
Abstract
Background:
Regulator of cullins-1 (ROC1) is a key subunit in the cullin–RING ligase (CRL) protein complex. Our previous study indicated that ROC1 was essential for bladder cancer cell survival and that ROC1 knockdown inhibited CRL activity, triggering G2 phase arrest and senescence. However, the role of ROC1 in the malignant progression of bladder cancer remained unknown.
Methods:
ROC1 expression in cancer cells was knocked down by siRNA silencing. The effects of ROC1 silencing were evaluated by in vitro assays for cell migration and by an in vivo mouse metastasis model. Epithelial–mesenchymal transition (EMT) induction was evaluated by immunofluorescence staining and western blotting of EMT-associated proteins. ROC1 expression in human tumours was further evaluated by immunohistochemical analysis.
Results:
ROC1 knockdown suppresses bladder cancer cell migration by inhibiting EMT. ROC1 knockdown inhibited EMT by inhibiting mammalian target of rapamycin (mTOR) activity via the accumulation of the mTOR-inhibitory protein DEPTOR, a CRL substrate. DEPTOR knockdown partially rescued ROC1 knockdown-inhibited EMT and the ROC1-induced inhibition of cancer cell migration. Furthermore, in vivo studies using a nude mouse metastasis model confirmed the in vitro data. Finally, tissue microarray analysis of clinical bladder cancer specimens indicated a positive correlation between ROC1 expression and EMT.
Conclusions:
ROC1 has an important role in the malignant progression of bladder cancer via the mTOR/DEPTOR pathway. ROC1 may serve as a novel therapeutic target for the treatment of muscle-invasive transitional cell carcinoma.
Similar content being viewed by others


Main
Bladder cancer is the fourth most common cancer and the eighth leading cause of cancer death among men worldwide (Siegel et al, 2015). Histologically, bladder transitional cell carcinoma (BTCC) is the most common bladder cancer subtype, accounting for ∼90% of all bladder cancers (Noon and Catto, 2013). Clinically, 20–30% of all newly diagnosed bladder cancers exhibit a type of muscle layer invasion known as muscle-invasive transitional cell carcinoma (MI-TCC). In addition, 10–30% of non-muscle-invasive bladder cancers will eventually progress to MI-TCC (Mitra and Cote, 2009). For patients with MI-TCC, cisplatin-based chemotherapy is usually recommended after surgery (Kaplan et al, 2014). However, the severe toxicity and relatively low anticancer efficiency of this chemotherapy regimen limits its wide application in the clinic (Noon and Catto, 2013; Kaplan et al, 2014). Furthermore, even with chemotherapy, progression and recurrence occur in the majority of MI-TCC patients, leading to poor prognosis. Therefore, improved understanding of the molecular mechanisms underlying bladder cancer progression and identification of novel anticancer targets for bladder cancer therapy are of great clinical significance and urgently needed.
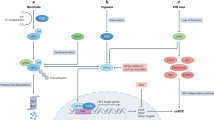
There is increasing interest in the role of cullin–RING ligases (CRL) in the development and progression of bladder cancer. CRL, also known as Skp1, Cullin and F-box protein, belongs to the largest family of E3 ubiquitin ligases (Skaar et al, 2014). It has been demonstrated that CRL mediates the proteasome-targeted degradation of ∼20% of ubiquitinated protein substrates (Skaar et al, 2014). Consequently, CRL has an important role in the ubiquitination of many proteins including cell cycle-related proteins, DNA replication proteins, signal transduction proteins and gene transcription factors (Lydeard et al, 2013; Skaar et al, 2014). CRL dysfunction has been linked to tumour development and progression (Lydeard et al, 2013), suggesting that it is a potential anticancer target. Indeed, the small molecule inhibitor MLN4924 inactivates CRL by inhibiting cullin activity, and effectively inhibits the growth of various cancer cells (Soucy et al, 2009; Gu et al, 2014).
