European-descent and Japanese populations, we aim to identify new genetic variants through a GWA study.
Materials and methods
Subjects
Detailed characteristics of the subjects analyzed in each stage of the study are described in Table 1 and Supplementary Note. Our CAD genetic studies were originally organized as part of an ongoing GWA study for cardiometabolic disorders among Japanese subjects. All participants are of Japanese ancestry. Cases were enrolled from clinical practices or annual medical checkups at medical institutions and university hospitals in accordance with the uniformly defined criteria. These criteria included (1) a validated history of either MI (typical electrocardiographic changes; increased serum levels of enzymes/biomarkers including creatine kinase, aspartate aminotransferase, and troponin; a ventricular wall motion abnormality detectable with echocardiography) or coronary revascularization (coronary artery bypass grafting or percutaneous coronary intervention), or (2) subjective symptoms of angina pectoris with ≥1 major coronary vessels showing ≥75% stenosis documented by coronary angiography. Cases were consecutively recruited during a given period from different clinics participating in this cross-sectional study and could be representative for an average CAD patient population in Japan. Controls were deemed free of MI by history, physical examination, and electrocardiogram, also randomly selected from a cross-sectional study of cardiovascular risk factors without matching of cases and controls in the individual recruitment areas; the ratios of cases to controls were not concordant among the areas (Table 1) and the associations were tested separately. In addition to the samples genotyped here, we incorporated genotype frequencies in the general Japanese population (n=964) derived from the Genome Medicine Database of Japan (GeMDBJ; http://gemdbj.nibio.go.jp), which was used as an in silico control panel, to increase the power of the GWA scan. Despite the recent changes in lifestyle, the prevalence estimate (<2%)12 and incidence of CAD in the general Japanese population was much lower than those in Europeans.13 CAD is one of the common diseases globally. Nevertheless, the in silico control panel seems to be useful in the case–control study, although we should take into account the possibility that some samples in this general Japanese population are falsely attributed.
All participants provided written informed consent, and the local ethics committees approved the protocols.
Genome-wide scan
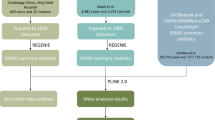
We designed a multistage GWA study of CAD in the Japanese population with three parallel tracks for SNP selection (Figure 1): genome-wide scan at stringent P-value threshold, genome-wide scan at less stringent threshold, plus targeted (or candidate-gene based) genotyping in stage 1 as separately described below. The discovery phase, stage 1, comprised stages 1a and 1b, which involved a combined panel of 1347 CAD cases and 1337 unaffected controls plus 964 general population subjects. In this discovery phase, we adopted a joint analysis strategy;14 that is, 806 cases and 1337 controls were genotyped with the Infinium HumanHap550 or Human610-Quad BeadArray (Illumina, San Diego, CA, USA) in stage 1a, and only the top hit SNPs were followed up for fast track in stage 1b (Supplementary Table I). Here, in stage 1a, five SNPs were genotyped with the TaqMan assay (Life Technologies, Carlsbad, CA, USA) on 12q24 (four SNPs) and 6p21 (one SNP) apart from the whole-genome genotyping assay in order to infer haplotypes using not imputed but assayed data of the SNPs that were in linkage disequilibrium (LD) to lead SNP (Supplementary Table II). SNPs showing significant (P<5 × 10−8) or suggestive (P<5 × 10−5 in the joint sample of stages 1a+1b) evidence of CAD association were forwarded to the replication phase, stage 2, which involved 3052 CAD cases and 6335 unaffected controls (Supplementary Table III). In stages 1b and 2, genotyping was performed with the TaqMan assay. A scheme for selecting SNPs genotyped in stages 1b and 2 is described in Supplementary Note.

