
Abstract
Citrin deficiency (CD) is a Mendelian disease due to biallelic mutations of SLC25A13 gene. Neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) is the major pediatric CD phenotype, and its definite diagnosis relies on SLC25A13 genetic analysis. China is a vast country with a huge population, but the SLC25A13 genotypic features of CD patients in our country remains far from being well clarified. Via sophisticated molecular analysis, this study diagnosed 154 new CD patients in mainland China and identified 9 novel deleterious SLC25A13 mutations, i.e. c.103A > G, [c.329 − 154_c.468 + 2352del2646; c.468 + 2392_c.468 + 2393ins23], c.493C > T, c.755 − 1G > C, c.845_c.848 + 1delG, c.933_c.933 + 1insGCAG, c.1381G > T, c.1452 + 1G > A and c.1706_1707delTA. Among the 274 CD patients diagnosed by our group thus far, 41 SLC25A13 mutations/variations were detected. The 7 mutations c.775C > T, c.851_854del4, c.1078C > T, IVS11 + 1G > A, c.1364G > T, c.1399C > T and IVS16ins3kb demonstrated significantly different geographic distribution. Among the total 53 identified genotypes, only c.851_854del4/c.851_854del4 and c.851_854del4/c.1399C > T presented different geographic distribution. The northern population had a higher level of SLC25A13 allelic heterogeneity than those in the south. These findings enriched the SLC25A13 mutation spectrum and brought new insights into the geographic distribution of the variations and genotypes, providing reliable evidences for NICCD definite diagnosis and for the determination of relevant molecular targets in different Chinese areas.
Similar content being viewed by others


Introduction
Citrin deficiency (CD) is a Mendelian disease entity due to biallelic mutations of SLC25A13 gene. With 18 exons and 17 introns, this gene is localized to chromosome 7q21.3, and encodes citrin, the liver-type mitochondrial aspartate/glutamate carrier isoform 2 (AGC2)1,2,3. Citrin has proven to be a dimeric protein with the molecular weight of 147.7 ± 1.7 kDa, and has a three-domain structure: a calcium-regulated N-terminal domain (residues 1–319, 36 kDa) with eight EF-hand motifs, a mitochondrial carrier domain (residues 320–612, 32 kDa) with six transmembrane helices, and a C-terminal domain (residues 613–675, 6 kDa) with an amphipathic helix being involved in the calcium-dependent regulation of the opening and closing of the AGC2 vestibule4.
Three age-dependent CD phenotypes have been described, which manifest in neonates or infants as neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD, OMIM #605814)5,6,7,8,9,10, in older children as failure to thrive and dyslipidemia caused by citrin deficiency (FTTDCD)4,11,12,13,14,15, and in adolescents and adults, usually between the ages of 11 and 79 years, as adult-onset citrullinemia type II (CTLN2, OMIM #603471)1,8,9,16. After the NICCD period, some individuals might directly step into FTTDCD and some could develop CTLN2 in one or more decades5,17,18,19,20.
The SLC25A13 genetic testing has been recognized as a reliable method for the definitive diagnosis of NICCD. However, about 10–15% of the SLC25A13 mutations could not be detected by conventional DNA analysis21,22, and the identification of these obscure mutations constitutes a challenge against the definite diagnosis of NICCD23,24. Although about 200 Chinese NICCD patients have been diagnosed via SLC25A13 analysis to date22,25,26,27, this number was rather limited, since China might have 85700 CD patients according to provisional epidemiological data19,27,28. Moreover, the SLC25A13 mutations worldwide demonstrate remarkable heterogeneity12,22,23,24,27,29,30,31,32,33,34,35,36,37,38. China is a vast country with a huge population, but the SLC25A13 genotypic features of CD patients, including the mutation spectrum and their geographic distribution, remains far from being well clarified in our country.
In this study, besides the conventional DNA analyses, cDNA cloning and in silico prediction as well as functional analysis were carried out to definitely diagnose pediatric CD patients. In addition, we investigated the geographic distribution of the SLC25A13 mutations and genotypes, and evaluated the allelic heterogeneity in an attempt to provide reliable evidences for the determination of relevant molecular diagnostic targets in different geographic areas of China.
Results
New CD patients and the establishment of a large pediatric cohort
As shown in Table 1, this study diagnosed 154 new CD patients via sophisticated molecular, functional and bioinformatic analysis. Along with the 120 cases reported previously23,24,27,33,34, a large cohort with a total of 274 CD patients were established by our group from July, 2005 to the end of February, 2016. To the best of our knowledge, this is the largest CD cohort described in official references to date. This pediatric CD cohort encompassed 117 females and 157 males, and involved 264 families from 26 provinces, autonomous regions and municipalities in China (Fig. 1).

