
Abstract
In the brain, programmed cell death (PCD) serves to adjust the numbers of the different types of neurons during development, and its pathological reactivation in the adult leads to neurodegeneration. Dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A (DYRK1A) is a pleiotropic kinase involved in neural proliferation and cell death, and its role during brain growth is evolutionarily conserved. Human DYRK1A lies in the Down syndrome critical region on chromosome 21, and heterozygous mutations in the gene cause microcephaly and neurological dysfunction. The mouse model for DYRK1A haploinsufficiency (the Dyrk1a+/− mouse) presents neuronal deficits in specific regions of the adult brain, including the substantia nigra (SN), although the mechanisms underlying these pathogenic effects remain unclear. Here we study the effect of DYRK1A copy number variation on dopaminergic cell homeostasis. We show that mesencephalic DA (mDA) neurons are generated in the embryo at normal rates in the Dyrk1a haploinsufficient model and in a model (the mBACtgDyrk1a mouse) that carries three copies of Dyrk1a. We also show that the number of mDA cells diminishes in postnatal Dyrk1a+/− mice and increases in mBACtgDyrk1a mice due to an abnormal activity of the mitochondrial caspase9 (Casp9)-dependent apoptotic pathway during the main wave of PCD that affects these neurons. In addition, we show that the cell death induced by 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine (MPTP), a toxin that activates Casp9-dependent apoptosis in mDA neurons, is attenuated in adult mBACtgDyrk1a mice, leading to an increased survival of SN DA neurons 21 days after MPTP intoxication. Finally, we present data indicating that Dyrk1a phosphorylation of Casp9 at the Thr125 residue is the mechanism by which this kinase hinders both physiological and pathological PCD in mDA neurons. These data provide new insight into the mechanisms that control cell death in brain DA neurons and they show that deregulation of developmental apoptosis may contribute to the phenotype of patients with imbalanced DYRK1A gene dosage.
Similar content being viewed by others


Main
The total number of neurons in the brain, and ultimately the size of this organ, depends both on the number of cells that are produced during neurogenesis and the number of neurons that die due to physiological programmed cell death (PCD). Dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A (DYRK1A) regulates brain growth in a dose-dependent manner,1 and indeed, loss-of-function mutations in DYRK1A (minibrain in Drosophila melanogaster) cause microcephaly and several neurological alterations in humans,2, 3, 4, 5 mice6 and flies.7 Accordingly, it has been proposed that haploinsufficiency of DYRK1A is the cause of the microcephaly and developmental delay associated to partial monosomy of chromosome 21 involving DYRK1A.8 Moreover, triplication of the gene has been associated to the developmental brain dysfunctions and age-associated neurodegeneration observed in Down syndrome.9, 10, 11
Anatomical analysis of adult Dyrk1a mutant mice that model human diseases involving an imbalance in DYRK1A gene dosage (the Dyrk1a+/− mouse and the mBACtgDyrk1a mouse, carrying one or three functional copies of Dyrk1a, respectively) revealed a positive correlation between Dyrk1a gene copy number, the overall size of the brain and the number of neurons in specific regions.1 DYRK1A regulates several fundamental neurodevelopmental processes, including proliferation, neuron differentiation and PCD.12 Overexpression of DYRK1A in neural precursors attenuates proliferation and promotes the differentiation of neurons in different model systems.13, 14, 15 Conversely, treatment of neural progenitors with DYRK1A kinase inhibitors increases proliferation.15 Although these data are consistent with some of the defects in cellularity identified in specific brain regions of Dyrk1a gene copy number mutants, they cannot explain the severe microcephaly evident in mice and humans carrying one functional copy of DYRK1A, or the overall macrocephaly in the mBACtgDyrk1a model carrying three Dyrk1a alleles.1, 5 Thus, deregulation of other DYRK1A functions might also contribute to the defects in brain cellularity in these Dyrk1a gene copy number mutants, such as those described in retinal neurons that restrain developmental PCD.16
Dopaminergic (DA) neurons in the substantia nigra (SN) and ventral tegmental area (VTA) have an important role in controlling fine motor actions, as well as in motivation and reward behaviours, and their loss is associated with Parkinson’s disease.17 In aged Dyrk1a+/− mice the SN is smaller and contains fewer DA neurons than in wild-type mice.18 These mutant animals are hypoactive, with altered gait dynamics, and as these defects are evident preweaning and in young animals,6, 18, 19 as well as in children with heterozygous mutations in DYRK1A,3, 4, 5 they might arise during development.
To provide insight into the aetiology of the neurological phenotype caused by DYRK1A haploinsufficiency, here we studied the development of mesencephalic DA (mDA) neurons in Dyrk1a+/− and mBACtgDyrk1a mouse models. The results obtained show that Dyrk1a copy number variation does not affect the generation of DA neurons, but rather it modifies the number of these neurons that undergo physiological PCD due to an inhibitory effect of the Dyrk1a kinase on the apoptotic activity of caspase9 (Casp9), the initiator caspase in the mitochondrial-dependent apoptotic pathway.20 Thus, deregulation of Casp9-dependent PCD during development may contribute to the brain size defects observed in aneuploidies involving DYRK1A.
As inappropriate re-activation of the mitochondrial-dependent apoptotic pathway in mature mDA neurons contributes to the neurodegeneration associated with Parkinson’s disease,21 we used the mBACtgDyrk1a mouse model to assess whether basal Dyrk1a-dependent inhibition of Casp9 apoptotic activity could restrain the neurodegeneration induced in vivo by the parkinsonian neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Our results show that the apoptotic response to the toxin in mBACtgDyrk1a mice is significantly attenuated, leading to an increase in the number of SN pars compacta DA neurons that resist the pathological insult.