Flowchart summarizing the multistage design of the present study. Independent SNPs (r2<0.8, in principle) were selected in the transition from stage 1a to stage 1b. The independency was secured between two parallel tracks for SNP selection in GWA scan (at stringent P-value threshold and at less stringent threshold) as well as within each track. Furthermore, on 6p21 and 12q24, we excluded SNPs that failed to show region-wide significant association with CAD after adjustment for lead SNPs: rs11752643 on 6p21 and rs3782886 on 12q24. Here, a region-wide significance level (P<4.0 × 10−5 for 6p21 and P<2.9 × 10−4 for 12q24) was set by Bonferroni correction (see Supplementary Table VI).
Data cleaning and analysis were performed using PLINK (version 1.06)15 as described elsewhere.16 The λ value for the genomic control was 1.15 in the stage 1a GWA scan samples and the test statistic was adjusted using the genomic-control method.17 To examine the potential influence of population structure on the association results for our GWA study, we further applied the EMMAX model,18 an efficient implementation of a variance component approach to the stage 1a results. Details of SNP genotyping and quality control are described in Supplementary Note.
Imputation of genotypes to the HapMap phase 2 (JPT+CHB) set was carried out using BEAGLE software (version 3.0.4).19 For 964 general Japanese population subjects from GeMDBJ in stage 1b, genotype results were available for SNPs assayed with the Infinium HumanHap550 (but not for imputed SNPs).
Replication study of candidate loci
Along with genome-wide exploration, we aimed to replicate the disease association of 29 SNPs from 29 candidate loci, which had been robustly confirmed in GWA studies of CAD in European-descent populations (Supplementary Table IV).3, 4, 5, 6, 7, 8 We performed screening of these SNPs in the discovery phase sample (Figure 1). In addition, as we found prominent CAD associations of variants other than those previously identified on 6p21 and 12q24 in the Japanese,20, 21 we investigated in the whole sample (4399 CAD cases and 7672 controls) the genetic impacts of 3 SNPs from 3 known genes in the LTA (lymphotoxin-α) cascade – rs1041981 in LTA, rs7291467 in LGALS2, and rs11066001 in BRAP.20, 21, 22
Statistical analyses
SNP association analysis
The SNPs were tested for CAD association by using the Cochran–Armitage trend test. In the replication phase (stage 2), the significance level was set at P<0.05 after adjustment for multiple testing with Bonferroni correction. To summarize the statistical evidence for each SNP across different cohorts and multistage panels, we used the Mantel–Haenszel and inverse variance methods, which were implemented in the rmeta package for the R software (version 2.10.0; http://cran.r-project.org/), and combined the strata according to enrollment sites (Supplementary Note). For novel loci, we considered associations genome-wide significant if they attained P<5 × 10−8. For the loci previously shown to have genome-wide significant associations, we considered a one-tailed P<0.05 (two-tailed P<0.1) as suggestive evidence of replication. In addition, we tested the association of selected SNPs with MI (as well as CAD) between a subgroup of cases with a validated history of MI (n=3003) and controls (n=7672).
Geographical variation
In the stage 1a GWA scan samples, no difference was evident by visual inspection of the multidimensional scaling plots among the enrollment sites – Tokyo, Nagoya, Osaka, and Shimane. However, considering the potential presence of geographical variation within regions of interest on chromosomes, which could result in population structure, we also examined allele frequency differences between the subsets of controls, each derived from separate geographical regions of Japan (Supplementary Note and Supplementary Table V). Statistical significance at SNPs with a P-value of <1 × 10−5 is defined to be suggestive, as reported by Wellcome Trust Case–Control Consortium for the British population (Supplementary Figure I).2
Haplotype phylogeny and positive selection on 12q24
A previous European GWA study claimed significant evidence supporting CAD association and signatures of natural selection in a 1.6-Mb interval on 12q24,9 near which the present Japanese GWA study also identified strong CAD associations at several SNP loci. Using a haplotype-based test, iHS,23 we tested the hypothesis that a long-range, evolutionarily derived haplotype, upon which CAD risk alleles could lie, arose from a positive selection in the Japanese (or East Asians) independently of Europeans.
