
Abstract
Death-associated protein kinase 1 (DAPK1) has been shown to have important roles in neuronal cell death in several model systems and has been implicated in multiple diseases, including Alzheimer’s disease (AD). However, little is known about the molecular mechanisms by which DAPK1 signals neuronal cell death. In this study, N-myc downstream-regulated gene 2 (NDRG2) was identified as a novel substrate of DAPK1 using phospho-peptide library screening. DAPK1 interacted with NDRG2 and directly phosphorylated the Ser350 residue in vitro and in vivo. Moreover, DAPK1 overexpression increased neuronal cell death through NDRG2 phosphorylation after ceramide treatment. In contrast, inhibition of DAPK1 by overexpression of a DAPK1 kinase-deficient mutant and small hairpin RNA, or by treatment with a DAPK1 inhibitor significantly decreased neuronal cell death, and abolished NDRG2 phosphorylation in cell culture and in primary neurons. Furthermore, NDRG2-mediated cell death by DAPK1 was required for a caspase-dependent poly-ADP-ribose polymerase cleavage. In addition, DAPK1 ablation suppressed ceramide-induced cell death in mouse brain and neuronal cell death in Tg2576 APPswe-overexpressing mice. Finally, levels of phosphorylated NDRG2 Ser350 and DAPK1 were significantly increased in human AD brain samples. Thus, phosphorylation of NDRG2 on Ser350 by DAPK1 is a novel mechanism activating NDRG2 function and involved in neuronal cell death regulation in vivo.
Similar content being viewed by others


Main
Death-associated protein kinase 1 (DAPK1) is a death domain-containing calcium/calmodulin-regulated serine/threonine kinase that was first identified by functional cloning based on its involvement in interferon-γ-induced apoptosis.1, 2 DAPK1 has been shown to promote apoptosis induced by various stimuli, including ceramide, and seems to have a particularly important role in the regulation of neuronal cell death.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 DAPK1 overexpression and activation increase neuronal cell death, and neurons without DAPK1 are less sensitive to apoptotic stimuli both in vitro and in vivo.8, 12, 13, 14, 15 Moreover, DAPK1 modulates brain damage through the N-methyl-d aspartate receptors, and inhibition of DAPK1 activity has neuroprotective effects in a mouse model of ischemia.16, 17, 18, 19 Furthermore, DAPK1 is activated by its dephosphorylation and promotes neuronal cell death in response to unliganded UNC5H2 or Neogenin.8, 13, 20 Although DAPK1 could be a key factor that induces neuronal cell death at multiple levels through regulating its substrates such as NR2B and p53,8, 10, 16 the molecular mechanisms of DAPK1 involved in neuronal apoptosis are not fully understood.
Recent studies have also indicated that DAPK1 may have a critical role in neurodegenerative diseases, including Alzheimer’s disease (AD). Mice deficient in DAPK1 kinase activity are more efficient learners and have better spatial memory compared with wild-type (WT) mice.21 Moreover, despite previous conflicting results, a recent large-scale genetic assay has identified DAPK1 as one of the genetic factors that may affect susceptibility to late-onset AD.22, 23, 24 Recently, our lab showed that DAPK1 expression is significantly upregulated in the hippocampi of AD patients compared with age-matched normal subjects.25 Furthermore, DAPK1 promotes tau protein accumulation and phosphorylation, and amyloidogenic APP processing.25, 26, 27, 28 Finally, DAPK1 kinase activity is responsible for Pin1 phosphorylation, which inhibits Pin1 activity.29 As Pin1 protects against AD by regulating phosphorylated tau and APP, DAPK1 deregulation may contribute to AD by inactivating Pin1 activity.30
In the article, N-myc downstream-regulated gene 2 (NDRG2) was identified as a novel DAPK1 substrate through a peptide library approach. DAPK1 was found to bind to and phosphorylate NDRG2. Moreover, NDRG2 might mediate and activate a DAPK1-dependent neuronal cell death. The significance of these findings was further substantiated that NDRG2 phosphorylation and DAPK1 levels were highly increased in human AD brains. Taken together, these results show, for the first time, that DAPK1 is a critical regulator of NDRG2 both in neuronal cell death and during the progression of AD.
Results
Identification of NDRG2 as a novel DAPK1 substrate
Although several DAPK1 substrates involved in apoptotic pathways have been identified, little is known about the direct substrates that are required for DAPK1 neuronal cell death. To address this question, we conducted unbiased proteomic and bioinformatics screens to identify possible DAPK1 substrates. A positional scanning peptide library assay was performed to identify the optimal phosphorylation motif for DAPK1, as reported.31, 32 DAPK1 showed a strong preference for substrates with Arginine residues at the -2 and -3 positions relative to the phosphorylation site (Supplementary Figure 1). Spot intensities from the peptide library screen were then quantified and converted into a matrix that could be used in the bioinformatic search engine Scansite to identify potential DAPK1 substrates. Supplementary Table 1 showed top-scoring putative DAPK1 substrates through the statistical criteria analysis in the SwissProt database. NDRG2 was found to be the most intriguing for the following reasons: (1) NDRG2 was scored in the top 0.1% in this screening. (2) NDRG2 overexpression has been shown to increase apoptosis;33, 34, 35 (3) NDRG2 expression is increased after focal cerebral ischemia;36 (4) NDRG2 expression levels are upregulated in human AD patient brains;37 (5) NDRG2 phosphorylation is increased in a neurodegenerative diseases;38 and (6) NDRG2 single-nucleotide polymorphisms occur in late-onset AD patients.39 These results suggest that NDRG2 may have an important role in the regulation of neuronal cell death and AD.
DAPK1 interacts with NDRG2 in vitro and in vivo
To confirm whether NDRG2 is a putative DAPK1 substrate, we first examined whether NDRG2 interacts with DAPK1. FLAG-DAPK1 was co-immunoprecipitated by anti-Myc-NDRG2 in 293T or SH-SY5Y cells (Figure 1a and Supplementary Figure 2a). Reverse co-immunoprecipitation also confirmed that DAPK1 bound to NDRG2 (Figure 1b and Supplementary Figure 2b). Moreover, endogenous DAPK1 and NDRG2 were formed a complex in mouse whole-brain lysates (Figure 1c). We further examined the DAPK1 and NDRG2 interaction in vitro using GST pull-down experiments.29 GST-NDRG2, but not control GST, bound to DAPK1 (Figure 1d). Moreover, both constitutively active mutant DAPK1ΔCaM and kinase-deficient mutant DAPK1K42A interacted with NDRG2 (Figure 1e), confirming that the kinase activity of DAPK1 is not required for NDRG2 binding. When HeLa or SH-SY5Y cells were co-transfected with GFP-NDRG2 and Myc-DAPK1 or its mutants and their subcellular localization was examined directly, NDRG2 colocalized in the cytoplasm with DAPK1 (Figure 1f and Supplementary Figure 2c). Thus, NDRG2 interacts with DAPK1 in vitro and in vivo, and both are colocalized in the cytoplasm.