The regulator of cullins-1 (ROC1), also known as RING box protein-1 (RBX1), is a key CRL subunit. ROC1 heterodimerises with distinct cullins to constitute CRL catalytic cores, but its function in cancer development and progression is poorly understood (Petroski and Deshaies, 2005; Skaar et al, 2014). ROC1 contains a small zinc-binding domain called the RING finger, is evolutionarily conserved from yeast to human and has an essential role in embryonic development (Petroski and Deshaies, 2005). Aberrant ROC1 expression leads to CRL dysfunction and embryonic lethality (Tan et al, 2009). In addition, ROC1 is essential for the maintenance of genome integrity, and several recent studies have emphasised its role in human cancers (Jia et al, 2011). Our previous studies indicate that ROC1 protein is overexpressed in bladder cancer and that ROC1 knockdown suppresses the growth of bladder cancer cells (Wang et al, 2013a, 2013b). Our initial mechanistic study indicated that ROC1 knockdown inhibits CRL activity and triggers the accumulation of specific substrates (such as p21 and p27), leading to G2 phase arrest and senescence (Wang et al, 2013a). However, the roles and underlying mechanisms of ROC1 in the malignant progression of bladder cancer remain largely unknown. Therefore, the present study was designed to investigate the effect of ROC1 knockdown on malignant bladder cancer progression and identify the signalling pathways involved. The study design included in vitro and in vivo experiments and tissue microarray analysis (TMA) of clinical cancer tissue samples. The results of this study may provide a basis for the future development of novel ROC1-based targeted therapies for bladder cancer.
Materials and methods
Cell culture and reagents
Human bladder cancer 253J and EJ cell lines were purchased from the Chinese Academy of Science (Shanghai, China) and cultured in RPMI 1640 (Gibco, Gaithersburg, MD, USA) containing 10% fetal bovine serum (FBS; Gibco) and 1% penicillin–streptomycin. Cells were grown at 37 °C in a humidified 5% CO2 environment. Rapamycin and dimethyl sulphoxide (DMSO) were purchased from Sigma-Aldrich (St Louis, MO, USA). Rapamycin was dissolved in DMSO and stored at −20 °C.
siRNAs and transfection
siRNA oligonucleotides for silencing various genes (such as ROC1 and DEPTOR) were obtained from Invitrogen (Shanghai, China), and the transfection procedure was performed according to the manufacturer’s instructions. siRNA sequences were as follows: ROC1, 5′-GACTTTCCCTGCTGTTACCTAA-3′; DEPTOR, 5′-GCCATGACAATCGGAAATCTA-3′; and scrambled control, 5′-ACGUGACACGUUCGGAGAA-3′.
Cell viability assay
Cell proliferation was assessed using the Cell Counting Kit-8 kit (Beyotime, Shanghai, China), which was carried out as previously described (Wang et al, 2013a).
Wound-healing assay
Cells were grown to complete confluence, and then a wound was made in the cell monolayer using a sterile 1-ml pipette tip. To minimise the potential proliferative differences in cells treated with siRNA or control, this assay was performed using serum-free medium. The wound was photographed under a microscope at 0 and 24 h, and the gap between the cells was measured. The rate of wound healing was calculated as follows: (rate of wound healing)=((gap width: 0 h)−(gap width: 24 h))/(gap width: 0 h). Mean values were obtained from at least three separate experiments.
Transwell migration assay
Cell migration activity was evaluated using the transwell (8-μm pore) migration chambers. After pretreated, cells (5 × 104 253 J cells or 1 × 104 EJ cells) suspended in 200 μl of serum-free medium were added to the upper chamber. The lower chamber was filled with 600 μl medium containing 20% FBS. After 24-h incubation, cell staining was performed using 0.1% crystal violet for 30 min. Non-migrated cells were carefully removed from the upper chamber filter using a cotton swab, and the cells that infiltrated the filter were counted in four random fields.
Immunofluorescence staining
Immunofluorescence staining was carried out as previously described (Wang et al, 2013a). Briefly, after fixation and permeation, cells were incubated with primary antibodies against E-cadherin (Cell Signaling Technology, Danvers, MA, USA) or vimentin (Santa Cruz, CA, USA), and then incubated with Alexa 548-conjugated anti-rabbit and FITC-conjugated anti-mouse IgG antibodies (Invitrogen, Carlsbad, CA, USA). Next, the cells were counterstained using 4,6-diamidino-2-phenylindole and analysed under a Zeiss LSM500 confocal microscope (Zeiss International, Oberkochen, Germany).
Western blotting analysis
Following gene transfection or drug treatments, cell lysates were prepared and quantified before performing further experiments. Western blotting analysis was carried out as previously described (Wang et al, 2013a). The following antibodies were used: anti-ROC1 (Abcam, Cambridge, MA, USA); anti-Phospho-4E-BP1 (Thr37/46) and anti-E-cadherin (Cell Signaling); anti-non-4E-BP1 and anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH; Epitomics, Hangzhou, China); anti-vimentin and anti-N-cadherin (Santa Cruz); and anti-DEPTOR (Sigma).