Results
Dyrk1a promotes the survival of mDA neurons during the physiological period of PCD
DA neurons in the SN and VTA originate from progenitors in the ventral neuroepithelium and they subsequently migrate radially to the mantle zone where they differentiate. The distribution of the neurons can be tracked by following that of tyrosine hydroxylase (TH), a marker of DA neurons that is initially detected in mice at around embryonic day (E)1022, 23 (see scheme in Figure 1a). As the distribution of Dyrk1a protein in the developing ventral mesencephalon is unknown, we used a Dyrk1a-specific antibody16, 24 to analyse the expression of the protein in this region at two developmental stages: at E12.5, during the phase of DA neurogenesis, and at postnatal day (P)0, when neurogenesis has been completed and the excess DA neurons generated are removed by PCD25 (see scheme in Figure 2a). In the embryo, Dyrk1a protein accumulates in the cytoplasm of post-mitotic cells within the intermediate zone and in cells that already express TH in the mantle zone (Figure 1b). By P0, Dyrk1a was also evident in differentiating neurons that express TH in the SN (Figure 1c) and VTA (data not shown). This distribution is compatible with Dyrk1a participating in the embryonic and postnatal development of mDA neurons, supporting the possibility that the deficit in SN DA neurons observed previously in 2-year-old Dyrk1a+/− mice arises during development.18

Expression of the Dyrk1a protein in the mouse ventral mesencephalon during development. (a) Schematic sagittal view of the mouse embryonic neural tube (E11.5 to E14.5) indicating the site of DA neurogenesis in the mesencephalon (yellow area) and the position occupied by DA progenitors (blue), immature DA neurons (TH− cell in yellow) and mature DA neurons (TH+ cells in red) in a transverse section at the level indicated by the black rectangle. (b and c) Confocal images showing co-localization of Dyrk1a (D1a in green) and TH (red) in the ventral mesencephalon of wild-type E12.5 embryos (b) and P0 mice (c). HIND, hindbrain; IZ, intermediate zone; MES, mesencephalon; MZ, mantle zone; TEL, telencephalon; VZ, ventricular zone. Bar=50 μm

TH+ cell counts in the mesencephalon of Dyrk1a mutant mice at different developmental stages. (a) Scheme indicating the period of DA neuron production (green) and of PCD (purple) in the mouse ventral mesencephalon.27 (b–g) Representative mesencephalic sections stained for TH from Dyrk1a+/+ (D1a+/+) and Dyrk1a+/− (D1a+/−) brains (b–d) and from wild-type (WT) and mBACtgDyrk1a (BACD1a) brains (e–g) at the developmental stages indicated, and total number of TH+ cells in the whole ventral mesencephalon (VTA+SN; (b and e), or in the VTA and the SN alone (c, d, f and g). Histogram values are the mean±S.E.M. *P≤0.05; **P≤0.01; ***P≤0.001; n=3 in (b, e and f); n=4 in (c and g); n=5 in (d). Bar=100 μm (b, c, e and f), 300 μm (d and g)
To test this possibility, we quantified the number of cells expressing TH in the mesencephalon of Dyrk1a+/+ and Dyrk1a+/− embryos at E14.5, soon after the period of DA neurogenesis in the ventral mesencephalon (Figure 2a). The number and distribution of TH+ neurons was similar in both these genotypes (Figure 2b), indicating that mDA neurons in Dyrk1a+/− embryos are produced normally. Moreover, these neurons seemed to differentiate correctly in the Dyrk1a+/− mutants, given that they expressed nuclear-orphan-receptor-1 (Nurr1), a transcription factor that is required for the maintenance of DA neurons,23 and that they innervated the striatum at E16.5, the correct stage of development26 (Supplementary Figure S1). Thus, the generation and specification of mDA neurons appear to be unaltered in Dyrk1a+/− mice.
The number of mesencephalic TH+ neurons in Dyrk1a+/− mutants remained normal at P0 (VTA+SN: Dyrk1a+/+=18 877±2364 and Dyrk1a+/−=18 434±697; P=0.86; n=4). However, when we assessed the number of these neurons in the mesencephalon of Dyrk1a+/+ and Dyrk1a+/− mice at P7, subsequent to the major programmed wave of DA neuron cell death (Figure 2a), fewer TH+ cells were detected in Dyrk1a mutant mice (37% less) than in wild-type mice (Figure 2c). Despite the PCD that occurs, the number of mesencephalic TH+ neurons increases during postnatal development,27 and indeed, TH+ cells were more abundant in young (8-week-old) Dyrk1a+/+ mice than at P7. A similar age-dependent increase in TH+ neurons was observed in 8-week-old Dyrk1a+/− mice, although there were fewer of these neurons in mutant animals (Figures 2c and d). Importantly, the reduction in TH+ cells at this age (38%) was similar to the reduction of SN DA neurons reported previously in aged Dyrk1a+/− mice.18 In both young Dyrk1a+/+ and Dyrk1a+/− mice the mesencephalic neurons that expressed the dopamine active transporter (DAT) also contained TH (Supplementary Figure S2). Moreover, we did not find differences between genotypes in the proportion of Nissl-stained neurons that contained TH (Dyrk1a+/+: SN=0.43±0.03; VTA=0.43±0.03 and Dyrk1a+/−: SN=0.46±0.01; VTA=0.46±0.04; n=4), indicating that Dyrk1a haploinsufficiency does not affect TH expression, but rather that it reduces the number of DA cells expressing this enzyme. Together, these results suggest that the loss of DA neurons in Dyrk1a+/− mice occurs during the major wave of developmental cell death in these cells.
To further assess how Dyrk1a influences DA neuron development, we performed a similar analysis in the mBACtgDyrk1a model that expresses three copies of the mouse Dyrk1a gene.1 As observed in the Dyrk1a+/− model, the number of TH+ cells in the mesencephalon of mBACtgDyrk1a mice was normal at E14.5 (Figure 2e) but not at P7, when there were significantly more DA neurons in the mutant (Figure 2f). However, and in contrast to what happens in the Dyrk1a+/− mouse, young mBACtgDyrk1a mice had normal numbers of DA neurons in both the SN and VTA (Figure 2g). The second wave of PCD in mDA neurons takes place during the second and third week of life25 (Figure 2a), when synaptogenesis is maximal in the developing striatum.28 Therefore, the excess of DA neurons in P7 mBACtgDyrk1a mice may be offset by the exacerbated wave of cell death during this period, possibly due to limited neurotrophic support of target neurons.27 This is supported by the fact that P21 mBACtgDyrk1a and wild-type animals have similar numbers of SN TH+ neurons (8588±813 versus 8388±659; P=0.85; n=4).
