
Abstract
Background:
Differences in genetics and receptor expression (phenotypes) of invasive ductal breast cancer (IDC) impact on prognosis and treatment response. Immunohistochemistry (IHC), the most used technique for IDC phenotyping, has some limitations including its invasiveness. We explored the possibility of contrast-enhanced positron emission tomography magnetic resonance (CE-FDG PET/MR) to discriminate IDC phenotypes.
Methods:
21 IDC patients with IHC assessment of oestrogen receptor (ER), progesterone receptor (PR), human epidermal growth factor-2 (HER2), and antigen Ki-67 (Ki67) underwent CE-FDG PET/MR. Magnetic resonance-perfusion biomarkers, apparent diffusion coefficient (ADC), and standard uptake value (SUV) were compared with IHC markers and phenotypes, using a Student’s t-test and one-way ANOVA.
Results:
ER/PR− tumours demonstrated higher Kepmean and SUVmax than ER or PR+ tumours. HER2− tumours displayed higher ADCmean, Kepmean, and SUVmax than HER2+tumours. Only ADCmean discriminated Ki67⩽14% tumours (lower ADCmean) from Ki67>14% tumours. PET/MR biomarkers correlated with IHC phenotype in 13 out of 21 patients (62%; P=0.001).
Conclusions:
Positron emission tomography magnetic resonance might non-invasively help discriminate IDC phenotypes, helping to optimise individual therapy options.
Similar content being viewed by others

Main
The mainstay of breast cancer treatment remains surgical resection, but the addition of systemic chemotherapy can reduce the risk of relapse (Santa-Maria et al, 2015). Neoadjuvant chemotherapy has been shown to have equivalent rates of disease-free survival (DFS) and overall survival (OS) when compared to adjuvant chemotherapy (Mauri et al, 2005). The benefit to neoadjuvant chemotherapy is the increased rate of breast-conserving surgery (Makris et al, 1998; Rastogi et al, 2008). The adjuvant breast and bowel project has shown that patients receiving neoadjuvant chemotherapy who had a complete pathologic response have a significantly improved DFS and OS. In the adjuvant breast and bowel project, the percentage of patients with a complete pathologic response was as much as 26% depending on the chemotherapy regimen (Rastogi et al, 2008), and according to a meta-analysis, rates of complete pathologic response ranged from 4 to 29% (Mauri et al, 2005). However, as many as 21% of patients in the adjuvant breast and bowel project had no clinical response (Rastogi et al, 2008).
It is also known that rates of response to neoadjuvant chemotherapy depend significantly on the biomarker profiles of the primary breast cancer (Precht et al, 2010; Esserman et al, 2012). Therefore, the ability to reliably identify the biomarker profile (phenotype) within the breast cancer may allow better selection of patients for neoadjuvant chemotherapy and selection of individualised therapy regimens, building the pillars for personalised medicine. For example, luminal A tumours have been shown to be less chemosensitive than other phenotypes of breast cancer, with some studies showing that adjuvant chemotherapy did not provide survival benefit for patients (Prat et al, 2015). Some studies have suggested that patients with luminal A breast cancer when treated with paclitaxel benefit only if the proliferation index is low. In addition, in patients with HER2+tumours, treatment with trastuzumab has shown to be beneficial with increases in complete response and event-free survival (Prat et al, 2015). These are just a few examples of how regimens targeted at tumour phenotype have proven beneficial.
This is particularly relevant in breast cancer due to its high level of heterogeneity (Shipitsin et al, 2007; Almendro and Fuster, 2011). Core biopsies are currently used to investigate cancer biomarkers, but only a small portion of the tumour can be sampled, which is not necessarily the predominant nor the most aggressive area, raising the possibility of under-estimating the most aggressive phenotype of cancer (Marusyk and Polyak, 2010). The sampling error of core biopsies can lead to treating patients who may not benefit from neoadjuvant chemotherapy or administering a chemotherapy regimen that is not tailored to the patient.
In this study, we explored the ability of contrast-enhanced breast [18F] FDG positron emission tomography magnetic resonance (PET/MR) to identify the phenotype of breast cancer.
Materials and methods
Patient enrolment
This Health Insurance Portability and Accountability Act compliant retrospective study was approved by the institutional review board. Patients gave written informed consent for study enrolment, including the possibility of using their imaging, genetics, and clinical data in future evaluations.
The authors had full control of the data and information submitted for publication and did not receive any financial support for this research study.
Inclusion criteria: (a) recent diagnosis of invasive ductal carcinoma, (b) biopsy of the primary breast cancer with available biomarkers, (c) absence of treatment, (d) age older than 18 years, (e) whole-body contrast-enhanced 18F FDG PET/MR that included breast MR perfusion, (f) breast cancer diameter of at least 20 mm, and (g) whole-body staging follow-up of ⩾12 months. We decided to choose a very conservative minimum size criterion, a tumour diameter of at least 20 mm, 5 times as thick as our DWI sequence, to improve the robustness of ADC measurements, to reduce the risk of averaging areas of possible geometric distortion, and to reduce the risk of partial volume effect being 10 times as big as the PET voxel size.
Exclusion criteria included: (a) pregnancy; (b) blood glucose levels >140 mg dl−1 (7.77 mmol l−1); (c) inadequate PET/MR images; (d) contraindication to MR imaging; and (e) inability to tolerate being in the PET/MR imager.