Imputation of classic human leukocyte antigen (HLA) types on 6p21
A number of SNPs in the extended major histocompatibility complex (MHC) region on 6p21 showed prominent CAD associations in stage 1a as well as suggestive evidence of geographical variation (Supplementary Figure I and Supplementary Table V). To examine the possibility that some classic HLA alleles were responsible for the association signals, we imputed classic HLA types for stage 1a samples using SNP genotypes and HLA alleles (at HLA-A, -B, -C, -DQA1, -DQB1, and -DRB1 loci) of the HapMap JPT (n=44) as a training set (see details in Supplementary Note),24 employing the BEAGLE software (version 3.0. 4).19 In addition, we investigated the accuracy of imputation by comparing the tag SNP genotypes with the data for direct genotyping of HLA alleles in an independent panel of Japanese samples (n=92), which were provided from the Cell Bank at the RIKEN BioResource Center through the National BioResource Project of the Ministry of Education, Culture, Sports, Science and Technology, Japan.25 Alleles at HLA-DRB1 and HLA-DQB1 were determined by the Luminex microbead method (Luminex, Austin, TX, USA).
Results
GWA analysis of CAD
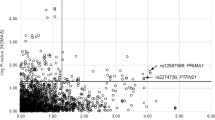
In stage 1a, we observed a cluster of strong association signals in two chromosomal regions (Figure 2 and Supplementary Table VI) 12q24 and 6p21. On 12q24, significant association signals were observed across a ∼0.7-Mb interval encompassing the BRAP and ALDH2 genes (Figure 3 and Supplementary Table VI). On 6p21, significant association was found across an ∼8-Mb interval encompassing the MHC region (Supplementary Figure II and Supplementary Table VI). In analyzing the joint sample of stages 1a+1b, we detected another CAD association near CDKN2A/B on 9p21, whereas SNPs from the other chromosomal regions did not remain after the multistage GWA scan.

Manhattan plots for stage 1 GWA study of CAD in the Japanese. SNPs showing P<5 × 10−8 are colored in red. The color reproduction of this figure is available at the European Journal of Human Genetics journal online.

Plots of CAD association for the 12q24 region. In the top panel, all genotyped SNPs in the current Japanese GWA scan (dot) that passed the quality control in stage 1a, and four additionally genotyped SNPs (cross) that are in strong LD with BRAP rs11066001 are plotted with their –log10(P) for CAD against chromosome positions (in Mb). In the middle panel, the –log10(P) for CAD associations with statistical adjustment for rs11066001 are shown. The bottom panel shows the genomic location of RefSeq genes with intron and exon structures (NCBI Build 36).
In the Japanese, the strongest evidence of CAD association (P=6.9 × 10−30 at rs3782886 in BRAP and P=1.6 × 10−34 at rs671 in ALDH2; summary statistics shown in Table 2) was identified via GWA scan in a long interval on 12q24 (∼0.7 Mb), within which previously reported candidate genes, BRAP and ALDH2,20, 26 were located (Figure 3). This 12q24 region was near the locus recently identified to associate with CAD and/or MI in Europeans;6, 9 a long-range haplotype (1.6 Mb) was hypothesized to have arisen from a positive selection specific to Europeans.9 Notably, a lead SNP in the Japanese GWA study– rs3782886 in BRAP – is polymorphic only in East Asians (Supplementary Figure III) and, conversely, a lead SNP in the European GWA study – rs3184504 in SH2B3 – is polymorphic only in Europeans.9 We retrieved a total of seven SNPs that were in strong LD (r2≥0.8) with rs3782886 and constituted the evolutionarily derived haplotype from the HapMap data (footnote to Supplementary Table II). This haplotype turned out to show CAD association and to have arisen in East Asians independently of Europeans from the phylogenetic viewpoint, in a manner similar to the one that we recently reported for a blood pressure association on 12q24 in East Asians.27
On 6p21, we investigated CAD association across an ∼8-Mb interval encompassing the MHC region with regard to HLA types (Figure 4 and Supplementary Note). The use of high-resolution HLA and SNP haplotype map24 enabled us to identify a significantly associated HLA haplotype, DRB1*1302–DQB1*0604 (Supplementary Tables VII and VIII). We then found that rs11752643 and rs2157339 could tag DQB1*0604 (r2=1.0 in JPT) and DRB1*1302 (r2=0.88 in JPT), respectively, and that these two SNPs were in LD with a candidate SNP rs1041981 in LTA.21 Furthermore, our construction of phylogeny involving the three SNPs (rs11752643, rs2157339, and rs1041981) and association analysis with the resultant haplotypes revealed that an HLA allele tagged by rs11752643 would constitute a principal association signal on 6p21 (Figure 4). Independently, we also found some effects driven by population structure on 6p21 (Supplementary Figure II and Supplementary Table V). This population structure highlighted a cluster of SNPs on 6p21 (Supplementary Table V); they could be categorized into >2 subgroups with regard to LD and only one of them (represented by rs11752643) appeared to be prominent in the test of CAD association (Figure 5). Indeed, with statistical adjustment for rs11752643, most of the association signals on 6p21 were noticeably weakened (Supplementary Figure II and Supplementary Table VI). We estimated the average effect size of rs11752643 to have an odds ratio (OR) of 1.26 (95% confidence interval (95% CI) 1.15–1.38, P=4.7 × 10−7; Table 2 and Supplementary Note).