Overall, our results indicate that changes in Dyrk1a dosage do not affect the generation and differentiation of mouse mDA neurons but they modify the number of these neurons that die during the main wave of DA PCD. Such cell death is increased in the loss-of-function mutant and decreased in the gain-of-function mutant. This protective effect of Dyrk1a might be mediated through its kinase activity, as harmine, an inhibitor of the enzymatic activity of DYRK1A,9 induces a dose-dependent reduction of TH+ neurons in primary mesencephalic cell cultures (Figure 3a).

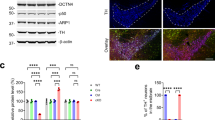
Effect of the DYRK1A inhibitor harmine on DA neuron survival and the levels of phosphorylated Casp9 in the developing ventral mesencephalon of Dyrk1a mutant mice. (a) Representative images of E12 mesencephalic cells after 4 days in culture (DIV) stained for TH (red), and the percentage of TH+ cells in cultures maintained in the presence or absence (NT) of harmine. (b) Scheme indicating the effect of Dyrk1a phosphorylation on the intrinsic cell death pathway. (c and d) Representative western blottings and their quantifications showing the levels of Dyrk1a (D1a) normalized to the amount of Vinculin (Vinc), and the proportion of Casp9 that is Thr125 phosphorylated (P(Thr125)) in total mesencephalic extracts from P2 Dyrk1a+/+ (D1a+/+) and Dyrk1a+/− (D1a+/−) mice (c), and from wild-type (WT) and mBACtgDyrk1a (BACD1a) mice (d). Mito, mitochondria. Histogram values are the mean±S.E.M. *P≤0.05; **P≤0.01 (n=3 in (a); n=4 in (c and d))
Dyrk1a promotes the survival of mDA neurons during development by restricting the activity of the intrinsic apoptotic pathway
The first wave of post-natal PCD in rodents involves the activation of the intrinsic (mitochondrial-dependent) cell death pathway.25 In this pathway, the release of cytochrome c from the mitochondria to the cytosol that is induced by apoptotic stimuli leads to Casp9 cleavage, which promotes cell death by activating downstream executioner caspases.20, 29 There is evidence that phosphorylation of the Thr125 residue in Casp9 prevents its cleavage in these circumstances and, hence, the subsequent activation of the intrinsic apoptotic pathway.30 DYRK1A is known to phosphorylate Casp9 at Thr125 (Laguna et al.16 and Seifert et al.31) and this phosphorylation is crucial to protect differentiating retinal neurons from mitochondrial-dependent cell death.16 To determine whether phosphorylation of Casp9 by Dyrk1a is involved in the protection of DA neurons in the developing ventral mesencephalon, we used a phospho-site-specific antibody and quantified the amount of Thr125-phosphorylated Casp9 in the ventral mesencephalon of Dyrk1a+/−, mBACtgDyrk1a and wild-type littermate mice during the main wave of developmental DA neuron cell death, by P2 (Figure 2a). In mesencephalic extracts derived from Dyrk1a+/− animals, there was significantly less Dyrk1a protein, which was correlated with a significant decrease in the pool of Thr125-phosphorylated Casp9 (Figure 3c). Conversely, there was an increase in the amount of Dyrk1a protein in mBACtgDyrk1a extracts that was correlated with an increase in the pool of phosphorylated Casp9 (Figure 3d).
We assessed the activity of the Casp9-dependent apoptotic pathway in the mesencephalon of postnatal Dyrk1a mutant mice, studying the distribution of the active (cleaved) forms of Casp9 and its downstream executioner caspase3 (Casp3) in the TH-immunostained mesencephalic region of Dyrk1a+/−, mBACtgDyrk1a and wild-type littermate mice. In wild-type animals, active Casp9 and Casp3 immunostaining can be observed in apoptotic TH+ cells at P0 (Figure 4a). As expected, there were significantly more apoptotic cells expressing active Casp9 in Dyrk1a+/− mice (Figure 4b) and fewer in mBACtgDyrk1a mice (Figure 4c). Consistently, there were more apoptotic cells expressing active Casp3 in Dyrk1a+/− animals (Figure 4d) and fewer in mBACtgDyrk1a animals (Figure 4e). Together, these results indicate that the defects in DA cellularity observed in P7 Dyrk1a mutant mice arise from the aberrant activity of Casp9-mediated apoptosis, and that Thr125 phosphorylation of Casp9 by Dyrk1a can regulate cell death in these neurons.

Numbers of apoptotic cells in the ventral mesencephalon of newborn Dyrk1a mutant mice. (a) Confocal images from brain sections of a P0 wild-type (WT) mouse showing TH expression in mesencephalic neurons that contain active Casp9 (A-Casp9) or active Casp3 (A-Casp3), and total numbers of A-Casp9+ cells (b and c) and A-Casp3+ cells (d and e) in the ventral mesencephalon (VTA+SN) of P0 Dyrk1a+/+ (D1a+/+) and Dyrk1a+/− (D1a+/−) mice (b and d), and of wild-type (WT) and mBACtgDyrk1a (BACD1a) mice (c and e). Histogram values are the mean±S.E.M. *P≤0.05; **P≤0.01; ***P≤0.001 (n=4 in (b and d); n=3 in (c and e))
Dyrk1a overexpression promotes the survival of SN pars compacta DA neurons in the MPTP-induced mouse model of Parkinson disease
There is evidence that the intrinsic apoptotic pathway participates in the neurodegeneration of SN pars compacta DA neurons in Parkinson’s disease. Indeed, dying neurons in post-mortem Parkinson’s tissue display morphological characteristics of apoptosis, including the activation of Casp9 and Casp3, cell shrinkage, chromatin condensation and DNA fragmentation.21, 32 Moreover, Dyrk1a protein is still found in the cytoplasm of TH+ neurons of the SN pars compacta after development (Figure 5a, upper panels). Taking into account the pro-survival action of Dyrk1a on developing DA neurons reported here (Figure 4), we hypothesized that Dyrk1a could restrain the neurodegenerative response in a parkinsonian context. To test this possibility we assessed the effect of the parkinsonian neurotoxin MPTP33 on adult mBACtgDyrk1a mice, which still contain relatively large amounts of Dyrk1a protein and Thr125-phosphorylated Casp9 in the ventral mesencephalon (Figure 5b), while establishing a normal distribution of Dyrk1a in their SN (Figure 5a).