DAPK1 interacts with NDRG2 in vitro and in vivo. (a and b) Co-immunoprecipitation of DAPK1 and NDRG2 in vivo. 293T cells expressing FLAG-vector or FLAG-DAPK1, and Myc-NDRG2 were subjected to immunoprecipitation with anti-Myc or FLAG antibody, followed by immunoblotting analysis with anti-FLAG or Myc antibody. (c) Endogenous binding of DAPK1 and NDRG2. Mouse whole-brain lysates were subjected to immunoprecipitation with an anti-DAPK1 antibody, followed by immunoblotting analysis with an anti-NDRG2 antibody. (d) DAPK1 and NDRG2 interaction in vitro. Glutathione-agarose beads containing GST or GST-NDRG2 were incubated with extracts of 293T cells expressing FLAG-vector control or FLAG-DAPK1. After washing, proteins pulled down by GST beads were subjected to immunoblotting analysis with an anti-FLAG antibody. (e) NDRG2 binding is not required the kinase activity of DAPK1. 293T cells expressing FLAG-DAPK1 or its mutants, DAPK1K42A or DAPK1ΔCaM were incubated with glutathione-agarose beads containing GST-NDRG2. After washing, binding proteins were subjected to immunoblotting analysis with an anti-FLAG antibody. (f) Colocalization of NDRG2 and DAPK1. HeLa cells were co-transfected with GFP-NDRG2 and either Myc-vector, Myc-DAPK1, Myc-DAPK1K42A or Myc-DAPK1ΔCaM, for 36 h followed by immunofluorescent staining. All experiments are representative of three independent experiments
To map the domains in NDRG2 and DAPK1 important for their interaction, a series of DAPK1 mutants was generated and used in vitro binding assay (Figure 2a). The DAPK1 fragments from 637 to 1423 efficiently bound to NDRG2, suggesting that the cytoskeleton-binding region (637–847, Cyto) is likely bound to NDRG2 (Figure 2b). Moreover, purified His-DAPK1-Cyto directly bound to GST-NDRG2 (Figure 2c) and GST-NDRG2 specifically bound to FLAG-DAPK1-Cyto (Figure 2d). Furthermore, all mutants with altered cytoskeleton-binding regions failed to interact with NDRG2 (Figure 2e). In addition, the GST-NDRG2 fragments (201–371) efficiently bound to DAPK1 (Figure 2f). These results indicate that the C-terminal domain of NDRG2 specifically binds to DAPK1 via its cytoskeleton-binding domain.

The identification of the domain in DAPK1 and NDRG2 required for their interaction. (a) Schematic representation of full-length DAPK1 and its truncated mutants. *: A lysine was replaced by an alanine (K42→A). (b) NDRG2 binding to the cytoskeleton-binding domain of DAPK1. Glutathione-agarose beads containing GST or GST-NDRG2 were incubated with extracts of 293T cells expressing FLAG-DAPK1 or its truncated mutants. After washing, proteins pulled down by GST beads were subjected to immunoblotting analysis with an anti-FLAG antibody. (c) The cytoskeleton-binding domain of DAPK1 directly interacts with NDRG2. Purified histidine-tag cytoskeleton-binding domain of DAPK1 was incubated with glutathione-agarose beads containing GST or GST-NDRG2. After washing, proteins pulled down by GST beads were subjected to immunoblotting analysis with an anti-His antibody. (d) The cytoskeleton-binding domain of DAPK1 is sufficient to interact with NDRG2. 293T cells expressing vector or the cytoskeleton-binding domain of DAPK1 were incubated with glutathione-agarose beads containing GST or GST-NDRG2. After washing, proteins pulled down by GST beads were subjected to immunoblotting analysis with an anti-FLAG antibody. (e) A deletion of the cytoskeleton-binding domain of DAPK1 fails to bind to NDRG2. 293T cells expressing FLAG-DAPK1 or its cytoskeleton-binding domain deletional mutants DAPK1ΔCyto, DAPK1K42AΔCyto or DAPK1ΔCaMΔCyto were incubated with glutathione-agarose beads containing GST or GST-NDRG2. After washing, binding proteins were subjected to immunoblotting analysis with an anti-FLAG antibody. (f) DAPK1 interacts with NDRG2 on C-terminus domain but not N-terminus domain. 293T cells expressing FLAG-DAPK1 were incubated with glutathione-agarose beads containing GST, GST-NDRG2, GST-NDRG2 (1–200) or GST-NDRG2 (201-371). After washing, binding proteins were subjected to immunoblotting analysis with an anti-FLAG antibody. All experiments are representative of three independent experiments
DAPK1 phosphorylates NDRG2 on Ser350 in vitro and in vivo
Given the specific interaction between DAPK1 and NDRG2, we next examined whether DAPK1 phosphorylates NDRG2. Recombinant DAPK1 was incubated with GST-NDRG2 and GST-myosin light chain (MLC) as a positive control.4 Both NDRG2 and MLC were phosphorylated by DAPK1 in a time-dependent manner (Figures 3a and b). DAPK1 efficiently phosphorylated NDRG2, whereas the Km value of MLC was slightly lower than NDRG2 in the steady-state kinetic parameters (Figure 3c). Importantly, DAPK1 phosphorylated only to the GST-NDRG2 fragment (201–371), but not the GST-NDRG2 fragment (1–200) (Figure 3d), suggesting that the putative phosphorylated site is located in the C-terminal domain of NDRG2. To determine which NDRG2 sites are phosphorylated by DAPK1, an Ala substitution was introduced into each of the six putative NDRG2 phosphorylation sites. Although DAPK1 efficiently phosphorylated NDRG2, Ser326A, Thr330A, Ser332A, Ser346A and Thr348A, it failed to phosphorylate Ser350A (Figure 3e). To detect phosphorylation of NDRG2 Ser350 in vivo, we raised a polyclonal antibody against NDRG2 pSer350. The antibody specifically recognized pSer350 NDRG2 but not the NDRG2 S350A mutant (Figure 3f and Supplementary Figure 3). NDRG2 Ser350 phosphorylation was increased in a time-dependent manner by DAPK1, but DAPK1 had no effect on Ser350A mutant (Figure 3g).

DAPK1 phosphorylates NDRG2 on Ser350 in vitro and in vivo. (a) DAPK1 phosphorylates NDRG2 in vitro. Purified recombinant DAPK1 was incubated with GST-NDRG2 or GST-MLC and 32P-ATP, detected by autoradiography. *: 32P-auto-phosphorylated recombinant truncated DAPK1. (b) Time-dependent phosphorylation of NDRG2 by DAPK1. Purified recombinant DAPK1 was incubated with GST-NDRG2 and 32P-ATP, detected by autoradiography. (c) Double reciprocal (Lineweaver–Burk) plots for analyzing the kinetic parameters of NDRG2 or phosphorylation catalyzed by DAPK1. The initial velocities (V0) were measured using purified recombinant DAPK1 at increasing substrate concentrations. (d) DAPK1 phosphorylates NDRG2 on C-terminus domain but not N-terminus domain. Purified recombinant DAPK1 was incubated with GST-NDRG2, GST-NDRG2 (1–200) or GST-NDRG2 (201–371), and 32P-ATP, detected by autoradiography. *: 32P-auto-phosphorylated recombinant truncated DAPK1. (e) DAPK1 phosphorylates NDRG2 on Ser350 in vitro. Purified recombinant DAPK1 was incubated with GST-NDRG2, GST-NDRG2-Ser326A, GST-NDRG2-Thr330A, GST-NDRG2-Ser332A, GST- NDRG2-Ser346A, GST-NDRG2-Thr348A, or GST-NDRG2-Ser350A, and 32P-ATP, detected by autoradiography. *: 32P-auto-phosphorylated recombinant truncated DAPK1. (f) DAPK1 phosphorylates NDRG2 but not NDRG2-Ser350A. Purified recombinant DAPK1 was incubated with GST, GST-NDRG2 or GST-NDRG2 S350A, and ATP, followed by immunoblotting analysis with an anti-pSer350 NDRG2 antibody. (g) Time-dependent phosphorylation of NDRG2 Ser350 by DAPK1. Purified recombinant DAPK1 was incubated with GST-NDRG2 or NDRG2 S350A, and ATP, followed by immunoblotting analysis with an anti-pSer350 NDRG2 antibody. (h) DAPK1 phosphorylates NDRG2 in vivo. 293T cells expressing Myc-vector, Myc-NDRG2 or Myc-NDRG2-Ser350A mutant, and FLAG-DAPK1 were subjected to immunoprecipitation with Myc antibody, followed by immunoblotting analysis with anti-pSer350 NDRG2 or anti-FLAG antibodies. (i) CIP treatment abolishes NDRG2 phosphorylation. 293T cells expressing Myc-vector, Myc-NDRG2 or Myc-NDRG2-Ser350A with FLAG-DAPK1 were subjected to immunoblotting analysis with an anti-pSer350 NDRG2 antibody in the presence or absence of CIP. *: Nonspecific band. (j) Endogenous phosphorylation of NDRG2 by DAPK1. Total brain lysates from WT or DAPK1 KO mice were subjected to immunoblotting analysis with anti-pSer350-NDRG2, anti-NDRG2, anti-DAPK1, or anti-actin antibodies. (k) DAPK1 kinase activity-dependent phosphorylation of NDRG2. 293T cells expressing FLAG-vector or FLAG-DAPK1K42A or FLAG-DAPK1ΔCaM with Myc-NDRG2 were subjected to immunoblotting analysis with an anti-pSer350 NDRG2 antibody. All experiments are representative of three independent experiments
Moreover, NDRG2, but not NDRG2 350A, was efficiently phosphorylated by DAPK1 in vivo (Figure 3h). This recognition was abolished by the dephosphorylation of NDRG2 with calf intestinal phosphatase (CIP) (Figure 3i). We next evaluated endogenous NDRG2 Ser350 phosphorylation levels in the brains of WT and DAPK1 knockout (KO) mice. Ser350 phosphorylation of endogenous NDRG2 was detected in WT brain lysates, but not in DAPK1 KO mice (Figure 3j). Finally, phosphorylated NDRG2 Ser350 levels were significantly increased in DAPK1ΔCaM-expressing cells compared with the vector control- or DAPK1K42A-expressing cells (Figure 3k). Taken together, these results show that DAPK1 phosphorylates NDRG2 on only a Ser350 motif in vitro and in vivo.