By the end of February in 2016, 274 Chinese patients from 26 provinces, municipalities and autonomous regions in China were diagnosed. This figure was generated by means of the software WPS Office PowerPoint 2016, which was freely available at http://www.wps.cn/product/wps2016/. The base map was created by incrementally assembling the outlines of the Chinese administrative regions, which could be downloaded via the URL link http://www.pptstore.net/ppt_yuansu/12145.html, as a free network resource.
Novel mutations and cDNA cloning analysis
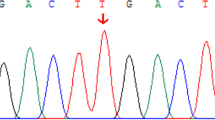
As shown in Table 2, 9 novel SLC25A13 mutations were identified in this study, and among them, c.103A > G (p.M35V), c.493C > T (p.Q165X), c.755−1G > C, c.845_c.848 + 1delG, c.933_c.933 + 1insGCAG, c.1381G > T (p.E461X), c.1452 + 1G > A and c.1706_1707delTA (p.S331fsX363) (Fig. 2a) were detected by Sanger sequencing of all the 18 SLC25A13 exons and their flanking sequences. To address the issue whether and how c.755−1G > C, c.845_c.848 + 1delG and c.933_c.933 + 1insGCAG affect the splicing process of the relevant pre-mRNA molecules, cDNA cloning analysis of the transcripts from the affected SLC25A13 alleles in PBLs were performed. They were found to give rise to the aberrant transcripts r.755_756delAG (p.252fs269X), r.845_848delG (p.D283fsX285) and r.933_934insGCAG (p.A312fsX317), respectively (Fig. 2b).

The figure (a) showed the segmental DNA sequencing results of the 8 novel micro mutations. The figure (b) illustrated the cDNA sequencing results of the aberrant transcripts r.845_848delG, r.755_756delAG and r.933_934insGCAG, respectively, which were transcribed from the mutated alleles harboring c.845_c.848 + 1delG (C0221), c.755 − 1G > C (C0257) and c.933_c.933 + 1insGCAG (C0364), respectively.
In patient C0360, four high-frequency mutation screening just detected the maternal mutation IVS16ins3kb. Further cDNA cloning analysis revealed that all the transcripts from the paternal allele featured exon 5 skipping (Table 3). The following screening of large insertion/deletion with primer set located in the adjacent intronic regions of exon 5 revealed a paternally-inherited unexpected PCR band of 1 kb in size, as illustrated in Fig. 3. Direct sequencing of this unexpected product revealed a 2646 bp deletion, involving the entire exon 5 and the adjacent intronic sequences, along with a 23 bp insertion 40 bp behind the breakpoint. According to the nomenclature rules39,40, this complex mutation was described as [c.329−154_c.468 + 2352del2646; c.468 + 2392_c.468 + 2393ins23], predictively leading to the production of a truncated citrin molecule p.E110fs127X.

(a) High-frequency mutation screening revealed the maternally-inherited mutation IVS16ins3kb. (b) Electrophoresis of the LA-PCR products with the primers covering exon 5 revealed an unexpected band of 1162 bp in size, which was inherited from the father. (c) Sanger sequencing of the 1162 bp product uncovered a 2646 bp deletion (sequences in blue), which spanned the entire exon 5 (capitals) and partial sequences of the introns 4 and 5. Meanwhile, a 23 bp insertion (sequences in red) which was 40nt behind the breakpoint was also discovered. Underlined were the primers for LA-PCR screening.
In patient C0388, the maternal SLC25A13 allele harbored two mutations [c.851_854del4; c.1452 + 1G > A], while the paternal mutation was not detected by direct DNA sequencing. The subsequent cDNA cloning analysis also shown that all the transcripts were from the maternal allele with r.851_854del4; moreover, half of them (12/21) demonstrated exon 14 skipping (r.1312_1452del, p.Ala438_Lys484del) (Table 3), indicating that the mutation c.1452 + 1G > A affected the splicing process of pre-mRNA molecules transcribed from the maternal allele.
Bioinformatic and functional findings of the novel missense mutation c.103A > G (p.M35V)
Alignment analysis in 11 different species indicated that the amino acid M35 in citrin protein is highly conserved (Supplemental Information 1). The probability value of disease-causing potential was >0.9999 upon MutationTaster analysis and strongly indicated its deleterious nature. Meanwhile, this mutation is predicted to be probably damaging by using PolyPhen-2 with a score of 0.989 (sensitivity: 0.72; specificity: 0.97), and have a deleterious effect by using PROVEAN with a score of −3.25.