Pathology
Core needle biopsies were performed under ultrasound guidance by a radiologist with more than 15 years of experience (OAC). A pathology technologist fixed them in 10% neutral buffered formalin (NBF) at the time of biopsy. All patients who underwent mastectomy after completion of neoadjuvant chemotherapy had the mastectomy specimens sent to the department of pathology immediately after resection. They were cut in 5 mm thick slices and fixed in NBF for 6–48 h. Haematoxylin and eosin and immunohistochemistry (IHC) studies were evaluated by one pathologist (ML) with more than 12 years of experience in breast pathology. Staining for oestrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor-2 (HER2) was performed as per AAP guidelines (Wolff et al, 2007; Hammond et al, 2010).
The Benchmark Ultra staining module (Ventana Medical Systems Inc, Tucson, AZ, USA) was employed for IHC assessment of ER, PR, and HER2. Paraffin fixed specimens were placed on positively charged slides after being cut into 5 μm-thick slices. For assessment of ER and PR, slices were incubated at 70 °C for 2 h; for HER2 assessment they were air-dried overnight at room temperature. The following primary monoclonal antibodies (Ventana Medical Systems Inc) were used: for ER, CONFIRM anti-ER (SP1, 1 μg ml−1); for PR, CONFIRM anti-PR (1E2, 1 μg ml−1); and for HER2, PATHWAY anti-HER2/neu (4B5, 6 μg ml−1). The pre-diluted monoclonal Clone 30-9 (Ventana Medical Systems Inc) was used for assessment of antigen Ki-67 (Ki-67) expression, and a count of more than 500 neoplastic cells was employed to measure the fraction of proliferating cells (KI-67 positive).
PET/MR protocol
All patients fasted for at least 6 h before imaging. Blood glucose levels were assessed with a blood glucometer (OneTouch Vita; LifeScan, Milpitas, CA, USA) before imaging to ensure it was <140 mg dl−1 (7.77 mmol l−1).
Positron emission tomography magnetic resonance imaging began 85±24 min after FDG injection (mean dose, 4.44 MBq per kilogram of body weight, range, 370–400 MBq). This incubation time is explained by the legal and IRB mandatory requirement of obtaining a standard of care PET-CT 60 min after FDG injection before acquiring any PET-MR. However, the reconstruction software automatically corrects for the delay between the time of FDG injection and that of PET data acquisition for each bed position.
Positron emission tomography magnetic resonance studies were acquired with a Biograph mMR imager (Siemens Healthcare). They included a whole-body PET/MR acquisition and a dedicated breast MR perfusion protocol. The whole-body PET/MR was performed with a 16-channel head and neck surface coil and three or four 12-channel body coils (depending on patient height) combined to form a multichannel whole-body coil by using total imaging matrix technology. Positron emission tomography magnetic resonance was acquired with the patient supine, from the mid-thigh towards the head. Positron emission tomography data were automatically attenuation corrected with attenuation maps generated from the two-point Dixon sequence. This portion of the exam was used only for measuring the SUVmax of the primary breast cancer; therefore, in Table 1 we provide only the pertinent parameters. After this acquisition, the patient was placed prone, and the dedicated breast study, including DWI and MR perfusion, was acquired with a dedicated four-channel breast coil. The scanner automatically generated both the ADC maps and the subtracted post-contrast images at each time point. The pertinent parameters are outlined in Table 1.
Image analysis
Images were analysed in consensus, by a nuclear medicine physician (AS) with 33 years of experience and by a radiologist with 17 years of experience in MR, on a dedicated workstation (SyngoVia, Siemens Healthcare). The automated PET encircling function from the mMR-general layout workflow was used to include the entire FDG avid tumour and obtain the SUVmax. For measuring the ADCmean, the same layout workflow was used; the entire tumour was manually encircled on the DWI and ADC images. Spatial correlation with subtracted MR perfusion images was employed to improve lesion identification, when necessary. For the MR perfusion analysis, a Tofts-based commercially available software (Tissue4D, Siemens Healthcare) was employed. The entire tumour was manually encircled on the earliest contrast-enhanced subtracted image set that best demonstrated the cancer against the adjacent normal tissues. Then, the following measurements were obtained: mean volume transfer coefficient (Ktrans), mean flux rate constant (Kep), mean extracellular volume ratio (Ve), and the initial area under the curve (iAUC).
Statistical analysis
The possible association between PET-MR biomarkers (ADCmean, Ktransmean, Kepmean, Vemean, iAUC, and SUVmax) and IHC expression of ER, PR, HER2, and Ki67 was assessed using two-sample Student’s t-tests, with a 0.05 significance level. We matched our PET/MR biomarkers against IHC expression of ER, PR, HER2, and Ki-67 on the biopsies from all patients. The biomarkers were also matched against a cohort that included the expression of the markers in the entire tumour in those with residual cancer at mastectomy plus the biopsy in those with a pathologic complete response at mastectomy.
The utility of biomarkers for predicting tumour phenotype was assessed by one-way ANOVA. Tumour phenotype prediction models were fit using linear discriminant analysis with leave-one-out cross-validation. The statistical significance of fitted models was assessed by permutation analysis. Two analyses were performed. First, we predicted biopsy phenotype for all patients. We also fit a second model, using full-tumour data when available, and biopsy data for patients who did not have detectable residual cancer. The statistical significance of the number of correct predictions was determined by simulation, conditional on the observed frequencies of subtypes in the data. Data were analysed using R (Version 3.1.0; R Development Core Team, 2014), with the MASS library extension (Venables et al, 2002).