Establishment of the EJ-EGFP-luciferase cell line and lentiviral transduction
EJ cells stably expressing the EGFP–luciferase fusion protein were generated using pLEX lentivirus vectors (Thermo Scientific Inc., Shanghai, China). Briefly, firefly luciferase and green fluorescent protein fusion genes (Fluc) driven by CMV promoter were cloned into the lentivirus vector. Stably transduced EJ cells were obtained via lentivirus infection and 2 weeks of 2 μg ml−1 puromycin selection.
In vivo metastasis assay
A systematic metastasis model of bladder cancer was established for the in vivo metastasis assay. Lenti-shROC1 containing short hairpin RNAs directed against human ROC1 and control Lenti-shCONT were used as previously described (Wang et al, 2013a). Briefly, EJ-EGFP-luciferase cells transfected for 120 h with Lenti-shROC1, or Lenti-shCONT were trypsinised, resuspended in PBS and then intravenously (i.v.) injected into the tail veins of nude mice (6 weeks old, male, athymic, BALB/C nu/nu; n=10 per group). Metastasis formation and growth were monitored via luciferase activity. The Luciferase Reporter Assay System E1500 (Promega, Madison, WI, USA) was used according to the manufacturer’s instructions to measure firefly luciferase activity. Three months after nude mice were inoculated with the Lenti-shROC1- or Lenti-shCONT-transfected cells, the mice were killed and metastatic tissues were examined. The animal study protocol (including animal handling and experimental procedures) was reviewed and approved by the Animal Care and Use Committee of Shanghai First people’s Hospital of Shanghai Jiao Tong University, Shanghai, China. The methods were carried out in accordance with the approved guidelines.
Tissue samples and immunohistochemistry
Bladder cancer tissue specimens were obtained from 45 patients who underwent surgery in the Shanghai Jiao Tong University Affiliated First People’s Hospital between January 2004 and May 2006. The patients included 31 males and 14 females between 45 and 86 years of age (median age of 62 years) who were diagnosed with primary BTCC. Twenty-six of these patients underwent transurethral resection, 10 underwent partial cystectomy and 9 underwent radical cystectomy. Tumour grade and stage were assessed according to the World Health Organization 1973 criteria and the American Joint Committee on Cancer 2002 TNM system. The protocol for the use of human surgical samples was reviewed and approved by the Medical Ethics Committee of Shanghai First People’s Hospital of Shanghai Jiao Tong University (Permit Number: 2013KY004). Tissue sample collection and processing were carried out in compliance with approved guidelines. Informed consent was obtained from each patient.
Immunohistochemistry was performed according to a previously described protocol (Wang et al, 2013a). Primary antibodies against ROC1 (Abcam), DEPTOR (Sigma) and E-cadherin (Boster, Wuhan, China) were used according to the manufacturers’ instructions.
Statistical analysis
Data were expressed as means±s.e.m. Statistical analyses were performed using the Bonferroni t-test after one-way analysis of variance for multi-group comparisons. Two-group comparisons were analysed using Student’s t-tests. The correlations between ROC1, DEPTOR and E-cadherin expression were analysed by Pearson’s χ2-tests. P<0.05 was considered statistically significant. All statistical evaluations were performed using SPSS 13.0 (SPSS, Inc., Chicago, IL, USA).
Results
ROC1 knockdown inhibits MI-TCC cell migration
To assess the role of ROC1 in MI-TCC cell migration, we knocked ROC1 expression down in two invasive bladder cancer cell lines (253J and EJ cells) using ROC1 siRNA and a scrambled siRNA control (siCONT) (Wang et al, 2013a). To minimise the potential influence of proliferation, our wound-healing and transwell migration assays were done after pretreated 24 h, when cell viability were not changed significantly (Figure 1A and B; Supplementary Figure S1A and S1B). Wound-healing assays using the transfected cells established that siROC1-treated cells migrated remarkably slower than siCONT-treated cells (Figure 1C and D). Transwell migration assays further demonstrated that cell motility was significantly impaired in siROC1-treated cells when compared with siCONT-treated cells (Figure 1E and F).

Inhibition of MI-TCC cell migration after ROC1 knockdown. (A, C and E) 253J cells; (B, D and F) EJ cells. Cells were transiently transfected with siROC1 or siCONT for 24 h and then subjected to cell viability CCK8 (A, B), wound-healing (C, D) or transwell migration assay (E, F). Representative results of three independent experiments are shown as means± s.e.m. *P<0.05.