Overview of SNPs and haplotypes explaining CAD association on 6p21. Genotypic association between SNPs across the 8 Mb extended major histocompatibility complex region and two HLA alleles of interest – DQB1*0604 at HLA-DQB1 (top panels; a–c) and DRB1*1302 at HLA-DRB1 (middle panels; d–f) – is shown for three HapMap populations: JPT (a, d), CEU (b, e), and YRI (c, f). The SNP positions across the 8 Mb region showing weak (r2<0.5; gray), moderate (0.5≤r2<0.8; blue), and strong (r2≥0.8; red) association with each HLA allele are depicted in the individual plots, above which the position of HLA-DQB1 and HLA-DRB1 is indicated by the triangle. In each plot, rs11752643 (tag SNP for DQB1*0604 in JPT (r2=1) and CEU (r2=1)), rs2157339 (tag SNP for DRB1*1302 in JPT (r2=0.88), CEU (r2=1), and YRI (r2=0.87)), and rs1041981 (nonsynonymous SNP in LTA) are circled in black, except that rs11752643 is not shown for YRI because of the lack of polymorphism. Data are derived from the study by de Bakker et al21 and HapMap (http://hapmap.ncbi.nlm.nih.gov/). Haplotype frequencies involving the three tested SNPs and CAD association in the largest district, Nagoya (g), and the phylogeny (h) are also shown. The color reproduction of this figure is available at the European Journal of Human Genetics journal online.

Relation of geographical variation (which may lead to population structure) with CAD association on 6p21. It is speculated that more than two independent subgroups of SNPs are present in the 6p21 region for geographical variation (a); and that one of them, including rs11752643, simultaneously accounts for CAD association and constitutes a genuine association signal (b). Pairwise LD coefficient r2 among 15 SNPs that show geographical variation on 6q21 in the Japanese is demonstrated in (c).
We made additional assessment of population structure in our GWA scan as indicated by a relatively high inflation factor, λ, of 1.15 in stage 1a. We found high reproducibility of association results between two different approaches, the genomic control method (PLINK)17 and the variance component model (EMMAX),18 indicating appropriate correction for the residual inflation of test statistic so as to constrain the risk of false positives and also to prevent the overcorrection that would remove true positives (data not shown).
Replication of previously reported SNPs
Among the CAD-associated loci previously reported in Europeans, strong association (after considering multiple testing, ie, P<0.05/29=0.0017) was replicated at the CDKN2A/B locus in the Japanese population (Table 2 and Supplementary Table IV). Despite finding significant association for rs4977574 at CDKN2A/B (P=1.4 × 10−5; Supplementary Table IV) in the candidate-gene-based genotyping, we proceeded with rs944797 instead of rs4977574 in the GWA scans from stages 1a+1b to stage 2; this decision was made considering the strong LD between the two SNPs (r2=0.94 in stage 1a). Suggestive evidence of replication (two-tailed P<0.1 in the case of concordant direction of association) was observed for five other loci – CELSR2-SORT1, 7q22, CNNM2-NT5C2, HHIPL1, and SMG6-SRR – for CAD association (Supplementary Table IV).