Expression of Dyrk1a and the levels of phosphorylated Casp9 in the ventral mesencephalon of adult mBACtgDyrk1a (BACD1a) mice. (a) Confocal images showing a similar Dyrk1a distribution in the SN of 8-week-old (8W) wild-type (WT) and BACD1a mice. Bar=100 μm. (b) Representative western blottings and their quantifications showing the levels of Dyrk1a (D1a) normalized to the amount of Vinculin (Vinc), and the proportion of Casp9 that is Thr125 phosphorylated (P(Thr125)) in total mesencephalic extracts prepared from 12- to 16-week-old WT and BACD1a littermates. Histogram values are the mean±S.E.M. *P≤0.05 (n=5)
Given the importance of the mitochondria in the pathogenesis of Parkinson’s disease32 and the multiple functions of DYRK1A,34 we assessed the status of the mitochondria in these mice. The relative levels of mitochondrial components, including cytochrome c oxidase complex subunits, were similar in the mesencephalon of adult mBACtgDyrk1a and wild-type mice (Supplementary Figure S3). Likewise, there were no differences in oxygen consumption in isolated brain mitochondria from either wild-type or mBACtgDyrk1a mice (Supplementary Figure S4a). With regard to the sensitivity of brain mitochondria to 1-methyl-4-phenylpyridinum ion, the active MPTP metabolite, similar inhibition of ADP-stimulated oxygen consumption supported by complex I substrate glutamate/malate (respiration, state 3) was observed in mBACtgDyrk1a and wild-type mitochondria. In addition, both genotypes produced similar amounts of reactive oxygen species and displayed similar levels of mitochondria antioxidant proteins (Supplementary Figures S4b and c). Together, these data indicate that mitochondrial biogenesis and function is well preserved in mBACtgDyrk1a mice, and that the inhibitory action of the parkinsonian toxin MPTP on the mitochondria is not affected in these animals.
We then challenged mBACtgDyrk1a mice and their wild-type littermates with a sub-acute regime of MPTP (30 mg/kg/day for five consecutive days: see schedule in Figure 6a), a protocol known to kill SN pars compacta DA neurons by activating mitochondrial-dependent apoptotic pathways in a time-dependent manner, peaking 4 days after the last MPTP injection.35, 36, 37 Basal levels of both Dyrk1a protein and phosphorylated Casp9 in these animals did not change after MPTP treatment (Supplementary Figure S5). Yet, at the peak of MPTP-induced apoptotic cell death, pyknotic cells and cells expressing active Casp9 were clearly detected in wild-type and mBACtgDyrk1a mice (Figure 6b). However, there were significantly fewer apoptotic active Casp9+ cells in mBACtgDyrk1a mice administered MPTP than in the equivalent wild-type mice (Figure 6b). Indeed, mBACtgDyrk1a mice exhibited a marked attenuation of MPTP-induced SN pars compacta DA cell death by day 21 post MPTP (Figure 6c), once the MPTP-induced lesion had been stabilized.36

MPTP intoxication of adult mBACtgDyrk1a mice. (a) Schedule of MPTP intoxication employed in experiments in (b and c). (b) Dual immunohistochemistry for TH and active Casp9 (A-Casp9) in the SN pars compacta of a wild-type (WT) animal 4 days after MPTP intoxication, showing a pyknotic cell (arrowhead) and an active Casp9+ (black) cell (arrow) expressing TH (brown), and the total numbers of active Casp9+ cells quantified in the SN pars compacta of WT and mBACtgDyrk1a (BACD1a) animals injected with saline (Sal.) or MPTP. (c) Representative images showing TH+ cells in the SN pars compacta of saline- or MPTP-treated WT and BACD1a animals 21 days after MPTP intoxication, and the quantification of the total TH+ cells in this region for the four experimental groups. Histogram values are the mean±S.E.M. N, not significant (P>0.05); *P≤0.05; **P≤0.01; ****P≤0.0001 (n=3; p-ANOVA=0.001 in (b) and n=5; p-ANOVA=0.01 in (c)). Bar=100 μm (b and c)
Together, these results show that moderate overexpression of Dyrk1a in mature mDA neurons attenuates the apoptotic response induced by MPTP intoxication, leading to an enhanced survival of SNpc DA neurons in this model.
Discussion
In this work we show that DYRK1A regulates Casp9-mediated cell death in mouse mDA neurons in vivo and in two different context: during the main wave of developmental PCD in these neurons and in the SN pars compacta of adult animals that have been challenged with the parkinsonian neurotoxin MPTP. In both contexts, the amount of Dyrk1a protein in the ventral mesencephalon is directly correlated to the levels of Casp9 phosphorylated at the inhibitory Thr125, and it is inversely correlated to the number of DA neurons that die by the intrinsic (Casp9-mediated) cell death pathway. There is evidence that basal DYRK1A phosphorylation of pro-Casp9 at Thr125 restrains Casp9-dependent apoptosis in the developing retina16 and in cultured cells under hyperosmotic stress.38 DYRK1A is synthesized as a constitutively active kinase39 and, therefore, it is assumed that its kinase activity is directly related to the amount of protein.9, 34 Our data suggest that Dyrk1a is the kinase that phosphorylates Casp9 in the developing and adult mesencephalon, and that this phosphorylation is not modified by MPTP intoxication. Accordingly, we propose a model in which the amount of Dyrk1a protein in brain DA neurons establishes the levels of inhibitory Thr125 phosphorylation of Casp9, which in turn defines the threshold of the apoptotic stimulus that is needed to irreversibly activate the caspase cell death cascade, either during development or in a pathological neurodegenerative condition in the adult.