ROC1 knockdown inhibits EMT in MI-TCC cells
EMT strongly influences the migratory ability of cancer cells (Lamouille et al, 2014). EMT is characterised by reduced expression of epithelial markers (such as E-cadherin) and upregulation of mesenchymal markers (such as N-cadherin and vimentin) (Lamouille et al, 2014). We investigated whether ROC1 knockdown influenced EMT by examining the expression levels of specific markers. Immunofluorescence microscopy analysis of ROC1-knockdown cells showed increased E-cadherin expression and decreased vimentin expression when compared with control cells (Figure 2A and B). Consistent with these observations, western blot analysis indicated that E-cadherin expression was increased by ROC1 knockdown, whereas the expression of N-cadherin and vimentin were decreased (Figure 2C and D). These data indicated that ROC1 knockdown inhibits EMT in 253J and EJ cells.

ROC1 knockdown induction of MI-TCC cell MET. ROC1 knockdown-induced MET in 253J (A) and EJ (B) cells. Cells were transfected with siROC1 or siCONT for 96 h, stained for E-cadherin (red) and vimentin (green), and then examined under a confocal microscope. Nuclear DNA was stained with DAPI (blue). Scale bar=10 μm. The expression of EMT-associated markers in 253J (C) and EJ (D) cells were examined using western blots; the quantification relative to GAPDH was accomplished with densitometric analysis by Image J software. Representative results of three independent experiments are shown.
ROC1 knockdown inhibits the mTOR pathway via DEPTOR protein accumulation
To investigate the mechanisms underlying ROC1 knockdown-inhibited EMT in bladder cancer cells, we explored the possible involvement of the mTOR pathway. As evidenced by reduced 4E-BP1 phosphorylation (p-4E-BP1), ROC1 knockdown resulted in mTOR inactivation in both 253J and EJ cells (Figure 3A and B). mTOR-induced phosphorylation of 4E-BP1 has previously been reported and this could serve as a classical marker of mTOR activation (Cao et al, 2015). To investigate which molecule was primarily responsible for suppressing mTOR activation, we analysed the expression of DEPTOR protein in ROC1-knockdown cells and controls. DEPTOR is a CRLβ-TrCP substrate and a naturally occurring mTOR inhibitor. DEPTOR protein levels in the ROC1-knockdown tumour cells were significantly elevated when compared with control cells (Figure 3A and B). To investigate DEPTOR’s contribution to ROC1 knockdown-inhibited EMT, we performed a simultaneous siRNA knockdown of DEPTOR and ROC1 expression in EJ cells and found that the DEPTOR knockdown markedly attenuated ROC1 knockdown-inhibited EMT. This was demonstrated by a reduction in E-cadherin expression and increasing N-cadherin expression (Figure 3C). Transwell migration assays demonstrated that DEPTOR knockdown partially restored migratory ability following ROC1 knockdown in EJ cells (Figure 3D). In addition, DEPTOR knockdown alone did not significantly impair cell motility when compared with siCONT-treated cells (Figure 3D). These results indicated that DEPTOR was essential for ROC1 knockdown-inhibited EMT and suggested the involvement of the mTOR pathway.

Effects of ROC1 knockdown on the mTOR pathway. mTOR pathway-associated protein expression in 253J (A) and EJ (B) cells was examined via western blot. EJ cells were transfected with siRNA for 96 h and then subjected to western blotting analysis (C) and transwell migration assays (D). Representative results of three independent experiments are shown. Columns, means of three independent experiments; bars, s.e.m. *P<0.05. Treated cells were subject to western blot analysis with GAPDH as a loading control, and the quantification relative to GAPDH was conducted by densitometric analysis using Image J software.
Rapamycin-induced mTOR pathway inhibition suppresses EMT in bladder cancer cells
To investigate the role of mTOR kinase in EMT regulation, we investigated rapamycin’s effects on EMT in bladder cancer cells. Cells were treated with various concentrations of rapamycin, and EMT marker expression was examined. After 72 h rapamycin treatment, mTOR kinase activity was significantly inhibited. This inhibition was reflected by a reduction in p-4E-BP1. EMT was also notably inhibited in rapamycin-treated cells. EMT inhibition was demonstrated by increased E-cadherin expression and decreased N-cadherin and vimentin expression. Rapamycin treatment also significantly suppressed 253J and EJ cell migration (Figure 4C and D). These results suggested that mTOR inhibition could effectively suppress EMT in bladder cancer cells.

The effects of rapamycin-induced inhibition of the mTOR pathway on EMT. (A, C) 253J cells. (B, D) EJ cells. Cells were treated with different concentrations of rapamycin (0, 50 and 100 nmol l−1) for 72 h and analysed via western blotting (A, B) or transwell migration assays (C, D). Representative results of three independent experiments are shown. Columns, means of three independent experiments; bars, s.e.m. *P<0.05. Treated cells were subject to western blot analysis with GAPDH as a loading control, and the quantification relative to GAPDH was conducted by densitometric analysis using Image J software.