DAPK1-induced NDRG2 phosphorylation promotes neuronal cell death
Given the specific phosphorylation of NDRG2 Ser350 by DAPK1, we next wanted to determine whether this phosphorylation has any biological significance or pathological consequence. Ceramide has important roles in the regulation of neuronal cell differentiation, and it is implicated in the pathological death of neurons that occurs in AD.40, 41 Moreover, ceramide is a potent DAPK1 activator and induces DAPK1 kinase activity-mediated cell death in multiple cell types including neuronal cells.12, 14, 42, 43, 44 Furthermore, amyloid-β oligomer (Aβ) toxicity has a critical role in the pathogenesis and progression of neuronal apoptosis and memory loss in AD.45, 46, 47 However, the molecular mechanisms by which DAPK1 regulates neuronal cell death by ceramide or Aβ have not been elucidated. When WT or DAPK1 KO mouse primary cortical neurons were treated with ceramide or Aβ, a live/dead cell assay revealed that ceramide- or Aβ-treated WT primary cortical neurons showed highly increased cell death (Figure 4a and Supplementary Figure 4a) and that DAPK1 ablation markedly attenuated ceramide- or Aβ-induced neuron death (Figures 4a, b Supplementary Figures 4a and b). Moreover, when WT mouse primary cortical neurons were pretreated with the DAPK1 inhibitor before co-treatment with ceramide or Aβ, the DAPK1 inhibitor significantly attenuated ceramide- or Aβ-induced neuron death (Figures 4c, d, Supplementary Figures 4c and d). Given that DAPK1 phosphorylates NDRG2 Ser350, we examined whether NDRG2 Ser350 phosphorylation affects ceramide-induced neuronal cell death. Although phosphorylation-deficient form, NDRG2S350A had no effect, its phosphorylation mimic forms, NDRG2S350D and NDRG2S350E significantly increased cell death compared with NDRG2 when WT or DAPK1 KO primary cortical neurons were transfected with NDRG2, NDRG2S350A, NDRG2S350D or NDRG2S350E (Figures 4e, f, Supplementary Figures 4e and f). Moreover, WT NDRG2 increased cell death compared with NDRG2S350A in WT primary cortical neurons, but not DAPK1 KO neurons (Figures 4e, f, Supplementary Figures 4e and f). Taken together, these results show that NDRG2 Ser350 phosphorylation increases its ability to induce neuronal cell death and may be dependent on DAPK1 kinase activity.

DAPK1 inhibition or NDRG2 dephosphorylation attenuates ceramide-induced primary neuronal cell death. (a) DAPK1 ablation attenuated ceramide-induced primary neuronal cell death. Primary cortical cultures derived from the cerebral cortex of day 15 (E15) WT or DAPK1 KO mice in DIV7 were treated ceramide (20 μM) for the 24 h, followed by color live-dead cell assay. Scale bar=50 μm. (b) Cells expressing red or green color were scored in 200 cells. Results shown are mean±error bar (**P<0.01). (c) DAPK1 inhibition suppressed ceramide-induced primary neuronal cell death. Primary cortical cultures derived from the cerebral cortex of day 15 (E15) WT mice in DIV7 were treated DAPK1 inhibitor (100 nM) for 12 h, and then treated ceramide (20 μM) for the 24 h, followed by color live-dead cell assay. Scale bar=50 μm. (d) Cells expressing red or green color were scored in 200 cells. Results shown are mean±error bar (**P<0.01). N.S, no significance. (e) NDRG2-Ser350D and NDRG2-Ser350E mutations increased primary neuronal cell death. DAPK1 KO mice primary cortical neurons in DIV7 were transfected with GFP or GFP-NDRG2, its mutants S350A, S350D, or S350E for 48 h and then treated ceramide (20 μM) for 10 h. Cell death was determined by TUNEL assay using the confocal microscope. Scale bar=50 μm. (f) TUNEL-positive neurons were scored in 200 cells. Results shown are mean±error bar (**P<0.01). All experiments are representative of three independent experiments
Next, the effects of DAPK1 on the ability of NDRG2 to induce cell death were further examined using cell culture systems. Treatment with 20 μM of ceramide for 10 h or Aβ for 24 h in DAPK1-overexpressing cells resulted in a twofold increase in cell death compared with the vector control (Supplementary Figures 5a and 6a). However, no difference could be detected between the vector and DAPK1-expressing cells at >40 μM of ceramide or 30 μM of Aβ (Supplementary Figures 5a and 6a). We therefore examined NDRG2-mediated cell death by DAPK1 using 20 μM of ceramide or Aβ. When SH-SY5Y cells were transfected with both DAPK1 and NDRG2, neuronal cell death was increased compared with cells expressing DAPK1 or NDRG2 alone (Figure 5a and Supplementary Figure 5b). NDRG2 was also capable of promoting DAPK1-mediated cell death in DAPK1 KO mouse embryonic fibroblasts (MEFs) (Supplementary Figure 5c). Next, we efficiently knocked down endogenous DAPK1 expression using lentiviral-mediated small hairpin RNA (shRNA) (Figure 5b). In contrast to the control cells, when DAPK1 expression was suppressed, the cell death was significantly decreased in NDRG2-overexpressing cells (Figure 5b). Moreover, the inhibition of DAPK1 using a DAPK1 inhibitor reduced ceramide-induced cell death when NDRG2 was overexpressed (Figure 5c). Furthermore, DAPK1K42A decreased, whereas DAPK1ΔCaM increased NDRG2-mediated cell death compared with the vector control (Figure 5d), suggesting that NDRG2-mediated cell death is due to DAPK1 activity. In addition, DAPK1-mediated neuronal cell death was reduced when the endogenous NDRG2 expression was inhibited (Figure 5e). Finally, NDRG2S350D and NDRG2S350E significantly increased cell death, although NDRG2S350A transfection had no effect compared with vector control, (Figure 5f). DAPK1 or NDRG2 overexpression/suppression had a similar effect on neuron cell death with Aβ treatment (Supplementary Figures 6b-f). As NDRG2S350A mutant did not increase DAPK1-mediated neuronal cell death (Figures 6b and d), these data indicate that DAPK1-dependent neuronal cell death may be mediated by NDRG2 phosphorylation at Ser350.