As depicted in Fig. 4, after growing for 96 hours, the growth ability of the pYX212-mutant (p.M35V) was not significantly different (P = 0.341) from that of the empty vector pYX212 (vector). However, both of them had significantly lower (P = 0.000) growth ability in comparison to pYX212-citrin (citrin). These findings indicated that the mutation p.M35V, as a lack-of-function variation, caused the elimination of the AGC2 function of citrin protein.

After growing for 96 hours, the growth ability of the yeast strain pYX212-mutant (p.M35V) was not significantly different (P = 0.341) from that transfected with the empty vector pYX212 (vector). However, both of them had significantly lower (P = 0.000) growth ability in comparison to pYX212-citrin (citrin), the yeast strain transfected with normal citrin recombinant. The results in each group were means ± SD of six repeated experiments.
SLC25A13 mutation spectrum and regional distribution
SLC25A13 mutations/variations were detected in 522 out of the 528 independent alleles in Table 2, with the diagnostic efficiency of 98.86%. The SLC25A13 spectrum was composed of 41 mutations/variations, including 12 missense mutations, 8 deletion, 10 nonsense, 4 splice-site, 3 insertion, 1 duplication, 1 pathogenic SNP29,41,42, 1 aberrant transcript42 and 1 complex mutation. The variations c.851_854del4 (58.33%), c.1638_1660dup (8.52%), IVS6 + 5G > A (7.58%) and IVS16ins3kb (10.04%) constituted the high-frequency mutations on top of the list. Three other mutations, IVS4ins6kb, IVS11 + 1G > A and c.1399C > T (p.R467X), were found at relative frequencies of 1–2%, and the remaining 34 mutations each had frequencies <1%.
Among all the 41 mutations/variations, as shown in Table 2, 16 ones were detected in the north, 16 in the border and 27 in the south. Meanwhile, there were 6 private mutations in the north, 8 in the border and 15 in the south. The comparisons of their relative frequencies among the three areas revealed that c.775C > T (p.Q259X), c.851_854del4, c.1078C > T (p.R360X), IVS11 + 1G > A, c.1364G > T (p.R455L), c.1399C > T (p.R467X) and IVS16ins3kb presented with significantly different geographic distribution with P all <0.05 (Table 2). In pairwise comparisons, the southern population had a higher relative frequency of c.851_854del4 than the border (P = 0.001) and northern (P = 0.000) populations. By contrast, the relative frequency of IVS16ins3kb was lower in the south than in the border (P = 0.013) and the north (P = 0.005). Besides, when compared with the south region, the relative frequencies of c.1399C > T (p.R467X) (P = 0.003) in the border and those of c.1364G > T (p.R455L) (P = 0.012) and IVS11 + 1G > A (P = 0.015) in the north were higher. No significant difference were observed on the geographic distribution of the mutations IVS6 + 5G > A and c.1638_1660dup among different areas.
SLC25A13 genotype and allelic heterogeneity
Except the 6 patients with only one mutation detected and the 13 ones with parents of different geographic origins, in this paper, the remaining 245 unrelated individuals were enrolled for the comparison of the genotype distribution. As shown in Table 4, there were 23 individuals from the north, 35 from the border and 187 from the south, respectively. Among a total of 53 genotypes, the four genotypes c.851_854del4/c.851_854del4, c.851_854del4/IVS16ins3kb, c.851_854del4/IVS6 + 5G > A and c.851_854del4/c.1638_1660dup were dominant with the relative frequencies of 42.04%, 10.61%, 7.76% and 6.94%, respectively. Despite of the marked diversity of the genotypes, only c.851_854del4/c.851_854del4 and c.851_854del4/c.1399C > T (p.R467X) demonstrated significantly different geographic distribution with P < 0.05. Further pairwise comparisons revealed that the relative frequency of c.851_854del4/ c.851_854del4 in the south was much higher than that in the north (P = 0.000).
The observed homozygosity calculated based on the genotype frequencies was 17.39% (4/23) in the north, 34.28% (12/35) in the border and 52.94% (99/187) in the south, respectively. In addition, the theoretical homozygosity value, which was calculated based on allele frequencies, was 15.24% in the north, 24.31% in the border and 44.81% in the south, respectively. In pairwise comparisons, both of the observed and theoretical homozygosity values were higher in the south than in the north, with the P values of 0.001 and 0.012, respectively. In other words, the northern population had a higher level of allelic heterogeneity at the SLC25A13 locus than the southern population.