ROC1 knockdown suppresses MI-TCC metastasis in vivo
For in vivo confirmation of our in vitro findings, we examined the effects of ROC1 knockdown using an athymic nude mouse metastasis model. EGFP–luciferase-labelled EJ cells were i.v. injected into athymic nude mice following ROC1 knockdown and systemic metastasis was assessed by detecting luciferase activity. All shCONT-cell-inoculated mice (10/10) exhibited pulmonary metastases. In contrast, only 60% (6/10) ROC1-knockdown mice exhibited lung metastasis nodules. ShROC1 cell injection resulted in a remarkable decrease in metastasis when compared with the control group (Figure 5A). We also detected the expression levels of DEPTOR and E-cadherin proteins in ROC1-knockdown tumour tissues and found that they were increased in comparison with the control group (Figure 5B). These data demonstrated that ROC1 knockdown suppressed EMT and MI-TCC metastasis in vivo.

Effects of ROC1 knockdown on metastasis in vivo. (A) Representative photographs of systematic metastasis detection in nude mice 12 weeks after the injection of shROC1 or shCONT cells. (B) IHC of lung metastasis nodules with the indicated antibodies in both groups. Columns, the mean luciferase activity of 10 mice; bars, s.e.m. ***P<0.01. Scale bar=50 μm.
ROC1 expression is positively associated with EMT in human BTCC tissues
Using bladder cancer TMA analysis, we evaluated the relationships between the pathological stages of bladder cancer and the expression levels of ROC1, DEPTOR and E-cadherin. Using the immunoreactivity score system, expression levels were divided into two categories (low or high). The TMA analysis indicated that ROC1 expression was increased, but that DEPTOR and E-cadherin expression were reduced in invasive bladder cancer tissues when compared with non-muscle-invasive bladder cancer tissues (Figure 6A). Furthermore, the TMA analysis indicated that ROC1 and DEPTOR expression levels were related to the tumour stage (Figure 6B). ROC1 expression exhibited significant negative correlations with DEPTOR (P=0.014, R=−0.378) and E-cadherin (P=0.001, R=−0.7756; Table 1). Collectively, cancer tissues with high ROC1 expression had lower DEPTOR and E-cadherin expression.

ROC1 expression in human bladder cancer tissues. (A) Immunohistochemical staining of ROC1, DEPTOR and E-cadherin in human bladder cancer. Representative immunohistochemistry images of non-muscle-invasive or muscle-invasive cancer are shown. (B) Correlation of the immunostaining intensities of ROC1, DEPTOR and E-cadherin with cancer invasiveness. Immunostaining intensity was divided into two groups (high and low), and the association of intensity with cancer invasiveness (non-muscle-invasive cancer or muscle-invasive cancer) was analysed by a χ2-test for each protein. The P-value for each association is shown. Scale bar=50 μm.
Discussion
This investigation made several discoveries concerning the role of ROC1 in the malignant progression of MI-TCC. First, we demonstrated that ROC1 knockdown inhibited the migration of bladder cancer cells by inhibiting EMT. Second, we showed that ROC1 knockdown inhibited mTOR activity via the accumulation of the mTOR-inhibitory protein DEPTOR. Third, we demonstrated that in vivo metastasis was suppressed by ROC1 knockdown. Finally, TMA analysis of clinical samples indicated that there was a positive correlation between ROC1 expression and EMT induction. These data suggest that ROC1 has an important role in bladder cancer progression. ROC1 protein could be an attractive anticancer target, and a treatment strategy targeting ROC1 may be capable of hindering the metastasis of bladder cancer.
Cancer progression is a complex, multistep process, and the acquisition of migratory ability is the prerequisite of metastasis (Hanahan and Weinberg, 2011). Studies have demonstrated that EMT induction is a pivotal cellular process that promotes the mobility of cancer cells and leads to the metastasis of epithelial cancers, including bladder cancer. EMT is characterised by the loss of epithelial and the acquisition of mesenchymal characteristics. This transition grants cancer cells the ability to migrate and invade distant sites (Lamouille et al, 2014). E-cadherin is the key mediator of cell–cell adhesion in epithelial tissues. In this study, we demonstrated that ROC1 knockdown suppressed cancer cell migration through EMT inhibition that was characterised by increased E-cadherin expression. Furthermore, using the EJ mouse metastasis model, we found that ROC1 played an important role in promoting cancer metastasis via similar mechanisms. Consistent with our findings, and further supporting the importance of CRL in the EMT process of cancer cells, the suppression of CRL activity by cullin4A silencing reversed EMT by regulating the transcriptional downregulation of ZEB1 (Wang et al, 2014). Notably, ROC1 knockdown was recently reported to inhibit cell migration via induction of RhoA accumulation in human vascular endothelium (Yao et al, 2014). These results suggest that inactivation of CRL could inhibit cell migration via different pathways.