From known variants in the LTA cascade, we tested association of rs7291467 (in LGALS2) and rs11066001 (in BRAP) with CAD and MI, as previously reported in the Japanese.20, 22 The strength of association of rs11066001 was almost equivalent to that of rs671 (in ALDH2), which showed the strongest association signal on 12q24, and more prominent with MI than non-MI CAD (OR=1.53, 95% CI 1.44–1.63 and P=6.9 × 10−40 for MI; OR=1.19, 95% CI 1.09–1.31 and P=9.0 × 10−5 for non-MI CAD). No significant association was found for rs7291467 regardless of the MI status (Supplementary Table IX).
Discussion
We conducted a GWA study on the Japanese population with greater coverage of common SNPs (87% of all phase I+II HapMap variants (minor allele frequency ≥0. 05) in CHB+JPT) and a larger number of cases (806 subjects) and unaffected controls (1337 subjects) in the initial screen than a previous study,21 although our sample size is rather modest by the current standard of GWA studies (and meta-analyses) of CAD or MI in populations of European descent.2, 3, 4, 5, 6, 7, 8 Three loci – 12q24, 6p21, and 9p21 – were successfully identified through the multistage scan; notably, the 6p21 locus has not been claimed to show significant CAD association in the GWA meta-analyses of Europeans.5, 7, 8 The 12q24 locus appeared to overlap with the one reported in Europeans, while there may be haplotypic heterogeneity.6, 9 In addition, the present genome-wide exploration identified significant association near CDKN2A/B in East Asians, as previously reported in European-descent populations.1, 2, 5, 7, 8 Of the three loci, which we claim to be associated with CAD in the Japanese GWA study (Table 2), the disease association at two loci – 12q24 and 9p21 – have been reported in several Asian populations10, 11, 26 and could be regarded confirmatory. Nevertheless, two novel findings are noted in our study: (1) the other locus within an HLA gene, HLA-DQB1, which is considered to explain the previous descriptions of CAD (in particular, MI) association signals on 6p21 in the Japanese21 and (2) a pronounced association with MI, as compared with CAD, for the variants on 12q24.
The 12q24 haplotype confers risk alleles for CAD much more significantly in the Japanese than in Europeans.6, 9 A previous Japanese study reported significant association with MI risk at SNPs (including rs3782886 and rs11066001) in BRAP, located on 12q24.20 The authors investigated BRAP (BRCA1-associated protein) as a candidate gene because of its potential involvement in the cytokine LTA cascade; that is, BRAP is a possible binding partner of galectin-2, which is encoded by LGALS2 that can bind to LTA. The same study group originally claimed significant association with MI risk at the LTA locus on 6p21 in the Japanese,21 as discussed below. Whether a single variant or multiple variants can be present on the relevant 12q24 haplotype remains unknown. In our study, association signals at two SNPs on 12q24 – rs11066001 (in BRAP) and rs671 (in ALDH2) – were in almost complete LD (r2=0.99 in the whole sample) and could not be distinguished from each other. It is possible that some molecular variant(s) in either of the two genes or other genes that are contained in the region covered by the long-range haplotype (1.5 Mb)27 underlie susceptibility to CAD. Besides BRAP, one such candidate is ALDH2, encoding the aldehyde dehydrogenase 2; the active and inactive subunits of the enzyme are encoded by two alleles of rs671, which showed the most significant evidence of association (P=1.6 × 10−34). Using a proteomic search, Chen et al28 found that enzyme activation of ALDH2 correlated with reduced ischemic heart damage in rodent models, in accordance with the results of CAD association. With the overlapping association signals being detected, further in-depth analysis of the 12q24 region will be required to dissect the phylogenetic relationship of CAD causality between the two ethnic groups.