NDRG2-dependent neuronal cell death is mediated by DAPK1 kinase activity. (a) SH-SY5Y cells were co-transfected with Myc-vector or Myc-NDRG2 and FLAG-DAPK1 and then treated ceramide (20 μM) for the 10 h, followed by Trypan blue staining. Results shown are mean±error bar, n=3. (*P<0.05, **P<0.01). (b) DAPK1 knockdowned SH-SY5Y cells were transfected with Myc-vector, Myc-NDRG2 or Myc-NDRG2 S350A and then treated ceramide (20 μM) for the 10 h, followed by Trypan blue staining. Results shown are mean±error bar, n=3. (*P<0.05, **P<0.01). N.S, no significance. (c) WT MEFs were transfected with Myc-vector or Myc-NDRG2 and then pretreated with 100 nM DAPK1 inhibitor for 12 h before the co-treatment with ceramide (20 μM) for 10 h, followed by Trypan blue staining. Results shown are mean±error bar, n=3. (*P<0.05, **P<0.01). (d) SH-SY5Y cells expressing Myc-NDRG2 or Myc-S350A, and FLAG-DAPK1, its mutants, DAPK1K42A or DAPK1ΔCaM were treated ceramide (10 μM) for 10 h, followed by Trypan blue staining. Results shown are mean±error bar, n=3 (**P<0.01). (e) NDRG2 knockdowned SH-SY5Y cells were transfected with FLAG-vector or FLAG-DAPK1 and then treated ceramide (20 μM) for the 10 h, followed by Trypan blue staining. Results shown are mean±error bar, n=3. (**P<0.01). (f) SH-SY5Y cells were transfected with Myc or Myc-NDRG2, its mutants S350A, S350D, or S350E and then treated ceramide (20 μM) for the 10 h, followed by Trypan blue staining. Results shown are mean±error bar, n=3. (**P<0.01). All experiments are representative of three independent experiments

DAPK1 and NDRG2 promote neuronal cell death through caspase-dependent pathway. (a) DAPK1 KO MEFs were co-transfected with Myc-vector, Myc-NDRG2 or Myc-S350A and FLAG-DAPK1 and then treated ceramide (20 μM) for the 10 h, followed by immunoblotting analysis with anti-PARP, anti-caspase-3, anti-p62 or anti-LC3 antibodies. (b) Quantitation of cleaved PARP. Results shown are mean±error bar, n=3. (**P<0.01). (c) NDRG2 knockdowned SH-SY5Y cells were transfected with FLAG-vector or FLAG-DAPK1 and then treated ceramide (20 μM) for the 10 h, followed by immunoblotting analysis with anti-PARP and anti-caspase-3 antibodies. (d) Quantitation of cleaved PARP. Results shown are mean±error bar, n=3. (**P<0.01). (e) DAPK1 KO MEFs were transfected with Myc or Myc-NDRG2, its mutants S350A, S350D or S350E and then treated ceramide (20 μM) for the 10 h, followed by immunoblotting analysis with anti-PARP and anti-caspase-3 antibodies. (f) Quantitation of cleaved PARP. Results shown are mean±error bar, n=3. (**P<0.01). All experiments are representative of three independent experiments
DAPK1 regulates NDRG2-derived cell death through a caspase-dependent pathway
Given that DAPK1 phosphorylates NDRG2 and increases NDRG2-derived cell death, we next wanted to determine the molecular mechanisms by which DAPK1-mediated NDRG2 phosphorylation regulates neuronal cell death. DAPK1-induced cell death has been characterized as both a caspase-dependent and autophagic cell death pathway depending on the stimuli or experimental condition.1 To examine how DAPK1-mediated NDRG2 function may induce cell death, DAPK1 KO MEFs were co-transfected with NDRG2 or NDRG2S350A, and DAPK1, and caspase activity was evaluated by monitoring the cleavage of poly-ADP-ribose polymerase (PARP) or caspase-3 after ceramide treatment. The levels of cleaved PARP or caspase-3 were markedly increased in both DAPK1- and NDRG2-overexpressing cells (Figures 6a, b and Supplementary Figure 7a). However, cleaved PARP or caspase-3 levels were not increased when both DAPK1 and NDRG2S350A were expressed (Figures 6a, b and Supplementary Figure 7a). Moreover, autophagy cell death markers, P62 and LC3 A/B were not increased or decreased in DAPK1 and NDRG2-overexpressing cells (Figure 6a), indicating that DAPK1-mediated NDRG2 function is required for the induction of a caspase-dependent pathway. Furthermore, knockdown of NDRG2 did not increase the levels of cleaved PARP or caspase-3 even DAPK1 was overexpressed (Figures 6c, d and Supplementary Figure 7b) and NDRG2S350D and NDRG2S350E, but not NDRG2S350A significantly upregulated cleaved PARP or caspase-3 levels (Figures 6e, f and Supplementary Figure 7c). In addition, caspase inhibition by Z-VAD-fmk, a pan-caspase inhibitor, abolished ceramide-induced PARP cleavage or caspase-3 activity when both DAPK1 and NDRG2 were co-expressed (Supplementary Figures 7d-f). Overall, these results suggest that NDRG2 is required for ceramide-induced programmed cell death, and that DAPK1 may regulate NDRG2-derived cell death through its phosphorylation and a caspase-dependent pathway.
DAPK1 regulates NDRG2 phosphorylation and neuronal cell death in AD
Given that DAPK1 activates NDRG2 function through Ser350 phosphorylation and that NDRG2 overexpression induces neuronal cell death, we investigated mouse hippocampal neuronal cell death in the WT and DAPK1 KO under ceramide treatment. We first confirmed that NDRG2 colocalized with NeuN, a neuron marker in the hippocampus regions of mouse or human brains (Supplementary Figure 8), as reported previously.37 Next, the levels of DAPK1 and NDRG2 pSer350 were increased in the hippocampal brain slices after ceramide or Aβ reatment (Supplementary Figures 9a and b). When WT or DAPK1 KO mouse brain slices were treated with 40 μM of ceramide, a TUNEL assay revealed that ceramide-treated WT brain slices showed highly increased cell death (Supplementary Figure 9c) and that DAPK1 ablation markedly attenuated ceramide-induced hippocampal neuron death (Supplementary Figures 9c and d). To examine the possible role of DAPK1 and NDRG2 on AD neuronal cell death in vivo, we crossed DAPK1 KO mice with Tg2576 mice overexpressing APP Swedish mutation.28 In the Tg2576/WT mice, the levels of NDRG2 phosphorylation on Ser350 were significantly higher than those of Tg2576/DAPK1 KO mice brain (Figure 7a and Supplementary Figure 10). Moreover, DAPK1 expression was increased in the Tg2576/WT mice as compared with non-transgenic (non-Tg) controls (Figure 7a and Supplementary Figure 10). Furthermore, the number of TUNEL-positive cells was significantly increased in the hippocampi of Tg2576/WT mice compared with non-Tg controls (Figures 7b and c). By contrast, in the Tg2576/DAPK1 KO mice, neuronal cell death was attenuated to those of the Tg2576/WT mice (Figures 7b and c). Taken together, these results show that DAPK1 ablation reduces NDRG2 phosphorylation levels and neuronal cell death in AD animal models.

NDRG2 Ser350 phosphorylation levels are significantly increased in the APP Tg mouse brain and human AD hippocampus. (a) Brain lysates from 8-month-old non-Tg control, Tg2576/WT or Tg2576/DAPK1 KO mice were subjected to immunoblotting analysis with anti-pSer350-NDRG2, anti-NDRG2, anti-DAPK1, anti-APP or anti-actin antibodies. (b) Apoptosis of paraffin-embedded brain sections from 8-month-old non-Tg control, Tg2576/WT or Tg2576/DAPK1 were examined by TUNEL assay. Scale bar=20 μm. (c) Summary of the percentage of TUNEL-positive cells from the hippocampus regions of non-Tg control, Tg2576/WT or Tg2576/DAPK1 mice. (d) Hippocampal tissues of 10 AD patients and 8 age-matched controls were harvested. Proteins from normal and AD hippocampus were used for immunoblotting analysis with anti-pSer350-NDRG2, anti-DAPK1, anti-pS396-Tau or anti-actin antibodies. (e) Anti-pSer350 intensities were quantified by computer densitometry (*P<0.05 versus normal control; ANOVA/Dunnett’s test). (f) Immunohistochemistry of paraffin-embedded brain serial sections from normal and AD hippocampus, comparing the levels of DAPK1 and phosphorylated NDRG2 on Ser350. x200 (insets); Scale bar=100 μm. All experiments are representative of three independent experiments. (g) Immunofluorescence using a monoclonal antibody specific for DAPK1 or a polyclonal antibody specific for phosphorylated NDRG2 Ser350 was conducted on hippocampus regions from human AD brain. Scale bar=50 μm. Representative images from five experiments are shown
Next, we investigated the potential association between late-onset AD and phosphorylated NDRG2 Ser350 expression in normal and human AD brains. The expression of NDRG2 pSer350 was increased more than threefold in the AD patients compared with the normal subjects (Figures 7d and e). Moreover, DAPK1 and phosphorylated tau Ser396 were highly overexpressed in the hippocampi of AD patients, as described previously (Figure 7d).25 Furthermore, neurons in normal hippocampus samples that contained low levels of DAPK1 also had low levels of NDRG2 Ser350 phosphorylation in immunohistochemistry assay (Figure 7f). In contrast, neurons from AD tissues that contained high levels of DAPK1 also had high levels of NDRG2 Ser350 phosphorylation (Figure 7f). In addition, in human AD brain, phosphorylated NDRG2 Ser350 and DAPK1 were colocalized in the cytoplasm of neuron (Figure 7g). These results indicate that NDRG2 Ser350 phosphorylation is upregulated in the hippocampi of AD patients, which further supports the idea that DAPK1 is a NDRG2 Ser350 kinase.