Results
We recruited 49 consecutive newly diagnosed non-operated patients with IDC (all women, age 18–79) between March 2012 and February 2013. Twenty-eight of these were excluded for the following reasons: 19 for non-availability of histology biomarkers and 9 for having a cancer <20 mm in maximal diameter. Therefore, our final population consisted of 21 patients. They underwent neoadjuvant chemotherapy that consisted of 5-Fluorouracil, epirubicin, and cyclophosphamide (FEC) in 16, FEC plus paclitaxel in 3, and paclitaxel in 2.
For all these 21 patients, core biopsy specimens were available. Entire tumour specimens from mastectomy were available for 16/21 patients; due to complete response to neoadjuvant chemotherapy in 5/21, no residual tumour was available. ER, PR, HER2, and Ki67 were assessed for 21/21 patients on biopsy specimens, and on 16/21 on the whole excised tumour.
On biopsy specimens, we found 14/21 ER+, 12/21 PR+, 10/21 HER2+, and 12/21 Ki-67 ⩽14% patients (Table 2). This resulted in 3/21 patients with a luminal A phenotype, 8/21 with a luminal B phenotype, 3/21 with a luminal HER2 phenotype, and 7/21 with a HER2 enriched phenotype (Table 2).
On whole-tumour assessment in the 16/21 patients with residual tumour after neoadjuvant chemotherapy, we found: 11/16 ER+, 9/16 PR+, 6/16 HER2+, and 10/16 Ki67 ⩽14% tumours (Table 2). Three of these 16 patients were Ki67>14% on biopsy but Ki67 ⩽14% on whole-tumour evaluation. In summary, upon entire residual tumour evaluation 5/16 patients had a luminal A phenotype, 5/16 a luminal B phenotype, 1/16 a luminal HER2 phenotype, and 5/16 a HER2 enriched phenotype.
Combining the IHC of residual cancer at mastectomy in 16 cases and that obtained on biopsy in the 5 cases who had complete response, we got these final results: 6/21 luminal A, 5/21 luminal B, 3/21 luminal Her2, and 7/21 Her2 enriched (Table 2).
Patients generally had high-grade tumours with 0/21 grade 1, 14/21 grade 2, and 7/21 grade 3. Based on biopsy and on entire tumour plus biopsy, for those with complete regression, the same results were obtained when evaluating for expression of ER, PR, and HER2. Both the mean flux rate constant (Kepmean) and maximum standard uptake value (SUVmax) differentiated ER/PR− and ER or PR+ tumours (Figures 1, 2, 3). Statistically significant higher values of Kepmean (P=0.011) and SUVmax (P=0.005) were found in ER/PR− tumours as compared to ER/PR+ tumours. No other imaging biomarkers were significantly different (Table 3).

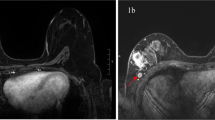
ER and/or PR and PET/MR biomarkers. Diagram displaying the correlation between ER and/or PR expression and Kep (A). Diagram displaying the correlation between ER and/or PR expression and SUVmax (B). ER+ and PR+ cancer: a high-resolution contrast-enhanced axial T1w VIBE (C) is shown with the corresponding axial Kep parametric map (D) and axial PET image (E). ER− and PR− cancer: a perfusion subtracted MRI (F) is shown with the corresponding parametric axial Kep parametric map (G) and axial PET image (H). The ER+ and PR+ cancer (arrow) is well depicted after contrast (C) and shows an extremely low Kep value, as demonstrated by the black colour (D). In Kep parametric maps, low values are displayed by dark colours (from black to dark tones of blue), meanwhile high Kep values are presented in light colours (from lighter tones of blues, to green and then yellow). The cancer also takes up low amounts of FDG (E). The ER− and PR− cancer (arrow) markedly enhances after contrast administration (F) and exhibits higher Kep values (G), as demonstrated by the light blue to green colour in the Kep parametric map. FDG uptake is pronounced (H).

Her2 and PET/MR biomarkers. Diagram demonstrating correlation between Her2 expression and Kep(A); diagram showing correlation between Her2 expression and ADC (B); diagram displaying correlation between Her2 expression and SUVmax (C). Her2+ cancer (D–F). A Kep parametric map (D) is shown with a corresponding ADC map (E) and axial PET (F). The Her2+ cancer (arrow) demonstrates high Kep values (light blue) (D), low ADC values (dark grey) (E), and high FDG uptake (F). Her2− cancer (G–I). A Kep parametric map (G) is shown with a corresponding ADC map (H) and axial PET (I). Her2− cancer displays low Kep values (dark blue) (G), high ADC values (light gray) (F), and low FDG uptake (I).

Ki67 and PET/MR biomarkers.Diagram displaying correlation between expression of Ki67 and ADC mean (A). Ki67− cancer (B and C). Axial high-resolution contrast-enhanced MRI (B) is shown with a corresponding ADC map (C). The Ki67− cancer (arrow) demonstrates low values on the ADC map (C). Ki67+ cancer (D and E). An axial high-resolution contrast-enhanced MRI (D) is shown with a corresponding ADC map (E). The Ki67+ cancer (arrow) exhibits higher ADC values (E).
Statistically significant higher values of ADCmean (P=0.009), Kepmean (P=0.028), and SUVmax (P=0.046) were measured in HER2 negative compared to HER2-positive tumours (Figure 2 and Table 3).
As far as Ki67 expression, ADCmean values were statistically significantly lower in Ki67⩽14% compared to Ki67>14% expression (P=0.025 on biopsy, P=0.011 on entire cancer plus biopsy; Table 3 and Figure 3). No other PET-MR biomarkers behaved differently in relation to Ki67 expression.