The second strongest association was identified via GWA scan for a cluster of SNPs in the extended MHC region on 6p21, where an HLA allele, DQB1*0604 (tagged by rs11752643), could show one of the most prominent association signals (Figure 5 and Supplementary Figure II). These findings have brought up an issue of disease association at LTA21 and candidate genes in LTA cascade, such as LGALS2.22 On 6p21, the association signal of rs1041981 (in LTA) has proven to come from that of rs11752643 via LD (D'=1.0, r2=0.03) in the present study (Figure 4). Also, we could not detect significant association at a SNP, rs7291467, previously reported in LGALS2 on 22q13 (OR 1.02, 95% CI 0.96–1.07, P=0.61). This is in good accordance with a pooled OR of 1.02 (95% CI, 0.99–1.06, P=0.23) that we estimated by meta-analysis involving all studies reported to date,29 except for the original one,22 with consideration of the winner's curse effect30 (Supplementary Figure IV). Together, the disease association for SNPs in LTA cascade genes may be less outstanding than what was originally expected, although further investigation is warranted to address this issue.
Because of the low frequency of DQB1*0604 allele (or a proxy SNP, rs11752643; 0.02 in HapMap CEU; Supplementary Table VIII and Supplementary Figure V), a large number of samples (n >9600) would be required to verify the equivalent strength of CAD association on 6p21 in Europeans. Despite the sufficient sample size (>14 000 cases in the discovery phase),7, 8 neither of two recent large-scale meta-analyses in Europeans could show significant association on 6p21, suggesting the absence of susceptibility locus, at least, with the equivalent effect size. Thus far, positive disease association with the DQB1*0604 allele has been reported only for myasthenia gravis.31 It is noteworthy that suggestive evidence of association has been recently observed between HLA-DRB1 and DQA1 loci and acute MI in a population-based Swedish cohort,32 whereas the reported risk alleles are not in LD with DQB1*0604.
Moreover, this study identified CAD association at another locus, near the CDKN2A/B gene, originally claimed by GWA scan in Europeans. The effect size for CAD was almost comparable between the populations: OR=1.25 and 1.29 for the Japanese and Europeans,5, 7, 8 respectively. Because of the limitation of discovery phase sample size (stages 1a+1b), statistical power is insufficient (power <0.7) to refute the disease association at 19 of 23 candidate loci previously identified in GWA studies of Europeans, except CDKN2A/B and 5 other loci showing suggestive evidence of replication (Supplementary Table IV).
Although GWA studies have thus far made major steps in unraveling the genetics of cardiovascular disease, biological explanations for the identified associations remain largely unknown. In this context, it has been hypothesized that different genetic risk factors may contribute to either CAD or MI given the complex nature of transition from a normal coronary artery to MI. Indeed, a recent GWA meta-analysis33 has addressed this issue and has indicated the presence of genetic predispositions leading to MI (in the presence of coronary atherosclerosis) as well as those shared between CAD and MI (promoting coronary atherosclerosis). The present findings of SNP–trait associations can support this notion; that is, the strength of association is prominent for MI at BRAP/ALDH2 on 12q24, whereas it is comparable between the disease entities at CDKN2A/B on 9q21 (Supplementary Table IX).
Our GWA study results have called our attention to sample admixture with geographical variation, a potential source of population structure. Because the bias introduced by population structure may not be as significant as once feared, performing GWA studies in admixed populations was suggested to be a reasonable strategy in increasing sample sizes with the necessity of independent replication.34 Nevertheless, as has been stated by previous GWA studies in Europeans,2 apparent disease associations in the few genomic regions identified as showing geographical differentiation need to be interpreted with caution. This seems to be the case with the 6p21 region, where more than two independent subgroups of SNPs are present for geographical variation and one of them, rs11752643, appears to simultaneously account for CAD association even after adjustment for geographical variation (Figure 5, Supplementary Note, Supplementary Figure II and Supplementary Table IX).
Just like most other GWA studies on CAD conducted to date, the present study has all the potential biases of cross-sectional analysis, most importantly a survival bias, as recently pointed out by prospective studies.35, 36 Also, although we attempted to explore CAD susceptibility loci with rather prominent effect sizes, the power cannot be necessarily sufficient; for example, the power to detect OR=1.2 is very low (Supplementary Table X). We should keep these limitations in mind when we interpret the genetic association results.