Discussion
Although the cumulative evidence indicates that DAPK1 may have a critical role in neuronal cell death and AD, the underlying mechanisms and overall impact on AD are still unknown. A key step toward understanding how DAPK1 regulates neuronal cell death and AD will be to develop systematic approaches for identifying the major DAPK1 substrates in AD and for determining their function and regulation by DAPK1. To address these questions, we used positional scanning peptide libraries to identify the optimal substrate phosphorylation motif for DAPK1. Based on these findings, we further identified numerous putative DAPK1 substrates using a proteome-wide search for sites that correspond to the DAPK1 phosphorylation motif. However, confirmation that DAPK1 substrates are, indeed, important in neuronal cell death and AD may require a substantial number of both in vitro and in vivo experiments. The rationale for selecting which targets to study first was based on several criteria, including whether they had been previously identified as well as whether they have known roles in the cell death and neuronal function involved in AD. As a result of these processes, we decided to first focus on NDRG2, as a novel DAPK1 substrate potentially involved in neuronal cell death and AD.
In this study, DAPK1 was shown to be a critical regulator of NDRG2 function and neuronal cell death in vitro and in vivo. First, DAPK1 was found to associate with NDRG2, and the two proteins colocalized in the cytoplasm of cells. Second, DAPK1 was found to phosphorylate only NDRG2 Ser350 in vitro and in vivo. Third, this phosphorylation increased NDRG2-mediated neuronal cell death. Fourth, Ser350 phospho-mimicking mutations fully activated NDRG2-mediated neuronal cell death, whereas Ser350 non-phosphorylation mutation completely inactivated its apoptotic function. Fifth, DAPK1 and NDRG2 promoted neuronal cell death through a caspase-dependent pathway. Sixth, DAPK1 ablation decreased NDRG2 Ser350 phosphorylation and neuronal cell death in the APP-overexpressing mouse brains. Finally, phosphorylated NDRG2 Ser350 and DAPK1 levels were highly upregulated in the brain tissues of AD patients. Thus, NDRG2 cellular function is fully activated by DAPK1-mediated Ser350 phosphorylation.
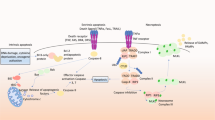
We found that the co-expression of DAPK1 and NDRG2 significantly increased neuronal cell death and that DAPK1K42A and NDRG2S350A failed to enhance cell death after ceramide treatment, suggesting that DAPK1 kinase activity is critical for neuronal cell death. Moreover, NDRG2-mediated cell death significantly decreased with knockdown of DAPK1 expression or DAPK1 inhibitor treatment. Furthermore, DAPK1-mediated NDRG2 phosphorylation promoted the cleavage of caspase-3 and PARP, suggesting that DAPK1 and NDRG2 might act to induce cell death through a caspase-dependent pathway. As DAPK1 is linked to both caspase-dependent and caspase-independent cell death in response to different stimuli1 and given that ceramide or Aβ increases DAPK1 activity,14, 43, 44, 48 our data suggest a model in which DAPK1 stimulates caspase-dependent neuronal cell death following ceramide or Aβ induction via NDRG2 phosphorylation at Ser350 (Supplementary Figure 11). It is known that ceramide stimulates generation of reactive oxygen species and promotes apoptosis by inhibition of the phosphoinositide 3-kinase (PI3K)/Akt pathway via PP2A, or activation of JNK or p38 MAPK cascades.49 Recent studies have reported that DAPK1 is involved in MAPKs signaling pathways under oxidative stress3 and its activity is critical for ceramide-induced apoptosis.12, 14, 43 In addition, PP2A-mediated dephosphorylation of DAPK1 can trigger UNC5H2-induced apoptosis.50 Moreover, NDRG2 activates JNK pathway and is a negative regulator of PI3K/Akt pathway by promoting the active dephosphorylated form of PTEN through the recruitment of PP2A.51, 52 Taking these findings together, it could be speculated that NDRG2 phosphorylation by DAPK1 is activated under ceramide treatment, leading to the downregulation of PI3K/Akt and/or activation of JNK pathways, thereby inducing apoptosis by activating Bad, GSK-3β and caspase-3.
The most interesting aspect of this study was the potential association between late-onset AD and phosphorylated NDRG2 Ser350 because DAPK1 is highly expressed in AD brains. Phosphorylated NDRG2 Ser350 levels were measured in the hippocampi of AD patients and normal controls because, in AD, the hippocampus is damaged early and severely compared with other parts of the cortex and because DAPK1 expression is restricted to the hippocampus of the adult brain.53 We found that hippocampal tissues from aged AD brains had increased levels of phosphorylated NDRG2 Ser350 protein along with high levels of DAPK1 relative to those from age-matched normal subjects. As neuronal loss is an essential event in AD progression and eventually leads to dementia, the critical role of DAPK1 and NDRG2 in neuronal cell death shown in this study substantiates the need for further studies on both proteins in neuronal cells. Future studies should include the development of Tg mice with inducible and brain-restricted DAPK1 expression and DAPK1 KO/Tg mice crossed with NDRG2 mouse models.
Taken together, these findings suggest that DAPK1 specifically phosphorylates NDRG2 Ser350 and this phosphorylation is an important caspase-dependent mechanism by which NDRG2 mediates cell death in neuronal cells. Furthermore, our results also show that phosphorylated NDRG2 Ser350 levels are increased in AD brains, and this may be a marker of AD pathology. Overall, our data suggest a model in which DAPK1 activates NDRG2 function via Ser350 phosphorylation followed by neuronal cell death through the cleavage of caspase-3 and PARP. Moreover, our studies suggest that DAPK1 represents a potential novel therapeutic approach for treating AD.
Materials and Methods
Materials
C6-ceramide, paraformaldehyde and dimethyl sulfoxide were purchased from Sigma (St. Louis, MO, USA). DAPK1 inhibitor (4Z)-4-(3-pyridylmethylene)-2-styryl-oxazol-5-one was obtained from Calbiochem (Billerica, MA, USA). Caspase-3 colorimetric assay kit was purchased from BioVision (Milpitas, CA, USA).
Preparation of Aβ solution
Synthetic Aβ25–35 peptides corresponding to WT human Aβ was obtained from Genemed Synthesis, Inc. (San Antonio, TX, USA). Aβ25–35 peptide stock solution was dissolved in 2 ml of Millipore (Billerica, MA, USA) SuperQ water; solutions were fractionated into five equal parts and lyophilized overnight. Each aliquot was dissolved in water (pH 7.4) to achieve the needed concentration.
Cell culture
The human cervix carcinoma cell line HeLa; the human neuroblastoma cell lines SH-SY5Y; the human embryonic kidney 293T were obtained from the American Type Culture Collection (Manassas, VA, USA). MEFs were obtained from the WT and DAPK1 KO embryos between embryonic day 12.5 and 14.5. HeLa, SH-SY5Y, 293T and MEF cells were cultured in Dulbecco’s modified Eagle’s medium high-glucose supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 μg/ml streptomycin (all from Life Technologies, Waltham, MA, USA). The cultures were maintained at 37 °C under 5% CO2.