Discussion
The first case of NICCD in mainland China was reported by our group in 200643. Since then, more and more Chinese CD patients were definitely diagnosed by SLC25A13 genetic analysis in our department23,24,27,33,34. During this process, conventional DNA analytic approaches such as PCR/LA-PCR, PCR-RFLP and Sanger sequencing played important roles. In this study, as shown in Table 2, the four mutations c.851_854del4, c.1638_1660dup, IVS6 + 5G > A and IVS16ins3kb together had a relative frequency of 84.47%, indicating that the screening of these high-frequency mutations should be initially performed for the rapid molecular diagnosis of CD patients. In addition, direct sequencing of the 18 SLC25A13 exons and their adjacent intronic regions could identified the remaining micro mutations, which accounted for 12.5%. Besides, the functional and bioinformatic tools also made substantial contribution to the pathogenicity confirmation of the novel missense mutation c.103A > G (p.M35V). As a result, the 154 new CD patients diagnosed in this paper, together with those reported in our department previously, constituted a 274-case cohort. So far as we know, this is the hitherto largest CD cohort in official references worldwide, laying a foundation for our subsequent clinical investigation. In particular, the 9 novel mutations identified in this study enriched the SLC25A13 mutation spectrum, and provided reliable laboratory evidences not only for the definite diagnosis of the corresponding individuals, but also for the genetic counseling of their families in the future.
It was noteworthy that, as an unique technique developed by our group42, the SLC25A13 cDNA cloning analysis using PBLs had proved to be a feasible tool for the detection of the large insertion/deletion mutations23,24,27. In this paper, this molecular tool also played unique roles in the definite diagnosis of NICCD patients. The aberrant transcripts detected by cDNA analysis proved that c.755 − 1G > C, c.845_c.848 + 1delG, c.933_c.933 + 1insGCAG and c.1452 + 1G > A all had influence on the splicing process of the pre-mRNA. In addition, by using this tool, the novel mutation [c.329 − 154_c.468 + 2352del2646; c.468 + 2392_c.468 + 2393ins23] in patient C0360 was identified as the third large mutation resulting in exon 5 skipping (r.329_468, p.E110fs127X) following the mutation IVS4ins6kb (GenBank accession number: KF425758)27 and c.329 − 1687_c.468 + 3865del24. Of particular note, in patient C0388, we confirmed that the maternally-inherited allele harbored two deleterious mutations based on the cDNA cloning results. Although the paternal mutation remained to be explored currently, the undetectable transcriptional product from the paternal allele clearly indicated the existence of a pathogenic mutation, providing direct laboratory evidences supporting the CD diagnosis. Unfortunately, due to the lack of fresh PBLs or liver specimens for further cDNA cloning analysis, mutations in 6 SLC25A13 alleles remained obscure in this study; however, the rate of unidentified mutations (1.14% of all mutated alleles) in our study is much lower than those in previous publications using the conventional molecular approaches only. SLC25A13 cDNA cloning analysis using PBLs should be taken as an important tool for the molecular diagnosis of CD patients.
Although the relative frequencies of c.1638_1660dup and IVS6 + 5G > A were uniform in different regions of China, some SLC25A13 mutations demonstrated different geographic distribution in this study. The relative frequencies of c.851_854del4 tended to decrease gradually from south to north, while that of IVS16ins3kb had an opposite tendency, as shown in the Table 2. The genetic variation following a continuous pattern from south to north might be attributed to the genetic flow occurred between distinct populations. Recent genetic studies have suggested that modern humans colonized East Asia via Southern and Northern routes on both sides of the Himalayas. Genetic flow between populations, which took place when the two migration routes overlapped, probably lasted a long time, resulting in the continuous pattern of genetic variation44,45. The continuous patterns of genetic variation at the SLC25A13 locus in the present study are compatible with this presumed migration model. Actually, previous haplotype study28 suggested that c.851_854del4 originated around the Guangxi and Yunan areas. Its higher frequency in south China can be explained as a result of the founder effect, while its lower frequency in the north, by genetic drift. Interestingly, besides the four common mutations, c.1399C > T (p.R467X) was relatively common in the border, while IVS11 + 1G > A and c.1364G > T (p.R455L) were relatively common in the north. This phenomenon might be attributed to different founding populations but a lower migration rate among different areas in mainland China. These mutations should be considered as targets when establishing a screening strategy for CD patients in relevant populations.