In conclusion, our GWA study has confirmed that three loci in three chromosomal regions are associated with CAD in the Japanese. These loci highlight the likely presence of risk alleles with largely two types of genetic effects – population specific and cosmopolitan – on susceptibility to CAD. The integration of multiethnic results will promote a better understanding of the global genetic architecture of CAD.
References
Schunkert H, Erdmann J, Samani NJ : Genetics of myocardial infarction: a progress report. Eur Heart J 2010; 31: 918–925.
Wellcome Trust Case Control Consortium: Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007; 447: 661–978.
Trégouët DA, König IR, Erdmann J et al: Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet 2009; 41: 283–285.
Erdmann J, Grosshennig A, Braund PS et al: New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet 2009; 41: 280–282.
Myocardial Infarction Genetics Consortium: Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet 2009; 41: 334–341.
Gudbjartsson DF, Bjornsdottir US, Halapi E et al: Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat Genet 2009; 41: 342–347.
Schunkert H, König IR, Kathiresan S et al: Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet 2011; 43: 333–338.
Coronary Artery Disease (C4D) Genetics Consortium: A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet 2011; 43: 339–344.
Soranzo N, Spector TD, Mangino M et al: A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet 2009; 41: 1182–1190.
Shen GQ, Li L, Rao S et al: Four SNPs on chromosome 9p21 in a South Korean population implicate a genetic locus that confers high cross-race risk for development of coronary artery disease. Arterioscler Thromb Vasc Biol 2008; 28: 360–365.
Saleheen D, Alexander M, Rasheed A et al: Association of the 9p21.3 locus with risk of first-ever myocardial infarction in Pakistanis: case-control study in South Asia and updated meta-analysis of Europeans. Arterioscler Thromb Vasc Biol 2010; 30: 1467–1473.
Moriyama Y, Okamura T, Inazu A et al: A low prevalence of coronary heart disease among subjects with increased high-density lipoprotein cholesterol levels, including those with plasma cholesteryl ester transfer protein deficiency. Prev Med 1998; 27: 659–667.
Okamura T : Dyslipidemia and cardiovascular disease: a series of epidemiologic studies in Japanese populations. J Epidemiol 2010; 20: 259–265.
Skol AD, Scott LJ, Abecasis GR, Boehnke M : Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet 2006; 38: 209–213.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Takeuchi F, Serizawa M, Yamamoto K et al: Confirmation of multiple risk loci and genetic impacts by a genome-wide association study of type 2 diabetes in the Japanese population. Diabetes 2009; 58: 1690–1699.
Devlin B, Roeder K : Genomic control for association studies. Biometrics 1999; 55: 997–1004.
Kang HM, Sul JH, Service SK et al: Variance component model to account for sample structure in genome-wide association studies. Nat Genet 2010; 42: 348–354.
Browning BL, Browning SR : A unified approach to genotype imputation and haplotype phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet 2009; 84: 210–223.
Ozaki K, Sato H, Inoue K et al: SNPs in BRAP associated with risk of myocardial infarction in Asian populations. Nat Genet 2009; 41: 329–333.
Ozaki K, Ohnishi Y, Iida A et al: Functional SNPs in the lymphotoxin-alpha gene that are associated with susceptibility to myocardial infarction. Nat Genet 2002; 32: 650–654.
Ozaki K, Inoue K, Sato H et al: Functional variation in LGALS2 confers risk of myocardial infarction and regulates lymphotoxin-alpha secretion in vitro. Nature 2004; 429: 72–75.
Pickrell JK, Coop G, Novembre J et al: Signals of recent positive selection in a worldwide sample of human populations. Genome Res 2009; 19: 826–837.
de Bakker PI, McVean G, Sabeti PC et al: A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet 2006; 38: 1166–1172.