Plasmid transfection
The expression constructs for WT and various mutants of DAPK1 with C-terminal FLAG tag were described.6 FLAG-DAPK1 deletion mutants were generated by PCR. The cytoskeleton deletion DAPK1 variants (DAPK1 ΔCyto: residues 648-841) were generated by PCR. Cells were transiently transfected with different plasmids using Lipofectamine 2000 (Life Technologies), PolyJet (Sigmagen, Rockville, MD, USA) or DNA-IN Neuro (GlobalStem, Gaithersburg, MD, USA) transfection reagents according to the manufacturer’s instruction. For generating stable NDRG2 overexpression SH-SY5Y cells, a NDRG2 clone that is constructed within lentiviral pByBE.1. Blastidine vector was transfected together with VSV-G- and gag-pol-expressing plasmids into 293FT cells. Sixty hours after transfection, virus-containing supernatants were used to infect the SH-SY5Y cells. A day after infection, the stable clones were selected using 1.5 μg/ml blastidine. For generating stable DAPK1 or NDRG2 knockdown SH-SY5Y cells, DAPK1 or NDRG2 shRNA clones, which are constructed within lentiviral pLKO.1-Puro vector, were transfected together with VSV-G- and gag-pol-expressing plasmids into 293FT cells. Sixty hours after transfection, virus-containing supernatants were used to infect the SH-SY5Y cells. A day after infection, the stable clones were selected using 1.5 μg/ml puromycin. The target sequences of DAPK1-specific shRNA was 5′-CAAGAAACGTTAGCAAATG-3′ and the target sequences of NDRG2-specific shRNA was 5′-CCGGCTGGAAGAGATGTCCTAATATCTCGAGATATTAGGACATCTCTTCCAGTTTTTTG-3′.
Primary cortical neuronal cell cultures in mice
Primary cultures of mouse embryonic cortical neurons were prepared as described previously.54, 55 Briefly, dissociated embryonic neurons from embryonic day 15 (E15) WT or DAPK1 KO C57BL/6 pregnant mice were resuspended in neurobasal medium supplemented with 2% B27 supplement, 0.5 mM glutamine and 1% penicillin–streptomycin (all from Life Technologies) and cultured at 37 °C under 5% CO2. After adaptation for 24 h, the culture medium was replaced with a neurobasal-A medium containing 2% B27 supplement, 0.5 mM glutamine and 1% penicillin–streptomycin. Primary cortical neuronal cells were used for ceramide or Aβ oligomer-induced neuronal cell death at day in vitro (DIV) 7.
Organotypic brain-slice cultures
Organotypic mouse brain-slice cultures were performed as described previously.25 Briefly, brains from 6-month-old WT and DAPK1 KO mice were removed and cut into coronal slices of 260-μm thicknesses with vibratome (Leica, Buffalo Grove, IL, USA). Slices containing the hippocampus were used for neuronal cell death using TUNEL assay. The slices were cultured onto organotypic Millicell cell culture inserts (Millipore) in six-well culture plates with culture medium.
Immunohistochemistry and immunostaining analyses
Immersion-fixed tissue (hippocampus) sections were embedded in paraffin. Coronal tissue sections were deparaffinized with xylene, rehydrated with descending grade of ethanol and incubated with 3% H2O2/PBS to quench the endogenous peroxidase activity. After antigen retrieval, sections were incubated in PBS blocking buffer with 5% horse serum, 5% bovine serum albumin and 0.1% v/v Tween 20. Primary antibodies were added in blocking buffer and incubated with sections overnight at 4 °C. For immunohistochemical analysis, secondary antibody was biotinylated goat-anti-rabbit or -mouse IgG. Sections were processed with ABC reagents by using a Vector ABC kit (Vector Laboratories, Burlingame, CA, USA). After washing, the horseradish peroxidase (HRP) reaction was detected with diaminobenzidine and H2O2. For immunofluorescent analysis, slides were incubated with Alexa488-labeled anti-mouse IgG and Alexa546-labeled anti-Rabbit IgG (Invitrogen, Carlsbad, CA, USA) diluted 1:200 for 2 h at room temperature. After washing with PBS, the nuclei were stained with DAPI for 10 min at room temperature.
Immunoblot analysis
After harvesting, the cells were lysed in ice-cold lysis buffer (50 mM Tris-HCl (pH 7.4), 50 mM NaCl, 1 mM EGTA, 1% Triton X-100, 1 mM DTT) supplemented with protease inhibitor cocktail and phosphatase inhibitor cocktail. Mouse and human brain extracts were prepared by homogenizing tissue in ice-cold lysis buffer (10 mM Tris-HCl (pH 7.4), 0.8 M NaCl, 1 mM EGTA, 10% sucrose, 1 mM DTT) supplemented with protease inhibitor cocktail and phosphatase inhibitor cocktail. The soluble protein concentration was determined by Bradford assay (Bio-Rad, Hercules, CA, USA). Protein samples (5–10 μg) were separated by SDS-PAGE using a 26-well electrophoresis system (Bio-Rad) and transferred to polyvinylidene fluoride membranes (PerkinElmer, Waltham, MA, USA). The membranes were then probed with various antibodies, followed by HRP-conjugated secondary antibodies. Antibody-bound proteins were detected using the Western Lightning Plus-ECL (PerkinElmer) or ECL Prime (GE Healthcare Life Sciences, Marlborough, MA, USA) chemiluminescence system. The following antibodies were used to probe different proteins: His, NDRG2 (Santa Cruz, Dallas, TX, USA); FLAG, DAPK1 (Sigma); β-actin, Myc, GFP, cleaved PARP, cleaved caspase-3, p62, LC3 A/B (Cell Signaling Technology, Danvers, MA, USA); PHF-13 (p-Ser396-Tau) (Anaspec, Fremont, CA, USA).
Phosphorylated Ser350-specific NDRG2 antibodies
Phosphorylated Ser350-specific NDRG2 antibodies were raised by immunizing rabbits with a KLH-coupled pSer350-containing NDRG2 peptide CSRSTLSQSSE (Neobio Lab, Woburn, MA, USA) and were affinity purified, as described.29
GST pull-down, in vitro kinase, and immunoprecipitation
GST pull-down, in vitro kinase and immunoprecipitation analyses were performed as described previously.29 Briefly, relevant proteins were expressed in 293T or Hela cells by transient transfection, followed by lysis or dilution in a buffer containing 50 mM Tris-HCl (pH 7.4), 50 mM NaCl, 1 mM EGTA, 1% Triton X-100, and 1 mM DTT supplemented with protease inhibitor cocktail and phosphatase inhibitor cocktail. The cellular supernatants were incubated with 1 μM GST or GST fusion proteins or specific antibodies for 1 h at 4 °C and 15 μl of glutathione-agarose beads or protein A agarose were then added, followed by further incubation for 2 h at 4 °C. The precipitated proteins were washed 4–6 times in the same buffer and subjected to immunoblotting analysis. For in vitro kinase assays, purified recombinant DAPK1 was incubated with GST-NDRG2 or MLC in the kinase reaction buffer at 25 °C for up to 30 min, detected by autoradiography. For measuring steady-state kinetic parameters, 1 μg of DAPK1 was used to catalyze increasing amounts of MLC or NDRG2 (0.7–8.4 μM) in 25 μl of kinase reaction mixture. In immunoprecipitation, relevant proteins were incubated with Myc or FLAG M2 bead or nonspecific IgG or monoclonal mouse anti-DAPK1 at 4 °C for 3 h, followed by the addition of 15 μl of Protein G-Sepharose (Sigma) for 2 h at 4 °C, followed by immunoblotting analysis.
Cell viability assays
Cell viability was assessed with a live/dead Viability Kit (Abcam, Cambridge, MA, USA) or Trypan blue staining according to the manufacturer’s instruction. Briefly, primary neuron at DIV 9 diluted with equal volumes of cell suspension and 2X live/dead dye solution, incubation for 10 min at room temperature in the dark. Cell viability was analyzed using fluorescent microscopy. For the Trypan blue staining, cell collections were diluted in Trypan blue dye (Sigma) by preparing a 1:1 dilution of the cell suspension using a 0.4% Trypan blue solution for 5 min. Cell viability was analyzed using hemocytometer.
TUNEL assay
TUNEL assay was carried out using in situ cell death detection kit, TMR red (Roche Group, Indianapolis, IN, USA) according to the manufacturer protocols. Briefly, primary neurons or brain slices treated with ceramide were incubated with TUNEL reagent (labeled with TMR red) at 37 °C for 1 h. TUNEL-positive cell density was calculated as number of apoptotic cells per square millimeter of section.