When alternative E-cadherin expression regulatory pathways were examined in 253J and EJ cells, we found that mTOR activity was strongly inhibited by ROC1 knockdown. The mTOR pathway has a pivotal role in the regulation of various cellular process, including cell death, apoptosis, autophagy and EMT (Betz and Hall, 2013). The mTOR pathway is a positive regulator of EMT (Gherardi et al, 2012; Lamouille et al, 2014). In the present study, ROC1 knockdown suppressed mTOR activity, as evidenced by p-4E-BP1 downregulation. Consistent with our findings, other recent studies have reported that mTOR activity is suppressed in cancer cells via CRL activity inhibition (Zhao et al, 2012; Li et al, 2014). However, the exact mechanisms underlying the changes in mTOR kinase regulation of EMT after ROC1 knockdown remain elusive. It has been reported that the mTOR pathway induces EMT by transactivating the RhoA and Rac1 signalling pathways (Gordon et al, 2014). One conceivable explanation may be that mTOR pathway inhibition after ROC1 knockdown alters cellular cytoskeletal arrangements and decreases the activation of RhoA and Rac1, consequently suppressing EMT in MI-TCC cells. However, further study is needed to establish the exact mechanism responsible for the ROC1-induced regulation of EMT in MI-TCC.
ROC1 knockdown triggers CRL inactivation and the accumulation of CRL substrates (such as p21 and p27), leading to cellular responses (such as G2 arrest and senescence) (Jia et al, 2011; Wang et al, 2013a). Soon after ROC1 knockdown (e.g., at 24 h, when cell viability has not yet been significantly altered) ROC1 knockdown-induced inhibition of mTOR in MI-TCC cells was dependent on DEPTOR accumulation. DEPTOR is a recently identified endogenous mTOR inhibitor that directly binds to mTOR and negatively regulates its activity (Peterson et al, 2009). DEPTOR is degraded by the CRLβ-TrCP E3 ubiquitin ligase (Duan et al, 2011; Gao et al, 2011; Zhao et al, 2011). With the cooperation of ROC1, the F-box protein β-TrCP binds to DEPTOR and promotes DEPTOR degradation in the CRL complex. Here we demonstrated that ROC1 knockdown triggered DEPTOR accumulation and subsequent mTOR inactivation. Correspondingly, blocking DEPTOR expression restored EMT induction upon ROC1 knockdown. We observed that simultaneous abrogation of ROC1 and DEPTOR only partially restored EMT induction. This indicates that DEPTOR is necessary but not sufficient for ROC1 knockdown-induced mTOR inactivation and EMT inhibition, and that other EMT-regulatory pathways may be involved. Therefore, additional investigation of the underlying mechanisms is needed.
DEPTOR directly binds mTOR and inhibits the activities of both mTORC1 and mTORC2 (Peterson et al, 2009). Rapamycin is a specific mTOR inhibitor, but has low anticancer efficacy in bladder cancer treatment due to selective inhibition of mTORC1 activity, which limits its clinical effectiveness (Sun, 2013; Ghosh et al, 2014). In the present study, we demonstrated that ROC1 knockdown induced DEPTOR accumulation. This could have important implications for the development of new bladder cancer treatment strategies. ROC1 knockdown-induced DEPTOR accumulation may inhibit the activity of both mTORC1 and mTORC2 and, therefore, may overcome the selective inhibition of mTORC1. Furthermore, ROC1 knockdown may induce the accumulation of other mTOR blockers that are CRL substrates, including HIF-1α, REDD1 and TSC2 (Jaakkola et al, 2001; Hu et al, 2008; Katiyar et al, 2009). Therefore, we speculate that RNA interference or pharmacological inhibition of ROC1 could be a novel approach for improving the responsiveness of bladder cancer patients to chemotherapy.