No grading differences were encountered between core biopsy and whole-cancer specimens. Grade 2 breast cancers had lower Kepmean (P=0.036) and lower SUVmax (P=0.042) than grade 3 tumours (Table 3). However, since there were no grade 1 tumours in our patients, we could not evaluate the differentiation of all grades.
No other imaging biomarkers showed a statistically significant correlation with breast cancer biomarkers. Specifically, the transfer constant (Ktransmean) was not significantly different for any of the tested molecular expressions.
Upon multivariate analysis utilising Kepmean, ADCmean, and SUVmax, we were able to correctly identify the tumour phenotype in 14/21 biopsies (67%; P=0.001). However, upon cross-validation, PET-MR biomarkers correctly predicted 8/21 phenotypes (38%), and this was not statistically significant (P=0.22; Table 4).
When we explored the capability of the PET-MR biomarkers against the phenotypes obtained from entire cancer specimens from 16 patients plus biopsies from the 5 cases who experienced complete remission, our model was capable of predicting the correct phenotype in 13 out of 21 patients (62%; P=0.001), and when we cross-validated these results, the model correctly predicted 10 out of 21 cases (48%). These results had statistical significance (P=0.04; Table 4).
Discussion
Utilising a multivariate analysis with (a) SUVmax, (b) ADCmean, and (c) Kepmean, we were able to correctly predict the phenotypes of breast cancer in 14/21 cases (67%) compared to biopsy and 13/21 (62%) compared to excision of the whole tumour plus biopsy in the five cases without residual disease at mastectomy. While numerous previous studies have looked at the correlations of PET and MR biomarkers with tumour genotypes and phenotypes, to our knowledge, this is the first study that attempts to predict tumour phenotype based on a multivariate analysis of both PET and MR biomarkers. This could be useful for future patient treatment or help patient selection for neoadjuvant therapy trials since selected therapy is often based on hormone receptor status, HER-2 status, and proliferation rate (Prat et al, 2015).
There was a statistically significant direct correlation of ADCmean with HER2-positive tumours (P=0.009), which is in agreement with several previous papers (Martincich et al, 2012; Choi et al, 2012a; Richard et al, 2013; Park et al, 2015; Lee et al, 2016). In our population, we found a direct correlation between ADCmean and Ki-67 ⩽14% on biopsy (P=0.025) and excision plus biopsy (P=0.011). However, published data on ADC and Ki-67 expression in breast cancer are conflicting (Jeh et al, 2011; Choi et al, 2012b; De Felice et al, 2014; Molinari et al, 2015; Mori et al, 2015). Specifically, Mori and colleagues showed a correlation of multiple different ADC parameters with Ki-67 at a cutoff of 14%, establishing a threshold cutoff of 1097 × 10−6 mm2 s−1 which was 82% sensitive and 71% specific for Ki-67 proliferation. However, they found that the high proliferation tumours had lower ADC as opposed to our findings where low proliferation tumours had a lower ADC value (Mori et al, 2015). On the other hand, DeFelice and Jeh found no correlation between ADC and Ki-67 expression (Jeh et al, 2011; De Felice et al, 2014). ADC is influenced by several factors, including grading, with a demonstrated direct correlation between ADC and higher tumour grade G3 (De Felice et al, 2014). In our study, the majority of the Ki-67 negative cancers were grade 2, and this might have contributed to the lower ADC observed in the setting of Ki-67 ⩽14%.
There was a statistically significant correlation of a high Kepmean with ER/PR negativity (P=0.011 when compared to ER or PR +) and HER-2 positivity (P=0.028). Enhancement characteristics have long been of interest in differentiating subtypes and phenotypes of breast cancer. As early as 2003, Szabo et al (2003) evaluated breast tumours on the basis of rim enhancement, washout, and early maximal enhancement, demonstrating that there was a correlation with these findings and markers of poor prognosis. More recently, studies have corroborated our findings of a correlation with Kep and Ki-67 proliferation (Kim et al, 2015) and ER negativity (Koo et al, 2012). Koo et al (2012) took the evaluation one step further and showed that triple-negative breast cancers had a higher mean Kep than luminal type breast cancers.
There was a statistically significant correlation of a high SUVmax with ER/PR-negative tumours (P=0.005) and HER-2 positivity (P=0.046). Prior studies have gone as far as to correlate a lower SUVmax with an improved DFS (Kadoya et al, 2013; Aogi et al, 2015; Garcia Vicente et al, 2015) and OS (Aogi et al, 2015; Garcia Vicente et al, 2015). This suggests a link between SUVmax and tumour biology and/or phenotype. Several prior studies have shown a correlation with either ER or PR negativity (Ueda et al, 2008; Groheux et al, 2011; De Cicco et al, 2013; Kadoya et al, 2013; Miyake et al, 2014; Yoon et al, 2014) and HER-2 enriched tumours (De Cicco et al, 2013) with an elevated SUVmax.