Caspase-3 activity assay
Caspase-3 activity assay was carried out using Caspase-3 colorimetric assay kit (BioVision) according to the manufacturer protocols. Briefly, cells treated with ceramide were incubated with reaction buffer and DEVD-pNA substrate at 37 °C for 1 h. Caspase-3 activity was measured at 405 nm in a microtiter plate reader.
Brain samples
Whole-brain tissues from WT and DAPK1 KO mice at 6 months of age and from non-Tg, Tg2576/WT and Tg2576/ DAPK1 KO mice at 8 months of age were harvested. WT, DAPK1 KO or Tg2576 mice were previously described.7, 25, 28, 56 WT or DAPK1 KO mice were maintained in pure C57BL/6 background. Tg2576 or Tg2576/DAPK1 KO mice were maintained in C57BL/6 × SJL hybrid background. Tg2576 mice in C57BL/6 × SJL F1 hybrid background were initially crossed with DAPK1 KO in C57BL/6 background to obtain Tg2576:DAPK1+/− and control non-Tg:DAPK1+/−. The mice analyzed for the study were derived from the cross of Tg2576:DAPK1+/− and non-Tg:DAPK1+/− in order to maintain the hemizygosity of Tg2576. After extraction, whole brains were snap frozen in liquid nitrogen-cold isopentane and stored at −80 °C until use. Total brain lysates were obtained from these mouse brain tissues. Brain hippocampus tissues of human Alzheimer’s patients and controls were obtained from the Neuropathology Core of the Massachusetts Alzheimer’s Disease Research Center (Boston, MA, USA) and the Harvard Brain Tissue Resource Center (Boston, MA, USA). Research was conducted in compliance with the policies and principles contained in the Federal Policy for the Protection of Human subjects. In most cases, brain hippocampus tissues obtained within 30 h post mortem were used; 10 from individuals with a clinical diagnosis of probable AD and 8 from individual without any neurological disorder (Supplementary Table 2).
Statistical analysis
The data are presented as means±S.E. of the three independent experiments. Statistical significances were calculated by one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison post-hoc test. GraphPad Prism version 4.0 (La Jolla, CA, USA) was used for statistical analyses.
Study approval
All animal generation, husbandry and experimental procedures were approved by the Beth Israel Deaconess Medical Center’s Institutional Animal Care and Use Committee, and conform to the NIH guidelines of the care and use of laboratory animals. All studies on human samples were approved by the Institutional Review Boards at the Beth Israel Deaconess Medical Center.
Abbreviations
- AD:
-
Alzheimer’s disease
- DAPK1:
-
death-associated protein kinase 1
- NDRG2:
-
N-myc downstream-regulated gene 2
- Aβ:
-
amyloid-β oligomer
- WT:
-
wild-type
- KO:
-
knockout
- MEF:
-
mouse embryonic fibroblast
- CIP:
-
calf intestinal phosphatase
- PARP:
-
poly-ADP-ribose polymerase
- S.E.:
-
standard error
References
Bialik S, Kimchi A . The death-associated protein kinases: structure, function, and beyond. Annu Rev Biochem 2006; 75: 189–210.
Deiss LP, Feinstein E, Berissi H, Cohen O, Kimchi A . Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev 1995; 9: 15–30.
Eisenberg-Lerner A, Kimchi A . DAP kinase regulates JNK signaling by binding and activating protein kinase D under oxidative stress. Cell Death Differ 2007; 14: 1908–1915.
Kuo JC, Lin JR, Staddon JM, Hosoya H, Chen RH . Uncoordinated regulation of stress fibers and focal adhesions by DAP kinase. J Cell Sci 2003; 116 (Pt 23): 4777–4790.
Bovellan M, Fritzsche M, Stevens C, Charras G . Death-associated protein kinase (DAPK) and signal transduction: blebbing in programmed cell death. FEBS J 2010; 277: 58–65.
Chen CH, Wang WJ, Kuo JC, Tsai HC, Lin JR, Chang ZF et al. Bidirectional signals transduced by DAPK-ERK interaction promote the apoptotic effect of DAPK. EMBO J 2005; 24: 294–304.
Gozuacik D, Bialik S, Raveh T, Mitou G, Shohat G, Sabanay H et al. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ 2008; 15: 1875–1886.
Fujita Y, Yamashita T . Role of DAPK in neuronal cell death. Apoptosis 2014; 19: 339–345.
Shu S, Pei L, Lu Y . Promising targets of cell death signaling of NR2B receptor subunit in stroke pathogenesis. Regen Med Res 2014; 2: 8.
Pei L, Shang Y, Jin H, Wang S, Wei N, Yan H et al. DAPK1-p53 interaction converges necrotic and apoptotic pathways of ischemic neuronal death. J Neurosci 2014; 34: 6546–6556.
He C, Stroink AR, Wang CX . The role of DAPK-BimEL pathway in neuronal death induced by oxygen-glucose deprivation. Neuroscience 2014; 258: 254–262.
Yamamoto M, Hioki T, Ishii T, Nakajima-Iijima S, Uchino S . DAP kinase activity is critical for C(2)-ceramide-induced apoptosis in PC12 cells. Eur J Biochem 2002; 269: 139–147.
Fujita Y, Taniguchi J, Uchikawa M, Endo M, Hata K, Kubo T et al. Neogenin regulates neuronal survival through DAP kinase. Cell Death Differ 2008; 15: 1593–1608.
Pelled D, Raveh T, Riebeling C, Fridkin M, Berissi H, Futerman AH et al. Death-associated protein (DAP) kinase plays a central role in ceramide-induced apoptosis in cultured hippocampal neurons. J Biol Chem 2002; 277: 1957–1961.
Schori H, Yoles E, Wheeler LA, Raveh T, Kimchi A, Schwartz M . Immune-related mechanisms participating in resistance and susceptibility to glutamate toxicity. Eur J Neurosci 2002; 16: 557–564.
Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM et al. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell 2010; 140: 222–234.
Shamloo M, Soriano L, Wieloch T, Nikolich K, Urfer R, Oksenberg D . Death-associated protein kinase is activated by dephosphorylation in response to cerebral ischemia. J Biol Chem 2005; 280: 42290–42299.
Velentza AV, Wainwright MS, Zasadzki M, Mirzoeva S, Schumacher AM, Haiech J et al. An aminopyridazine-based inhibitor of a pro-apoptotic protein kinase attenuates hypoxia-ischemia induced acute brain injury. Bioorg Med Chem Lett 2003; 13: 3465–3470.
Liu SB, Zhang N, Guo YY, Zhao R, Shi TY, Feng SF et al. G-protein-coupled receptor 30 mediates rapid neuroprotective effects of estrogen via depression of NR2B-containing NMDA receptors. J Neurosci 2012; 32: 4887–4900.
Llambi F, Lourenco FC, Gozuacik D, Guix C, Pays L, Del Rio G et al. The dependence receptor UNC5H2 mediates apoptosis through DAP-kinase. EMBO J 2005; 24: 1192–1201.
Yukawa K, Tanaka T, Bai T, Li L, Tsubota Y, Owada-Makabe K et al. Deletion of the kinase domain from death-associated protein kinase enhances spatial memory in mice. Int J Mol Med 2006; 17: 869–873.
Li Y, Grupe A, Rowland C, Nowotny P, Kauwe JS, Smemo S et al. DAPK1 variants are associated with Alzheimer's disease and allele-specific expression. Hum Mol Genet 2006; 15: 2560–2568.
Li H, Wetten S, Li L St, Jean PL, Upmanyu R, Surh L et al. Candidate single-nucleotide polymorphisms from a genome-wide association study of Alzheimer disease. Arch Neurol 2008; 65: 45–53.
Laumet G, Chouraki V, Grenier-Boley B, Legry V, Heath S, Zelenika D et alSystematic analysis of candidate genes for Alzheimer's disease in a French, genome-wide association studyJ Alzheimer's Dis 2010; 20 (4): 1181–1188.
Kim BM, You MH, Chen CH, Lee S, Hong Y, Hong Y et al. Death-associated protein kinase 1 has a critical role in aberrant tau protein regulation and function. Cell Death Dis 2014; 5: e1237.