The altered size of certain regions in the brain of Dyrk1a+/− and mBACtgDyrk1a mutant mice is prominent in some cases (i.e., the hippocampus and the ventral thalamic postero lateral and postero medial nuclei) but not in others (i.e., the cerebral cortex). The prominently affected regions are smaller and with less neurons in the Dyrk1a+/− model, and they are larger and with more neurons in the mBACtgDyrk1a model.1 Therefore, deregulation of Casp9-dependent developmental cell death could contribute to the defects in cellularity in these brain regions. Nonetheless, alterations in the cytoarchitecture of the brain are complex in Dyrk1a mutants, with regions showing higher densities of neurons in the Dyrk1a+/− haploinsufficient model and lower densities in the mBACtgDyrk1a triploid model.1, 6 These observations indicate that in addition to PCD, other developmental processes that regulate neural progenitor proliferation and differentiation must also be disturbed in specific domains of the developing brain in Dyrk1a mutants.12
The excess of DA neurons observed in the mBACtgDyrk1a overexpression model after the main postnatal wave of PCD was not longer evident at P21. As the second wave of DA neuron PCD in the mouse takes place during the second and third week of life,25 this observation raises the possibility that the second wave of cell death was exacerbated in mBACtgDyrk1a mice as an attempt to counterbalance the excess of DA neurons and adjust the number of projection neurons to their targets. During the second wave of PCD, at P14, the levels of Thr125-phosphorylated Casp9 in the ventral mesencephalon of mBACtgDyrk1a mice are still higher than in the wild types (Supplementary Figure 6a), and consequently mBACtgDyrk1a mutants at this stage have less cells expressing active Casp9 (Supplementary Figure 6b). However, and contrary to what we have observed in newborn mBACtgDyrk1a mice, the number of apoptotic cells expressing active Casp3 was slightly increased in P14 mBACtgDyrk1a mice (Supplementary Figure 6b). Together, these observations suggest that the second wave of DA neuron PCD involves, in addition to the Casp9-mediated pathway, another caspase-dependent cell death pathway. Indeed, there is evidence that cell death of DA differentiating neurons induced by deprivation of growth factors is mediated by a non-classical mitochondria-independent cell death pathway that involves the activation of several caspases and the death receptor-caspase8 pathway.40 The existence of such mechanisms may explain why the defects in brain size and cellularity are more severe in Dyrk1a+/− mice than in mBACtgDyrk1a mice.1
SN pars compacta DA neurons in adult mBACtgDyrk1a mice were protected against MPTP-induced neurodegeneration, suggesting that therapies aimed at preventing Casp9 cleavage could ameliorate or delay neurodegeneration in Parkinson’s disease. This concept is supported by studies showing a certain degree of protection against MPTP intoxication through the administration of the pan-caspase inhibitor Q-VD-OPH,41 the expression in transgenic mice of the general caspase inhibitor protein baculoviral p35,42 or by adenoviral gene transfer of the X-chromosome-linked inhibitor of apoptosis.43 Although neuroprotection in these studies was analysed shortly after MPTP administration, the results shown here indicate that inhibition of Casp9 cleavage can protect DA cells from MPTP-induced cell death over longer periods of time. Unfortunately, the beneficial effect of caspase inhibitors in Parkinson’s disease has been challenged by the negative results of clinical trials into the use of caspase inhibitors,44 which could be due to the difficulties to design clinical trials relevant to neuroprotection, not only given the heterogeneity of the disease but also due to the absence of in vivo indicators of neuronal dysfunction to monitor the possible effects of the drug.
In addition to Casp9, DYRK1A was shown to phosphorylate α-synuclein in vitro and acute overexpression of both DYRK1A and α-synuclein enhances the formation of intracellular inclusions in immortalized hippocampal H19-7 cells, which is associated to a decreased survival of these cells.45 The neuroprotection offered against MPTP and the fact that 6-month-old mBACtgDyrk1a mice have a normal complement of DA neurons indicate that triplication of the mouse Dyrk1a gene alone does not compromise the viability of SN pars compacta DA neurons, but instead it protects these cells against pathological insult. Based on the effect of DYRK1A on α-synuclein phosphorylation, it has been suggested that DYRK1A might favour the formation of Lewy bodies in Parkinson’s disease and in Down syndrome.10 However, the possible effect of trisomy 21 in Lewy body formation has been challenged by clinical studies showing that parkinsonian signs are less evident in Down syndrome individuals than in patients with advanced Alzheimer’s disease46 and also by pathological observations revealing the absence of Lewy bodies in the SN of adults with Down syndrome.47 Although the neuroprotection offered by Dyrk1a described here could differ in a context of complete trisomy 21 in humans, the results of this work raise concern about possible deleterious effects of excessive therapeutic DYRK1A inhibition to improve cognitive performance in adults with Down syndrome.9
In summary, our data shows that normal levels of DYRK1A kinase activity are necessary for the survival of mDA neurons during development and in the adult brain after re-activation of the intrinsic cell death pathway in a pathological situation. These results could be of interest in the context of Parkinson’s disease and challenge the general association of DYRK1A overexpression with neurodegenerative diseases. In addition, our data provide insights into the aetiology of the microcephaly and motor gait dysfunctions in children with heterozygous mutations in the DYRK1A gene.
Materials and Methods
Unless otherwise stated, all materials were obtained from Sigma-Aldrich (Tres Cantos, Madrid, Spain).
Animals
The generation of the Dyrk1a+/− mouse and the mBACtgDyrk1a mouse was described previously.1, 6 Both Dyrk1a mutants were maintained on their original genetic backgrounds and genotyped by PCR:1, 6 Dyrk1a+/− mice by repeated backcrossing of Dyrk1a+/− males to C57BL/6Jx129S2/SvHsd F1 females (Harlan Laboratories, Bicester, UK) and mBACtgDyrk1a mice by repeated backcrossing of transgenic males to C57BL6/J females (Charles River Laboratories, L'Arbresle, France). Fertility of Dyrk1a+/− mice is compromised when the mutants were crossed with C57BL6/J mice, which precluded having both Dyrk1a mutants on the same genetic background. The day of the vaginal plug was defined as E0, and the day of birth was defined as P0. We used wild-type littermates as controls and males for the studies in adult animals, except for the respiratory and reactive oxygen species production assays that were performed on both males and females.
All the experimental procedures were carried out in accordance with the European Union guidelines (Directive 2010/63/EU) and the followed protocols were approved by the ethics committees of the Parc de Recerca Biomèdica de Barcelona (PRBB) and Research Institute-University Hospital Vall d’Hebron (VHIR).