In our TMA analysis, ROC1 expression was associated with bladder cancer invasion. ROC1 expression exhibited a significant negative correlation with DEPTOR (P=0.009, R=−0.378) and E-cadherin (P=0.001, R=−0.7756) levels. As bladder tumours with high ROC1 expression tended to have lower DEPTOR (60%) and E-cadherin (56.7%) expression levels, low E-cadherin expression was correlated with the malignant progression of bladder cancer. This result agrees with results reported by previous studies (Lamouille et al, 2014; Knowles and Hurst, 2015). We speculate that ROC1 could be a promising prognostic factor for bladder cancer progression. However, the sample numbers of the current study are limited, and, therefore, the statistical significance found in this study may not be stable. Furthermore, correlation of ROC1 expression with patient prognosis was not investigated. Therefore, future studies are needed to investigate whether or not ROC1 is a promising prognostic factor for bladder cancer progression. These studies should be conducted using a greater sample size and a longer follow-up period.
In conclusion, the present study demonstrated that ROC1 has an important role in bladder cancer progression by regulating the mTOR/DEPTOR pathway. This suggests that ROC1 could be a novel anticancer target for bladder cancer therapy. Further studies clarifying the detailed mechanisms underlying the role of ROC1 in bladder cancer are needed.
Change history
02 February 2016
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Betz C, Hall MN (2013) Where is mTOR and what is it doing there? J Cell Biol 203 (4): 563–574.
Cao Y, Liu X, Lu W, Chen Y, Wu X, Li M, Wang XA, Zhang F, Jiang L, Zhang Y, Hu Y, Xiang S, Shu Y, Bao R, Li H, Wu W, Weng H, Yen Y, Liu Y (2015) Fibronectin promotes cell proliferation and invasion through mTOR signaling pathway activation in gallbladder cancer. Cancer Lett 360 (2): 141–150.
Duan S, Skaar JR, Kuchay S, Toschi A, Kanarek N, Ben-Neriah Y, Pagano M (2011) mTOR generates an auto-amplification loop by triggering the betaTrCP- and CK1alpha-dependent degradation of DEPTOR. Mol Cell 44 (2): 317–324.
Gao D, Inuzuka H, Tan MK, Fukushima H, Locasale JW, Liu P, Wan L, Zhai B, Chin YR, Shaik S, Lyssiotis CA, Gygi SP, Toker A, Cantley LC, Asara JM, Harper JW, Wei W (2011) mTOR drives its own activation via SCF(betaTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol Cell 44 (2): 290–303.
Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G (2012) Targeting MET in cancer: rationale and progress. Nat Rev Cancer 12 (2): 89–103.
Ghosh M, Brancato SJ, Agarwal PK, Apolo AB (2014) Targeted therapies in urothelial carcinoma. Curr Opin Oncol 26 (3): 305–320.
Gordon BS, Kazi AA, Coleman CS, Dennis MD, Chau V, Jefferson LS, Kimball SR (2014) RhoA modulates signaling through the mechanistic target of rapamycin complex 1 (mTORC1) in mammalian cells. Cell Signal 26 (3): 461–467.
Gu Y, Kaufman JL, Bernal L, Torre C, Matulis SM, Harvey RD, Chen J, Sun SY, Boise LH, Lonial S (2014) MLN4924, an NAE inhibitor, suppresses AKT and mTOR signaling via upregulation of REDD1 in human myeloma cells. Blood 123 (21): 3269–3276.
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144 (5): 646–674.
Hu J, Zacharek S, He YJ, Lee H, Shumway S, Duronio RJ, Xiong Y (2008) WD40 protein FBW5 promotes ubiquitination of tumor suppressor TSC2 by DDB1-CUL4-ROC1 ligase. Genes Dev 22 (7): 866–871.
Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ (2001) Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292 (5516): 468–472.
Jia L, Bickel JS, Wu J, Morgan MA, Li H, Yang J, Yu X, Chan RC, Sun Y (2011) RBX1 (RING box protein 1) E3 ubiquitin ligase is required for genomic integrity by modulating DNA replication licensing proteins. J Biol Chem 286 (5): 3379–3386.
Kaplan AL, Litwin MS, Chamie K (2014) The future of bladder cancer care in the USA. Nat Rev Urol 11 (1): 59–62.
Katiyar S, Liu E, Knutzen CA, Lang ES, Lombardo CR, Sankar S, Toth JI, Petroski MD, Ronai Z, Chiang GG (2009) REDD1, an inhibitor of mTOR signalling, is regulated by the CUL4A-DDB1 ubiquitin ligase. EMBO Rep 10 (8): 866–872.
Knowles MA, Hurst CD (2015) Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer 15 (1): 25–41.
Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 15 (3): 178–196.
Li H, Tan M, Jia L, Wei D, Zhao Y, Chen G, Xu J, Zhao L, Thomas D, Beer DG, Sun Y (2014) Inactivation of SAG/RBX2 E3 ubiquitin ligase suppresses KrasG12D-driven lung tumorigenesis. J Clin Invest 124 (2): 835–846.