Several prior studies have attempted to correlate different MR and PET biomarkers with breast cancer phenotypes including a high median ADC with HER-2 enriched tumours (Martincich et al, 2012), differences in ADC between multiple phenotypes (Richard et al, 2013; Lee et al, 2016), a low SUVmax for luminal A tumours (Miyake et al, 2014), and increased SUVmax with triple-negative breast cancers (Groheux et al, 2011; Yoon et al, 2014). While we agree that evaluating for the phenotype of a malignancy is important, rather than comparing the phenotypes by single parameters, we propose evaluating for phenotypes based on a multivariate algorithm. In our analysis, we compared different MR biomarkers and SUVmax values with particular receptors, then used a multivariate analysis to determine luminal types based on the receptors exhibited. For example, an ADC value that correlates with HER-2 positivity along with a mean Kep and SUVmax which correlate with ER or PR positivity lend itself to a luminal HER-2 tumour.
In our analysis, we were in agreement with the phenotype of breast cancer based on biopsy in 16/21 (67%) cases. However, after cross validation only 8/21 phenotypes (38%) were predicted, and this was not statistically significant (P=0.22). However, based on excised specimens in those with residual cancer plus biopsy for those with complete response, we correctly predicted the phenotype in 13 of 21 patients (62%), and these predictions retained statistical significance after cross-validation predicting 10 out of 21 cases (48%; P=0.004). Although these initial results are encouraging, the prediction rate is still low and they cannot be translated into clinical practice. The incapability to be correct in predicting the remaining cases could be potentially explained by the heterogeneity of the primary breast cancer and by the necessity to rely on biopsy for the 5 cases that experienced complete response after neoadjuvant chemotherapy. In these 5 cases, we had to use the IHC performed on the small biopsied portion of the cancer for the analysis. PET/MR, assessing the entire tumour, can potentially provide a more comprehensive assessment of its biology compared to biopsy, which is limited to a small area of the cancer, not always representative of the entire neoplastic biology.
A significant limitation to our study is the small number of cases. Therefore, our data should be considered as a preliminary investigation. A larger sample size would increase our power for identifying more useful MR and PET biomarkers. Other studies showed a statistically significant correlation between Ki-67 level and SUVmax (Buck et al, 2002; De Cicco et al, 2013; Yue et al, 2015); the lack of correlation in our study might be related to differences in population and/or sample size. Additional biomarkers as supported by a larger study may lead to improved phenotypic identification by imaging. With data from more patients we could build a more reliable and accurate algorithm that could be validated prospectively.
Our findings also relied on regions of interest (ROIs), which included the whole tumour rather than spot ROIs. Spot ROIs may better account for tumour heterogeneity, especially in the case of large tumours. Although PET/MR is capable of providing such spot ROIs, they might need to be checked against the exact same corresponding area within the sliced tumour.
In a study by Arponen and colleagues, they demonstrated that by placing ROIs in the area of the tumour with the lowest ADC they could show a significant correlation with low ADC and axillary lymph node metastases, lymphovascular invasion, and high tumour grade. This was in addition to the correlation that was seen in both whole-tumour ROIs and spot ROIs with low ADC that included PR positivity and stage (Arponen et al, 2015). PET/MR derived colour-coded biomarker maps are amenable to a section-by-section analysis to highlight the differences in phenotype expression in different areas of each section, pointing out the most aggressive ones that need to be selectively biopsied. The targeting of biopsies for the areas of the primary breast cancer with more concerning features by MR biomarkers may be a future direction. A comparison between random and targeted biopsies could be evaluated for clinically significant heterogeneity. This likely explains the differences in Ki67 expression between biopsy and surgical pathology that occurred in 3 cases. However, a different response to therapy of different neoplastic clones cannot be completely ruled out as an explanation for this discordance. Evaluation of untreated cancers might overcome this possible limitation. However as per current NCCN guidelines, neoadjuvant chemotherapy is administered to stage IIA, IIB and III cancers that fulfil criteria for breast-conserving surgery except for tumour size (National Comprehensive Cancer Network, 2016).
This preliminary study does have additional limitations. We used FDG since it is the most commonly clinically available radiopharmaceutical and to make our results potentially verifiable by other groups. Receptor-specific radiopharmaceuticals were not used because they are not widely available. This would have potentially rendered our study irreproducible, more expensive (our study was self-funded), and moreover a single receptor-specific radiopharmaceutical might have highlighted only one feature of the receptor expression profile of the cancers. However, we are aware that other radiopharmaceuticals showed potential to characterise breast cancer better that FDG (van de Wiele et al, 2002; Van Den Bossche et al, 2006; Siwowska and Muller, 2015; Accardo et al, 2016; Park et al, 2016).
Moreover, we acquired PET/MR 85±24 min after FDG injection, instead of the usual incubation period of 60 min. Our longer incubation time is explained by the legal obligation and IRB requirements to perform a standard of care PET-CT at 60 min after FDG injection, before acquiring a PET-MR study. However, it is unlikely that this influenced the FDG values we measured. In fact, the PET-MR reconstruction software, approved for clinical use worldwide, automatically corrects for the delay between the time of FDG injection and the time of PET data acquisition for each bed position. Moreover, several studies have demonstrated comparable SUV values between PET-CT and subsequently acquired PET-MR (Pace et al, 2014; Atkinson et al, 2016; Pujara et al, 2016).
The main technical limitation was the use of a PET/MR compatible body coil for PET attenuation correction. In subsequently acquired patients, including follow-ups on this same cohort, we used a dedicated PET/MR compatible breast coil that was not available at the start of the project. However, to ensure consistency, we preferred not to combine patients acquired with different coils. For a preliminary feasibility study, we believe that the findings of properly identifying the breast cancer phenotype in 62–67% of cases is promising regarding the potential of CE-FDG PET/MR to non-invasively assess whole-tumour phenotypes.