Iwakawa M, Goto M, Noda S et al: DNA repair capacity measured by high throughput alkaline comet assays in EBV-transformed cell lines and peripheral blood cells from cancer patients and healthy volunteers. Mutat Res 2005; 588: 1–6.
Takagi S, Iwai N, Yamauchi R et al: Aldehyde dehydrogenase 2 gene is a risk factor for myocardial infarction in Japanese men. Hypertens Res 2002; 25: 677–681.
Kato N, Takeuchi F, Tabara Y et al: Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nat Genet 2011; 43: 531–538.
Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D : Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 2008; 321: 1493–1495.
Li W, Xu J, Wang X et al: Lack of association between lymphotoxin-alpha, galectin-2 polymorphisms and coronary artery disease: a meta-analysis. Atherosclerosis 2010; 208: 433–436.
Zollner S, Pritchard JK : Overcoming the winner's curse: estimating penetrance parameters from case-control data. Am J Hum Genet 2007; 80: 605–615.
Deitiker PR, Oshima M, Smith RG, Mosier DR, Atassi MZ : Subtle differences in HLA DQ haplotype-associated presentation of AChR alpha-chain peptides may suffice to mediate myasthenia gravis. Autoimmunity 2006; 39: 277–288.
Björkbacka H, Lavant EH, Fredrikson GN et al: Weak associations between human leucocyte antigen genotype and acute myocardial infarction. J Intern Med 2010; 268: 50–58.
Reilly MP, Li M, He J et al: Identification of ADAMTS7 as a novel locus for coronary atherosclerosis and association of ABO with myocardial infarction in the presence of coronary atherosclerosis: two genome-wide association studies. Lancet 2011; 377: 383–392.
Hao K, Chudin E, Greenawalt D, Schadt EE : Magnitude of stratification in human populations and impacts on genome wide association studies. PLoS One 2010; 5: e8695.
Karvanen J, Silander K, Kee F et al: The impact of newly identified loci on coronary heart disease, stroke and total mortality in the MORGAM prospective cohorts. Genet Epidemiol 2009; 33: 237–246.
Ripatti S, Tikkanen E, Orho-Melander M et al: A multilocus genetic risk score for coronary heart disease: case-control and prospective cohort analyses. Lancet 2010; 376: 1393–1400.
Acknowledgements
We thank all the people who have continuously supported the Hospital-based Cohort Study at the National Center for Global Health and Medicine, the Amagasaki Study and the Kyushu University Fukuoka Cohort Study, and the KING Study. We also thank Drs Suminori Kono, Ken Sugimoto, Kei Kamide, and Chikanori Makibayashi, and the many physicians of the participating hospitals and medical institutions for their assistance in collecting the DNA samples and accompanying clinical information. This work was supported by grants for the Core Research for Evolutional Science and Technology (CREST) from the Japan Science Technology Agency; the Program for Promotion of Fundamental Studies in Health Sciences, National Institute of Biomedical Innovation Organization (NIBIO); and KAKENHI (Grant-in-Aid for Scientific Research) on Priority Areas ‘Applied Genomics’ from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Takeuchi, F., Yokota, M., Yamamoto, K. et al. Genome-wide association study of coronary artery disease in the Japanese. Eur J Hum Genet 20, 333–340 (2012). https://doi.org/10.1038/ejhg.2011.184
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.184
Keywords
This article is cited by
-
Genetic associations of cardiovascular risk genes in European patients with coronary artery spasm
Clinical Research in Cardiology (2024)
-
The role of aldehyde dehydrogenase 2 in cardiovascular disease
Nature Reviews Cardiology (2023)
-
Uncovering myocardial infarction genetic signatures using GWAS exploration in Saudi and European cohorts
Scientific Reports (2023)
-
East Asian variant aldehyde dehydrogenase type 2 genotype exacerbates ischemia/reperfusion injury with ST-elevation myocardial infarction in men: possible sex differences
Heart and Vessels (2022)
-
Plasminogen Activator Inhibitor-1 Polymorphisms and Risk of Coronary Artery Disease: Evidence From Meta-Analysis and Trial Sequential Analysis
Biochemical Genetics (2022)