MPTP treatment
To induce sub-acute MPTP toxicity, 14- to 24-week-old wild-type or mBACtgDyrk1a male mice received one daily i.p. injection of MPTP-HCl (30 mg/kg of free base) on five consecutive days, as described previously.48 MPTP-treated mice and saline-injected control animals were then killed at the time after the last injection indicated (Figure 6a).
Tissue processing for histology and immunostaining
To obtain embryonic and P0 tissue, whole heads were fixed by immersion in 4% paraformaldehyde (PFA) for 24 h at 4 °C, cryoprotected with 30% sucrose in PBS, embedded in Tissue-Tek O.C.T. (Sakura Finetek Europe B.V., Alphen aan den Rijn, The Netherlands), frozen in isopentane at −30 °C and sectioned on a cryostat. Cryosections (16 μm) were collected on Starfrost precoated slides (Knittel Glasser, Braunschweig, Germany) and distributed serially. Postnatal and adult (2- to 6-month-old) mice were deeply anaesthetized in a CO2 chamber and transcardially perfused with 4% PFA. The brains were removed, post-fixed and cryoprotected as indicated above, and cryotome (40 μm) sections were then distributed serially. For Dyrk1a immunostaining, the post-fixed brains were sectioned directly on a vibratome (40 μm).
Immunostainings
Immunohistochemistry on brain sections was performed following the avidin–biotin–peroxidase method (Vectastain ABC kit, Vector Labs, Burlingame, CA, USA). Briefly, sections were incubated in PBS at 37 °C for 15 min and then endogenous peroxidase activity was blocked with 3% H2O2 and 10% methanol in PBS for 30 min at room temperature (RT). The sections were then blocked for 2 h at RT in PBS containing 0.2% Triton-X100 (Merck, Madrid, Spain) and 10% fetal bovine serum (FBS; Life Technologies S.A., Alcobendas, Spain) and probed for 12 to 48 h at 4 °C with the primary antibodies diluted in Antibody Buffer (AB: PBS containing 0.2% Triton-X100 and 5% FBS). After washing, the sections were incubated with the corresponding biotinylated secondary antibody diluted in AB, washed and incubated with an avidin–biotin–peroxidase solution according to the manufacturer’s indications. Peroxidase activity was visualized with 0.03% diaminobenzidine (DAB) and 0.003% H2O2. All samples were counterstained with Nissl (0.1% Cresyl violet acetate) to visualize non-immunopositive cells.
For dual immunohistochemistry with α-active-Casp3 or α-active-Casp9 and α-TH antibodies using the avidin–biotin–peroxidase method, we performed a sequential labelling. Cryosections were first incubated with α-active-Casp3 or α-active-Casp9, and antibody binding was detected using the appropriate biotinylated secondary antibody and the biotin–avidin complex (Vectastain, Vector Labs), which was visualized with 0.03% DAB and 0.2% nickel ammonium sulphate. Sections were then treated for 30 min with 10% methanol and 3% H2O2 in PBS to inhibit the residual peroxidase activity and they were incubated with the TH antibody. The following steps were performed as described before.
Secondary Alexa Fluor-conjugated antibodies (Life Technologies S.A.) were used for dual immunofluorescence and Hoechst was used to stain the nuclei following the same protocol as for immunohistochemistry but without blocking endogenous peroxidase activity.
A complete list of the antibodies used in immunohistochemistry and immunofluorescence can be found in Supplementary Table 1.
Cell counts on tissue sections
The TH+ cells were quantified in brain sections counterstained with Nissl using stereological methods. Sections from animals of different genotypes were processed in parallel to avoid daily variations in immunostaining. The TH+ cells in E14.5 tissue was estimated by counting the immunopositive cells in dissectors that covered 25% of the reference area in every four serially distributed sections of the ventral mesencephalon. The TH+ cell number in the SN and VTA of P7, P21 and adult samples was counted in dissectors covering 15% of the reference area in every three serial sections. The proportion of TH+ cells relative to Nissl stained neurons was quantified by counting both types of cells in dissectors covering the 10% of the reference area every six serial sections. The coefficient of error tolerated was <10% and the dissectors with the upper right corner outside the sampled area were not included in the total number of dissectors. The nuclei that contacted one of the two predetermined adjacent sides of the rectangular dissector frame were not included in the count. The number of active Casp3+ and active Casp9+ cells were calculated by counting the immunopositive cells in the entire region labelled with the anti-TH staining in one of every six serially distributed sections at P0, and in one every three serially distributed sections from the P14 mice and the adult saline and MPTP-treated animals.
Mesencephalic primary cultures
Mouse primary cultures enriched in DA neurons were prepared as described elsewhere.49 Briefly, the ventral mesencephalon was isolated from E12.5 C57BL6/J embryos and the mechanically dissociated cells were seeded on cover slides coated with poly-D-lysine and laminin, and cultured in the presence of a mixture of F12/DMEM media (Life Technologies S.A.) supplemented with 0.25% bovine serum albumin, N1 additives and antibiotics (Life Technologies S.A.). After 4 days in culture, the cells were treated with harmine (harmine-HCl 1–10 μM dissolved in H2O MilliQ; TCI Europe N. V., Zwijndrecht, Belgium) or vehicle (H2O MilliQ) overnight, fixed and immunostained. The coverslips were sampled systematically, counting the number of TH+ cells in at least 12 fields per coverslip and in two coverslips per condition. Cell counts were performed in three independent cultures.
Western blotting
Protein extracts (60 μg) were resolved by SDS-PAGE and transferred onto a nitrocellulose membrane (Hybond-ECL, Amersham Biosciences, Little Chalfont, UK) that was probed with antibodies whose binding was detected by infrared fluorescence using the LI-COR Odyssey IR Imaging System V3.0 (LI-COR Biosciences, Cambridge, UK). A complete list of the antibodies used to probe western blottings is provided in Supplementary Table 2.
Statistical analysis
The data were analysed by the two-tailed Student’s t-test or by ANOVA followed by a two-tailed Student’s t-test, and they are presented as the mean±S.E.M. Any differences were considered significant at P-values≤0.05. The n indicated in the figure legend refers to the minimum number of biological samples (embryos, mice or cell cultures) of the same condition (genotype) that were analysed in each experiment: *P≤0.05; **P≤0.01; ***P≤0.001; ****P≤0.0001.