Change history
28 March 2017
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Accardo A, Galli F, Mansi R, Del Pozzo L, Aurilio M, Morisco A, Ringhieri P, Signore A, Morelli G, Aloj L (2016) Pre-clinical evaluation of eight DOTA coupled gastrin-releasing peptide receptor (GRP-R) ligands for in vivo targeting of receptor-expressing tumors. EJNMMI Res 6 (1): 17.
Almendro V, Fuster G (2011) Heterogeneity of breast cancer: etiology and clinical relevance. Clin Transl Oncol 13 (11): 767–773.
Aogi K, Kadoya T, Sugawara Y, Kiyoto S, Shigematsu H, Masumoto N, Okada M (2015) Utility of (18)F FDG-PET/CT for predicting prognosis of luminal-type breast cancer. Breast Cancer Res Treat 150 (1): 209–217.
Arponen O, Sudah M, Masarwah A, Taina M, Rautiainen S, Kononen M, Sironen R, Kosma VM, Sutela A, Hakumaki J, Vanninen R (2015) Diffusion-weighted imaging in 3.0 Tesla breast MRI: diagnostic performance and tumor characterization using small subregions vs. whole tumor regions of interest. PLoS One 10 (10): e0138702.
Atkinson W, Catana C, Abramson JS, Arabasz G, McDermott S, Catalano O, Muse V, Blake MA, Barnes J, Shelly M, Hochberg E, Rosen BR, Guimaraes AR (2016) Hybrid FDG-PET/MR compared to FDG-PET/CT in adult lymphoma patients. Abdom Radiol (NY) 41 (7): 1338–1348.
Buck A, Schirrmeister H, Kuhn T, Shen C, Kalker T, Kotzerke J, Dankerl A, Glatting G, Reske S, Mattfeldt T (2002) FDG uptake in breast cancer: correlation with biological and clinical prognostic parameters. Eur J Nucl Med Mol Imaging 29 (10): 1317–1323.
Choi BB, Kim SH, Kang BJ, Lee JH, Song BJ, Jeong SH, Yim HW (2012a) Diffusion-weighted imaging and FDG PET/CT: predicting the prognoses with apparent diffusion coefficient values and maximum standardized uptake values in patients with invasive ductal carcinoma. World J Surg Oncol 10: 126.
Choi SY, Chang YW, Park HJ, Kim HJ, Hong SS, Seo DY (2012b) Correlation of the apparent diffusion coefficiency values on diffusion-weighted imaging with prognostic factors for breast cancer. Br J Radiol 85 (1016): e474–e479.
De Cicco C, Gilardi L, Botteri E, Fracassi SL, Di Dia GA, Botta F, Prisco G, Lombardo D, Rotmensz N, Veronesi U, Paganelli G (2013) Is [18F] fluorodeoxyglucose uptake by the primary tumor a prognostic factor in breast cancer? Breast 22 (1): 39–43.
De Felice C, Cipolla V, Guerrieri D, Santucci D, Musella A, Porfiri LM, Meggiorini ML (2014) Apparent diffusion coefficient on 3.0 Tesla magnetic resonance imaging and prognostic factors in breast cancer. Eur J Gynaecol Oncol 35 (4): 408–414.
Esserman LJ, Berry DA, Cheang MC, Yau C, Perou CM, Carey L, DeMichele A, Gray JW, Conway-Dorsey K, Lenburg ME, Buxton MB, Davis SE, van't Veer LJ, Hudis C, Chin K, Wolf D, Krontiras H, Montgomery L, Tripathy D, Lehman C, Liu MC, Olopade OI, Rugo HS, Carpenter JT, Livasy C, Dressler L, Chhieng D, Singh B, Mies C, Rabban J, Chen YY, Giri D, Au A, Hylton N Investigators IST (2012) Chemotherapy response and recurrence-free survival in neoadjuvant breast cancer depends on biomarker profiles: results from the I-SPY 1 TRIAL (CALGB 150007/150012; ACRIN 6657). Breast Cancer Res Treat 132 (3): 1049–1062.
Garcia Vicente AM, Soriano Castrejon A, Lopez-Fidalgo JF, Amo-Salas M, Munoz Sanchez Mdel M, Alvarez Cabellos R, Espinosa Aunion R (2015) Basal (1)(8)F-fluoro-2-deoxy-D-glucose positron emission tomography/computed tomography as a prognostic biomarker in patients with locally advanced breast cancer. Eur J Nucl Med Mol Imaging 42 (12): 1804–1813.
Groheux D, Giacchetti S, Moretti JL, Porcher R, Espie M, Lehmann-Che J, de Roquancourt A, Hamy AS, Cuvier C, Vercellino L, Hindie E (2011) Correlation of high 18F-FDG uptake to clinical, pathological and biological prognostic factors in breast cancer. Eur J Nucl Med Mol Imaging 38 (3): 426–435.
Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, Fitzgibbons PL, Francis G, Goldstein NS, Hayes M, Hicks DG, Lester S, Love R, Mangu PB, McShane L, Miller K, Osborne CK, Paik S, Perlmutter J, Rhodes A, Sasano H, Schwartz JN, Sweep FC, Taube S, Torlakovic EE, Valenstein P, Viale G, Visscher D, Wheeler T, Williams RB, Wittliff JL, Wolff AC American Society of Clinical Oncology College of American Pathologists (2010) American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer (unabridged version). Arch Pathol Lab Med 134 (7): e48–e72.