Wu PR, Tsai PI, Chen GC, Chou HJ, Huang YP, Chen YH et al. DAPK activates MARK1/2 to regulate microtubule assembly, neuronal differentiation, and tau toxicity. Cell Death Differ 2011; 18: 1507–1520.
Duan DX, Chai GS, Ni ZF, Hu Y, Luo Y, Cheng XS et alPhosphorylation of tau by death-associated protein kinase 1 antagonizes the kinase-induced cell apoptosisJ Alzheimer's Dis 2013; 37 (4): 795–808.
Kim BM, You MH, Chen CH, Suh J, Tanzi RE, Lee TH . Inhibition of death-associated protein kinase 1 attenuates the phosphorylation and amyloidogenic processing of amyloid precursor protein. Hum Mol Genet 2016 (e-pub ahead of print; doi:10.1093/hmg/ddw114).
Lee TH, Chen CH, Suizu F, Huang P, Schiene-Fischer C, Daum S et al. Death-associated protein kinase 1 phosphorylates Pin1 and inhibits its prolyl isomerase activity and cellular function. Mol Cell 2011; 42: 147–159.
Lee TH, Pastorino L, Lu KP . Peptidyl-prolyl cis-trans isomerase Pin1 in ageing, cancer and Alzheimer disease. Expert Rev Mol Med 2011; 13: e21.
Hutti JE, Shen RR, Abbott DW, Zhou AY, Sprott KM, Asara JM et al. Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKepsilon promotes cell transformation. Mol Cell 2009; 34: 461–472.
Hutti JE, Jarrell ET, Chang JD, Abbott DW, Storz P, Toker A et al. A rapid method for determining protein kinase phosphorylation specificity. Nat Methods 2004; 1: 27–29.
Liu N, Wang L, Li X, Yang Q, Liu X, Zhang J et al. N-Myc downstream-regulated gene 2 is involved in p53-mediated apoptosis. Nucleic Acids Res 2008; 36: 5335–5349.
Kim HS, Kim MJ, Lim J, Yang Y, Lee MS, Lim JS . NDRG2 overexpression enhances glucose deprivation-mediated apoptosis in breast cancer cells via inhibition of the LKB1-AMPK pathway. Genes Cancer 2014; 5: 175–185.
Cao W, Zhang JL, Feng DY, Liu XW, Li Y, Wang LF et al. The effect of adenovirus-conjugated NDRG2 on p53-mediated apoptosis of hepatocarcinoma cells through attenuation of nucleotide excision repair capacity. Biomaterials 2014; 35: 993–1003.
Li Y, Shen L, Cai L, Wang Q, Hou W, Wang F et al. Spatial-temporal expression of NDRG2 in rat brain after focal cerebral ischemia and reperfusion. Brain Res 2011; 1382: 252–258.
Mitchelmore C, Buchmann-Moller S, Rask L, West MJ, Troncoso JC, Jensen NA . NDRG2: a novel Alzheimer's disease associated protein. Neurobiol Dis 2004; 16: 48–58.
Herskowitz JH, Seyfried NT, Duong DM, Xia Q, Rees HD, Gearing M et al. Phosphoproteomic analysis reveals site-specific changes in GFAP and NDRG2 phosphorylation in frontotemporal lobar degeneration. J Proteome Res 2010; 9: 6368–6379.
Mukherjee S, Kim S, Gibbons LE, Nho K, Risacher SL, Glymour MM et al. Genetic architecture of resilience of executive functioning. Brain Imaging Behav 2012; 6: 621–633.
Mielke MM, Lyketsos CG . Alterations of the sphingolipid pathway in Alzheimer's disease: new biomarkers and treatment targets? Neuromol Med 2010; 12: 331–340.
Panchal M, Gaudin M, Lazar AN, Salvati E, Rivals I, Ayciriex S et al. Ceramides and sphingomyelinases in senile plaques. Neurobiol Dis 2014; 65: 193–201.
Jin Y, Blue EK, Gallagher PJ . Control of death-associated protein kinase (DAPK) activity by phosphorylation and proteasomal degradation. J Biol Chem 2006; 281: 39033–39040.
Widau RC, Jin Y, Dixon SA, Wadzinski BE, Gallagher PJ . Protein phosphatase 2A (PP2A) holoenzymes regulate death-associated protein kinase (DAPK) in ceramide-induced anoikis. J Biol Chem 2010; 285: 13827–13838.
Shohat G, Spivak-Kroizman T, Cohen O, Bialik S, Shani G, Berrisi H et al. The pro-apoptotic function of death-associated protein kinase is controlled by a unique inhibitory autophosphorylation-based mechanism. J Biol Chem 2001; 276: 47460–47467.
Hardy J, Selkoe DJ . The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002; 297: 353–356.
Abad MA, Enguita M, DeGregorio-Rocasolano N, Ferrer I, Trullas R . Neuronal pentraxin 1 contributes to the neuronal damage evoked by amyloid-beta and is overexpressed in dystrophic neurites in Alzheimer's brain. J Neurosci 2006; 26: 12735–12747.
Biswas SC, Shi Y, Vonsattel JP, Leung CL, Troy CM, Greene LA . Bim is elevated in Alzheimer's disease neurons and is required for beta-amyloid-induced neuronal apoptosis. J Neurosci 2007; 27: 893–900.
Cheung YT, Zhang NQ, Hung CH, Lai CS, Yu MS, So KF et al. Temporal relationship of autophagy and apoptosis in neurons challenged by low molecular weight beta-amyloid peptide. J Cell Mol Med 2011; 15: 244–257.
Jazvinscak Jembrek M, Hof PR, Simic G . Ceramides in Alzheimer's disease: key mediators of neuronal apoptosis induced by oxidative stress and Abeta accumulation. Oxid Med Cell Longev 2015; 2015: 346783.
Guenebeaud C, Goldschneider D, Castets M, Guix C, Chazot G, Delloye-Bourgeois C et al. The dependence receptor UNC5H2/B triggers apoptosis via PP2A-mediated dephosphorylation of DAP kinase. Mol Cell 2010; 40: 863–876.
Nakahata S, Ichikawa T, Maneesaay P, Saito Y, Nagai K, Tamura T et al. Loss of NDRG2 expression activates PI3K-AKT signalling via PTEN phosphorylation in ATLL and other cancers. Nat Commun 2014; 5: 3393.
Park Y, Shon SK, Kim A, Kim KI, Yang Y, Cho DH et al. SOCS1 induced by NDRG2 expression negatively regulates STAT3 activation in breast cancer cells. Biochem Biophys Res Commun 2007; 363: 361–367.
Yamamoto M, Takahashi H, Nakamura T, Hioki T, Nagayama S, Ooashi N et al. Developmental changes in distribution of death-associated protein kinase mRNAs. J Neurosci Res 1999; 58: 674–683.
Niethammer M, Smith DS, Ayala R, Peng J, Ko J, Lee MS et al. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron 2000; 28: 697–711.
Saha RN, Ghosh A, Palencia CA, Fung YK, Dudek SM, Pahan K . TNF-alpha preconditioning protects neurons via neuron-specific up-regulation of CREB-binding protein. J Immunol 2009; 183: 2068–2078.
Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996; 274: 99–102.
Acknowledgements
We thank S-H Min for providing a NDRG2 construct and A Kimchi for providing DAPK1 KO mice. Human brain tissues were provided by the Neuropathology Core of the Massachusetts Alzheimer Disease Research Center (P50AG05134) and the Harvard Brain Tissue Resources Center (R24MH068855). The work was supported by grants from the Korea Healthcare Technology R&D Project, Ministry for Health & Welfare Affairs, Republic of Korea (HI08C2149) to BM Kim and NIH grant (R00AG033104), the Alzheimer’s Association (NIRG-12-258863), the American Federation for Aging Research, and the Massachusetts Alzheimer’s Disease Research Center (P50AG005134) to TH Lee.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by L Greene
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
You, MH., Kim, B., Chen, CH. et al. Death-associated protein kinase 1 phosphorylates NDRG2 and induces neuronal cell death. Cell Death Differ 24, 238–250 (2017). https://doi.org/10.1038/cdd.2016.114
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.114