The methodology followed to isolate brain mitochondria and to perform the experiments with isolated mitochondria are indicated in the Supplementary Material and Methods.
Abbreviations
- Casp3:
-
caspase3
- Casp9:
-
caspase9
- DA:
-
dopaminergic
- E:
-
embryonic day
- mDA:
-
mesencephalic dopaminergic
- MPTP:
-
1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine
- PCD:
-
programmed cell death
- P:
-
postnatal day
- SN:
-
substantia nigra
- TH:
-
tyrosine hydroxylase
- VTA:
-
ventral tegmental area
References
Guedj F, Pereira PL, Najas S, Barallobre MJ, Chabert C, Souchet B et al. DYRK1A: a master regulatory protein controlling brain growth. Neurobiol Dis 2012; 46: 190–203.
Moller RS, Kubart S, Hoeltzenbein M, Heye B, Vogel I, Hansen CP et al. Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am J Hum Genet 2008; 82: 1165–1170.
van Bon BW, Hoischen A, Hehir-Kwa J, de Brouwer AP, Ruivenkamp C, Gijsbers AC et al. Intragenic deletion in DYRK1A leads to mental retardation and primary microcephaly. Clin Genet 2011; 79: 296–299.
Yamamoto T, Shimojima K, Nishizawa T, Matsuo M, Ito M, Imai K . Clinical manifestations of the deletion of Down syndrome critical region including DYRK1A and KCNJ6. Am J Med Genet A 2011; 155A: 113–119.
Courcet JB, Faivre L, Malzac P, Masurel-Paulet A, Lopez E, Callier P et al. The DYRK1A gene is a cause of syndromic intellectual disability with severe microcephaly and epilepsy. J Med Genet 2012; 49: 731–736.
Fotaki V, Dierssen M, Alcantara S, Martinez S, Marti E, Casas C et al. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol Cell Biol 2002; 22: 6636–6647.
Tejedor F, Zhu XR, Kaltenbach E, Ackermann A, Baumann A, Canal I et al. minibrain: a new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron 1995; 14: 287–301.
Chettouh Z, Croquette MF, Delobel B, Gilgenkrants S, Leonard C, Maunoury C et al. Molecular mapping of 21 features associated with partial monosomy 21: involvement of the APP-SOD1 region. Am J Hum Genet 1995; 57: 62–71.
Becker W, Sippl W . Activation, regulation, and inhibition of DYRK1A. FEBS J 2011; 278: 246–256.
Wegiel J, Gong CX, Hwang YW . The role of DYRK1A in neurodegenerative diseases. FEBS J 2011; 278: 236–245.
Haydar TF, Reeves RH . Trisomy 21 and early brain development. Trends Neurosci 2012; 35: 81–91.
Tejedor FJ, Hammerle B . MNB/DYRK1A as a multiple regulator of neuronal development. FEBS J 2011; 278: 223–235.
Yabut O, Domogauer J, D'Arcangelo G . Dyrk1A overexpression inhibits proliferation and induces premature neuronal differentiation of neural progenitor cells. J Neurosci 2010; 30: 4004–4014.
Park J, Oh Y, Yoo L, Jung MS, Song WJ, Lee SH et al. Dyrk1A phosphorylates p53 and inhibits proliferation of embryonic neuronal cells. J Biol Chem 2010; 285: 31895–31906.
Hammerle B, Ulin E, Guimera J, Becker W, Guillemot F, Tejedor FJ . Transient expression of Mnb/Dyrk1a couples cell cycle exit and differentiation of neuronal precursors by inducing p27KIP1 expression and suppressing NOTCH signaling. Development 2011; 138: 2543–2554.
Laguna A, Aranda S, Barallobre MJ, Barhoum R, Fernandez E, Fotaki V et al. The protein kinase DYRK1A regulates caspase-9-mediated apoptosis during retina development. Dev Cell 2008; 15: 841–853.
Tritsch NX, Sabatini BL . Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 2012; 76: 33–50.
Martinez de Lagran M, Bortolozzi A, Millan O, Gispert JD, Gonzalez JR, Arbones ML et al. Dopaminergic deficiency in mice with reduced levels of the dual-specificity tyrosine-phosphorylated and regulated kinase 1A, Dyrk1A(+/−). Genes Brain Behav 2007; 6: 569–578.
Fotaki V, Martinez De Lagran M, Estivill X, Arbones M, Dierssen M . Haploinsufficiency of Dyrk1A in mice leads to specific alterations in the development and regulation of motor activity. Behav Neurosci 2004; 118: 815–821.
Riedl SJ, Salvesen GS . The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol 2007; 8: 405–413.
Vila M, Przedborski S . Targeting programmed cell death in neurodegenerative diseases. Nat Rev Neurosci 2003; 4: 365–375.
Marin F, Herrero MT, Vyas S, Puelles L . Ontogeny of tyrosine hydroxylase mRNA expression in mid- and forebrain: neuromeric pattern and novel positive regions. Dev Dyn 2005; 234: 709–717.
Ang SL . Transcriptional control of midbrain dopaminergic neuron development. Development 2006; 133: 3499–3506.
Ferron SR, Pozo N, Laguna A, Aranda S, Porlan E, Moreno M et al. Regulated segregation of kinase Dyrk1A during asymmetric neural stem cell division is critical for EGFR-mediated biased signaling. Cell Stem Cell 2010; 7: 367–379.
Burke RE . Postnatal developmental programmed cell death in dopamine neurons. Ann N Y Acad Sci 2003; 991: 69–79.
Voorn P, Kalsbeek A, Jorritsma-Byham B, Groenewegen HJ . The pre- and postnatal development of the dopaminergic cell groups in the ventral mesencephalon and the dopaminergic innervation of the striatum of the rat. Neuroscience 1988; 25: 857–887.
Jackson-Lewis V, Vila M, Djaldetti R, Guegan C, Liberatore G, Liu J et al. Developmental cell death in dopaminergic neurons of the substantia nigra of mice. J Comp Neurol 2000; 424: 476–488.
Hattori T, McGeer PL . Synaptogenesis in the corpus striatum of infant rat. Exp Neurol 1973; 38: 70–79.
Fuchs Y, Steller H . Programmed cell death in animal development and disease. Cell 2011; 147: 742–758.