Jeh SK, Kim SH, Kim HS, Kang BJ, Jeong SH, Yim HW, Song BJ (2011) Correlation of the apparent diffusion coefficient value and dynamic magnetic resonance imaging findings with prognostic factors in invasive ductal carcinoma. J Magn Reson Imaging 33 (1): 102–109.
Kadoya T, Aogi K, Kiyoto S, Masumoto N, Sugawara Y, Okada M (2013) Role of maximum standardized uptake value in fluorodeoxyglucose positron emission tomography/computed tomography predicts malignancy grade and prognosis of operable breast cancer: a multi-institute study. Breast Cancer Res Treat 141 (2): 269–275.
Kim JY, Kim SH, Kim YJ, Kang BJ, An YY, Lee AW, Song BJ, Park YS, Lee HB (2015) Enhancement parameters on dynamic contrast enhanced breast MRI: do they correlate with prognostic factors and subtypes of breast cancers? Magn Reson Imaging 33 (1): 72–80.
Koo HR, Cho N, Song IC, Kim H, Chang JM, Yi A, Yun BL, Moon WK (2012) Correlation of perfusion parameters on dynamic contrast-enhanced MRI with prognostic factors and subtypes of breast cancers. J Magn Reson Imaging 36 (1): 145–151.
Lee HS, Kim SH, Kang BJ, Baek JE, Song BJ (2016) Perfusion parameters in dynamic contrast-enhanced mri and apparent diffusion coefficient value in diffusion-weighted MRI:: association with prognostic factors in breast cancer. Acad Radiol 23 (4): 446–456.
Makris A, Powles TJ, Ashley SE, Chang J, Hickish T, Tidy VA, Nash AG, Ford HT (1998) A reduction in the requirements for mastectomy in a randomized trial of neoadjuvant chemoendocrine therapy in primary breast cancer. Ann Oncol 9 (11): 1179–1184.
Martincich L, Deantoni V, Bertotto I, Redana S, Kubatzki F, Sarotto I, Rossi V, Liotti M, Ponzone R, Aglietta M, Regge D, Montemurro F (2012) Correlations between diffusion-weighted imaging and breast cancer biomarkers. Eur Radiol 22 (7): 1519–1528.
Marusyk A, Polyak K (2010) Tumor heterogeneity: causes and consequences. Biochim Biophys Acta 1805 (1): 105–117.
Mauri D, Pavlidis N, Ioannidis JP (2005) Neoadjuvant versus adjuvant systemic treatment in breast cancer: a meta-analysis. J Natl Cancer Inst 97 (3): 188–194.
Miyake KK, Nakamoto Y, Kanao S, Tanaka S, Sugie T, Mikami Y, Toi M, Togashi K (2014) Journal Club: diagnostic value of (18)F-FDG PET/CT and MRI in predicting the clinicopathologic subtypes of invasive breast cancer. AJR Am J Roentgenol 203 (2): 272–279.
Molinari C, Clauser P, Girometti R, Linda A, Cimino E, Puglisi F, Zuiani C, Bazzocchi M (2015) MR mammography using diffusion-weighted imaging in evaluating breast cancer: a correlation with proliferation index. Radiol Med 120 (10): 911–918.
Mori N, Ota H, Mugikura S, Takasawa C, Ishida T, Watanabe G, Tada H, Watanabe M, Takase K, Takahashi S (2015) Luminal-type breast cancer: correlation of apparent diffusion coefficients with the Ki-67 labeling index. Radiology 274 (1): 66–73.
National Comprehensive Cancer Network (2016) NCCN Clinical Practice Guidelines in Oncology: Breast Cancer (Version 2.2016).
Pace L, Nicolai E, Luongo A, Aiello M, Catalano OA, Soricelli A, Salvatore M (2014) Comparison of whole-body PET/CT and PET/MRI in breast cancer patients: lesion detection and quantitation of 18F-deoxyglucose uptake in lesions and in normal organ tissues. Eur J Radiol 83 (2): 289–296.
Park JH, Kang MJ, Ahn JH, Kim JE, Jung KH, Gong G, Lee HJ, Son BH, Ahn SH, Kim HH, Shin HJ, Moon DH, Kim SB (2016) Phase II trial of neoadjuvant letrozole and lapatinib in Asian postmenopausal women with estrogen receptor (ER) and human epidermal growth factor receptor 2 (HER2)-positive breast cancer [Neo-ALL-IN]: highlighting the TILs, ER expressional change after neoadjuvant treatment, and FES-PET as potential significant biomarkers. Cancer Chemother Pharmacol 78 (4): 685–695.
Park SH, Choi HY, Hahn SY (2015) Correlations between apparent diffusion coefficient values of invasive ductal carcinoma and pathologic factors on diffusion-weighted MRI at 3.0 Tesla. J Magn Reson Imaging 41 (1): 175–182.
Prat A, Pineda E, Adamo B, Galvan P, Fernandez A, Gaba L, Diez M, Viladot M, Arance A, Munoz M (2015) Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 24 (Suppl 2): S26–S35.
Precht LM, Lowe KA, Atwood M, Beatty JD (2010) Neoadjuvant chemotherapy of breast cancer: tumor markers as predictors of pathologic response, recurrence, and survival. Breast J 16 (4): 362–368.
Pujara AC, Raad RA, Ponzo F, Wassong C, Babb JS, Moy L, Melsaether AN (2016) Standardized uptake values from PET/MRI in metastatic breast cancer: an organ-based comparison with PET/CT. Breast J 22 (3): 264–273.
R Development Core Team (2014) R: A language and environment for statistical computing. R Foundation for Statistical Computing: Vienna, Austria.