Lydeard JR, Schulman BA, Harper JW (2013) Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep 14 (12): 1050–1061.
Mitra AP, Cote RJ (2009) Molecular pathogenesis and diagnostics of bladder cancer. Annu Rev Pathol 4 (1): 251–285.
Noon AP, Catto JW (2013) Bladder cancer in 2012: challenging current paradigms. Nat Rev Urol 10 (2): 67–68.
Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM (2009) DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137 (5): 873–886.
Petroski MD, Deshaies RJ (2005) Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol 6 (1): 9–20.
Siegel RL, Miller KD, Jemal A (2015) Cancer statistics, 2015. CA Cancer J Clin 65 (1): 5–29.
Skaar JR, Pagan JK, Pagano M (2014) SCF ubiquitin ligase-targeted therapies. Nat Rev Drug Discov 13 (12): 889–903.
Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Cullis CA, Doucette A, Garnsey JJ, Gaulin JL, Gershman RE, Lublinsky AR, McDonald A, Mizutani H, Narayanan U, Olhava EJ, Peluso S, Rezaei M, Sintchak MD, Talreja T, Thomas MP, Traore T, Vyskocil S, Weatherhead GS, Yu J, Zhang J, Dick LR, Claiborne CF, Rolfe M, Bolen JB, Langston SP (2009) An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458 (7239): 732–736.
Sun SY (2013) mTOR kinase inhibitors as potential cancer therapeutic drugs. Cancer Lett 340 (1): 1–8.
Tan M, Davis SW, Saunders TL, Zhu Y, Sun Y (2009) RBX1/ROC1 disruption results in early embryonic lethality due to proliferation failure, partially rescued by simultaneous loss of p27. Proc Natl Acad Sci USA 106: 6203–6208.
Wang W, Liu Z, Qu P, Zhou Z, Zeng Y, Fan J, Liu Y, Guo Y, Qiu J (2013a) Knockdown of regulator of cullins-1 (ROC1) expression induces bladder cancer cell cycle arrest at the G2 phase and senescence. PloS One 8 (5): e62734.
Wang W, Qiu J, Liu Z, Zeng Y, Fan J, Liu Y, Guo Y (2013b) Overexpression of RING box protein-1 (RBX1) associated with poor prognosis of non-muscle-invasive bladder transitional cell carcinoma. J Surg Oncol 107 (7): 758–761.
Wang Y, Wen M, Kwon Y, Xu Y, Liu Y, Zhang P, He X, Wang Q, Huang Y, Jen KY, LaBarge MA, You L, Kogan SC, Gray JW, Mao JH, Wei G (2014) CUL4A induces epithelial-mesenchymal transition and promotes cancer metastasis by regulating ZEB1 expression. Cancer Res 74 (2): 520–531.
Yao WT, Wu JF, Yu GY, Wang R, Wang K, Li LH, Chen P, Jiang YN, Cheng H, Lee HW, Yu J, Qi H, Yu XJ, Wang P, Chu YW, Yang M, Hua ZC, Ying HQ, Hoffman RM, Jeong LS, Jia LJ (2014) Suppression of tumor angiogenesis by targeting the protein neddylation pathway. Cell Death Dis 5: e1059.
Zhao Y, Xiong X, Jia L, Sun Y (2012) Targeting Cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis 3: e386.
Zhao Y, Xiong X, Sun Y (2011) DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(betaTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol Cell 44 (2): 304–316.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81402379), the Shanghai Municipal Natural Science Fund of Medicine (No. 134119a2801) and the Medical Scientific Research Foundation of Jiangsu Province, China (No. Z201526).
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Wang, W., Chen, H., Liu, Z. et al. Regulator of cullins-1 expression knockdown suppresses the malignant progression of muscle-invasive transitional cell carcinoma by regulating mTOR/DEPTOR pathway. Br J Cancer 114, 305–313 (2016). https://doi.org/10.1038/bjc.2015.444
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.444
Keywords
This article is cited by
-
ROC1 promotes the malignant progression of bladder cancer by regulating p-IκBα/NF-κB signaling
Journal of Experimental & Clinical Cancer Research (2021)
-
Regulator of cullins-1 (ROC1) negatively regulates the Gli2 regulator SUFU to activate the hedgehog pathway in bladder cancer
Cancer Cell International (2021)
-
ROC-1, P21 and CAIX as markers of tumor aggressiveness in bladder carcinoma in Egyptian patients
Diagnostic Pathology (2020)
-
The functional analysis of Cullin 7 E3 ubiquitin ligases in cancer
Oncogenesis (2020)

{kind=link}