Allan LA, Clarke PR . Apoptosis and autophagy: Regulation of caspase-9 by phosphorylation. FEBS J 2009; 276: 6063–6073.
Seifert A, Allan LA, Clarke PR . DYRK1A phosphorylates caspase 9 at an inhibitory site and is potently inhibited in human cells by harmine. FEBS J 2008; 275: 6268–6280.
Perier C, Bove J, Vila M . Mitochondria and programmed cell death in Parkinson’s disease: apoptosis and beyond. Antioxid Redox Signal 2012; 16: 883–895.
Langston JW, Ballard PA Jr . Parkinson's disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N Engl J Med 1983; 309: 310.
Aranda S, Laguna A, de la Luna S . DYRK family of protein kinases: evolutionary relationships, biochemical properties, and functional roles. FASEB J 2011; 25: 449–462.
Vila M, Jackson-Lewis V, Vukosavic S, Djaldetti R, Liberatore G, Offen D et al. Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl- 4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Proc Natl Acad Sci USA 2001; 98: 2837–2842.
Perier C, Tieu K, Guegan C, Caspersen C, Jackson-Lewis V, Carelli V et al. Complex I deficiency primes Bax-dependent neuronal apoptosis through mitochondrial oxidative damage. Proc Natl Acad Sci USA 2005; 102: 19126–19131.
Perier C, Bove J, Wu DC, Dehay B, Choi DK, Jackson-Lewis V et al. Two molecular pathways initiate mitochondria-dependent dopaminergic neurodegeneration in experimental Parkinson’s disease. Proc Natl Acad Sci USA 2007; 104: 8161–8166.
Seifert A, Clarke PR . p38alpha- and DYRK1A-dependent phosphorylation of caspase-9 at an inhibitory site in response to hyperosmotic stress. Cell Signal 2009; 21: 1626–1633.
Lochhead PA, Sibbet G, Morrice N, Cleghon V . Activation-loop autophosphorylation is mediated by a novel transitional intermediate form of DYRKs. Cell 2005; 121: 925–936.
Yu LY, Saarma M, Arumae U . Death receptors and caspases but not mitochondria are activated in the GDNF- or BDNF-deprived dopaminergic neurons. J Neurosci 2008; 28: 7467–7475.
Yang L, Sugama S, Mischak RP, Kiaei M, Bizat N, Brouillet E et al. A novel systemically active caspase inhibitor attenuates the toxicities of MPTP, malonate, and 3NP in vivo. Neurobiol Dis 2004; 17: 250–259.
Viswanath V, Wu Y, Boonplueang R, Chen S, Stevenson FF, Yantiri F et al. Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson's disease. J Neurosci 2001; 21: 9519–9528.
Eberhardt O, Coelln RV, Kugler S, Lindenau J, Rathke-Hartlieb S, Gerhardt E et al. Protection by synergistic effects of adenovirus-mediated X-chromosome-linked inhibitor of apoptosis and glial cell line-derived neurotrophic factor gene transfer in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. J Neurosci 2000; 20: 9126–9134.
Nicholson DW . From bench to clinic with apoptosis-based therapeutic agents. Nature 2000; 407: 810–816.
Kim EJ, Sung JY, Lee HJ, Rhim H, Hasegawa M, Iwatsubo T et al. Dyrk1A phosphorylates alpha-synuclein and enhances intracellular inclusion formation. J Biol Chem 2006; 281: 33250–33257.
Vieregge P, Ziemens G, Freudenberg M, Piosinski A, Muysers A, Schulze B . Extrapyramidal features in advanced Down’s syndrome: clinical evaluation and family history. J Neurol Neurosurg Psychiatry 1991; 54: 34–38.
Hestnes A, Daniel SE, Lees AJ, Brun A . Down's syndrome and Parkinson's disease. J Neurol Neurosurg Psychiatry 1997; 62: 289.
Jackson-Lewis V, Przedborski S . Protocol for the MPTP mouse model of Parkinson's disease. Nat Protoc 2007; 2: 141–151.
Farkas LM, Dunker N, Roussa E, Unsicker K, Krieglstein K . Transforming growth factor-beta(s) are essential for the development of midbrain dopaminergic neurons in vitro and in vivo. J Neurosci 2003; 23: 5178–5186.
Acknowledgements
This work was supported by the Spanish Ministry of Economy and Competitiveness (MINECO) (grant numbers SAF-2010-17004 and CSIC-201020I003 to M.L.A.), the Generalitat de Catalunya (grant number 2009SGR1464), the Fondo de Investigación Sanitaria-Instituto de Salud Carlos III, Spain (to MV, JB and CP), and the I3 Program from the MINECO (to CP). MJB was supported by the CIBERER and by the program JAE DOC-CSIC/European Social Fund, and AL was supported by an FI grant (AGAUR, Generalitat de Catalunya). We express our gratitude to Erika Ramírez and Annabelle Parent for their technical assistance, to Sílvia Miralles, Ma Carmen Badosa and the personnel of the PRBB and PRAAL-PCB animal facilities for taking care of the mice, and to Mark Sefton for editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Verkhratsky
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Barallobre, M., Perier, C., Bové, J. et al. DYRK1A promotes dopaminergic neuron survival in the developing brain and in a mouse model of Parkinson’s disease. Cell Death Dis 5, e1289 (2014). https://doi.org/10.1038/cddis.2014.253
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.253
This article is cited by
-
Suppression of DYRK ortholog expression affects wax ester fermentation in Euglena gracilis
Journal of Applied Phycology (2018)
-
Zebrafish knockout of Down syndrome gene, DYRK1A, shows social impairments relevant to autism
Molecular Autism (2017)
-
Neuronal survival in the brain: neuron type-specific mechanisms
Cell Death & Disease (2017)
-
Effects of Alpha-Synuclein on Primary Spinal Cord Neurons Associated with Apoptosis and CNTF Expression
Cellular and Molecular Neurobiology (2017)
-
Overexpression of the DYRK1A Gene (Dual-Specificity Tyrosine Phosphorylation-Regulated Kinase 1A) Induces Alterations of the Serotoninergic and Dopaminergic Processing in Murine Brain Tissues
Molecular Neurobiology (2017)