Rastogi P, Anderson SJ, Bear HD, Geyer CE, Kahlenberg MS, Robidoux A, Margolese RG, Hoehn JL, Vogel VG, Dakhil SR, Tamkus D, King KM, Pajon ER, Wright MJ, Robert J, Paik S, Mamounas EP, Wolmark N (2008) Preoperative chemotherapy: updates of National Surgical Adjuvant Breast and Bowel Project Protocols B-18 and B-27. J Clin Oncol 26 (5): 778–785.
Richard R, Thomassin I, Chapellier M, Scemama A, de Cremoux P, Varna M, Giacchetti S, Espie M, de Kerviler E, de Bazelaire C (2013) Diffusion-weighted MRI in pretreatment prediction of response to neoadjuvant chemotherapy in patients with breast cancer. Eur Radiol 23 (9): 2420–2431.
Santa-Maria CA, Camp M, Cimino-Mathews A, Harvey S, Wright J, Stearns V (2015) Neoadjuvant therapy for early-stage breast cancer: current practice, controversies, and future directions. Oncology (Williston Park) 29 (11): 828–838.
Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebryiskaya T, Beroukhim R, Hu M, Halushka MK, Sukumar S, Parker LM, Anderson KS, Harris LN, Garber JE, Richardson AL, Schnitt SJ, Nikolsky Y, Gelman RS, Polyak K (2007) Molecular definition of breast tumor heterogeneity. Cancer Cell 11 (3): 259–273.
Siwowska K, Muller C (2015) Preclinical development of small-molecular-weight folate-based radioconjugates: a pharmacological perspective. Q J Nucl Med Mol Imaging 59 (3): 269–286.
Szabo BK, Aspelin P, Kristoffersen Wiberg M, Tot T, Bone B (2003) Invasive breast cancer: correlation of dynamic MR features with prognostic factors. Eur Radiol 13 (11): 2425–2435.
Ueda S, Tsuda H, Asakawa H, Shigekawa T, Fukatsu K, Kondo N, Yamamoto M, Hama Y, Tamura K, Ishida J, Abe Y, Mochizuki H (2008) Clinicopathological and prognostic relevance of uptake level using 18F-fluorodeoxyglucose positron emission tomography/computed tomography fusion imaging (18F-FDG PET/CT) in primary breast cancer. Jpn J Clin Oncol 38 (4): 250–258.
van de Wiele P, Dierckx R, Scopinaro F, Waterhouse R, Annovazzi A, Kolindou A, Signore A (2002) Nuclear medicine imaging for prediction or early assessment of response to chemotherapy in patients suffering from breast carcinoma. Breast Cancer Res Treat 72 (3): 279–286.
Van Den Bossche B, Van Belle S, De Winter F, Signore A, Van de Wiele C (2006) Early prediction of endocrine therapy effect in advanced breast cancer patients using 99mTc-depreotide scintigraphy. J Nucl Med 47 (1): 6–13.
Venables WN, Ripley BD, Venables WN (2002) Modern Applied Statistics with S 4th edn Springer: New York.
Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, Dowsett M, Fitzgibbons PL, Hanna WM, Langer A, McShane LM, Paik S, Pegram MD, Perez EA, Press MF, Rhodes A, Sturgeon C, Taube SE, Tubbs R, Vance GH, van de Vijver M, Wheeler TM, Hayes DF American Society of Clinical Oncology/College of American Pathologists (2007) American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch Pathol Lab Med 131 (1): 18–43.
Yoon HJ, Kang KW, Chun IK, Cho N, Im SA, Jeong S, Lee S, Jung KC, Lee YS, Jeong JM, Lee DS, Chung JK, Moon WK (2014) Correlation of breast cancer subtypes, based on estrogen receptor, progesterone receptor, and HER2, with functional imaging parameters from (6)(8)Ga-RGD PET/CT and (1)(8)F-FDG PET/CT. Eur J Nucl Med Mol Imaging 41 (8): 1534–1543.
Yue Y, Cui X, Bose S, Audeh W, Zhang X, Fraass B (2015) Stratifying triple-negative breast cancer prognosis using 18F-FDG-PET/CT imaging. Breast Cancer Res Treat 153 (3): 607–616.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Catalano, O., Horn, G., Signore, A. et al. PET/MR in invasive ductal breast cancer: correlation between imaging markers and histological phenotype. Br J Cancer 116, 893–902 (2017). https://doi.org/10.1038/bjc.2017.26
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.26
Keywords
This article is cited by
-
Prediction of therapy response of breast cancer patients with machine learning based on clinical data and imaging data derived from breast [18F]FDG-PET/MRI
European Journal of Nuclear Medicine and Molecular Imaging (2024)
-
Joint EANM-SNMMI guideline on the role of 2-[18F]FDG PET/CT in no special type breast cancer
European Journal of Nuclear Medicine and Molecular Imaging (2024)
-
The influence of receptor expression and clinical subtypes on baseline [18F]FDG uptake in breast cancer: systematic review and meta-analysis
EJNMMI Research (2023)
-
Preoperative prediction of Ki-67 status in invasive breast carcinoma using dynamic contrast-enhanced MRI, diffusion-weighted imaging and diffusion tensor imaging
Egyptian Journal of Radiology and Nuclear Medicine (2023)
-
Towards a fast PET/MRI protocol for breast cancer imaging: maintaining diagnostic confidence while reducing PET and MRI acquisition times
European Radiology (2023)