Moreover, although a diversity of genotypes with a total number of 53 was discovered in the large CD cohort, c.851_854del4/c.851_854del4 was the unique genotype with higher relative frequency in the south than in the north (49.20% vs 4.35%), as shown in Table 4. This could be explained once again by the aforementioned founder effect of the c.851_854del4 mutation, which might occurred in a far remote ancestor in the south; Furthermore, subsequent homozygosity comparison demonstrated that, different from the CD patients from the south who had higher homozygosity, patients in the north showed higher allelic heterogeneity at the SLC25A13 locus. This finding suggested that some CD patients might be missed while the CD prevalence be underestimated in the north, when the same high-frequency mutations as in the south were choosed as the molecular targets for the detection of CD patients and SLC25A13 carriers in the north area. Therefore, the exploration of additional SLC25A13 mutations should be regarded as an important issue in the north area in terms of CD molecular diagnosis and epidemic survey.
In summary, via sophisticated molecular, functional and in silico analysis of SLC25A13 gene and its cDNA, this paper reported 154 new CD patients and identified 9 novel pathogenic mutations. The SLC25A13 mutation spectrum in the hitherto largest CD cohort of 274 cases and their different geographic distribution formed a substantial contribution to the in-depth understanding of the genotypic feature of CD patients in China, and provided reliable evidences for the development of molecular diagnostic strategies in different Chinese areas.
Methods
Subjects and Ethics
This research enrolled a total of 274 CD patients diagnosed by our group in the past over 10 years, including 154 new CD cases which were diagnosed by sophisticated molecular, functional and bioinformatic analysis of SLC25A13 gene and its cDNA, as described below, from February, 2013 to the end of February, 2016. Our study adheres to the ethical guidelines of the World Medical Association Declaration of Helsinki (WMADH 2008) and was approved by the Medical Ethical Committee of the First Affiliated Hospital, Jinan University. SLC25A13 analyses were conducted with the written informed consents from the guardians of the patients.
SLC25A13 Mutation Analysis
Genomic DNA of the patients suspected to have CD and their parents was extracted from EDTA-anticoagulant peripheral venous blood. The 4 high-frequency mutations c.851_854del4, c.1638_1660dup, IVS6 + 5G > A and IVS16ins3kb were initially screened by using PCR/LA-PCR and PCR-RFLP, respectively. All the 18 SLC25A13 exons and their adjacent intronic regions were amplified by PCR and analyzed by Sanger sequencing in patients with just one mutation was detected. Following that, if there was a SLC25A13 mutation remained obscure, IVS4ins6kb, another large insertion with relative high frequency in Chinese, would be screened by LA-PCR as in our previous publications27.
Reverse transcription-PCR (RT-PCR) and cDNA cloning analysis
RT-PCR and cDNA cloning analysis were subsequently carried out in patients still with undetected mutation by all the approaches above, and in those with novel mutations that might affect the splicing of pre-mRNA molecules. In brief, total RNA was extracted from peripheral blood lymphocytes (PBLs), which were collected from 2 ml fresh EDTA-anticoagulant peripheral venous blood. Then RT-PCR was performed to synthesized cDNA following the kit manufacture’s protocol (Invitrogen, USA). With the cDNA as template, nest-PCR was then performed for the target products, and the purified nest-PCR products were cloned into PMD-18T vector (Takara, Japan) and transformed into DH5α Escherichia coli competent cells. The positive clones were tested by LA-PCR with the universal primer set RVM (GAGCGGATAACAATTTCACACAGG) and M13-47 (CAGCACTGACCCTTTTGGGACCGC) and then sequenced, as previously described42.
In silico analyses
The conservative property of the amino acid affected by the novel missense mutation was analyzed by the software Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/)46. The amino acid sequences of human citrin were comparatively aligned with other homologous proteins from 10 different eukaryotic species, including chimpanzee, mouse, rat, dog, cow, pig, opossum, chicken, xenopus tropicalis and caenorhabditis elegans. All of these amino acid sequences were obtained from the NCBI database (www.ncbi.nlm.nih.gov). Then the pathogenicity of the missense mutation was predicted by the softwares PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/)47,48, mutationTaster (http://mutationtaster.org/ MutationTaster/index.html)49 and PROVEAN (http://provean.jcvi.org/index.php)50,51, respectively.
Functional study
A diploid AGC1-disrupted yeast model, BYagc1Δ, which was constructed in our previous publication34, was used to evaluate the functional effect of the novel missense mutation. The normal citrin-coding sequence (NM_014251.2) was amplified and the novel missense mutation was introduced into the wild type SLC25A13 cDNA by overlap-extension PCR. These products were purified and cloned into the vector pYX212 (Novagen, USA) to constitute the plasmid pYX212-citrin and pYX212-mutant, respectively. The BYagc1Δ strains were then transfected with the recombinant plasmids and the empty vector pYX212, respectively. The positive clones were screened using the uracil minus medium SD-URA and cultured in SA medium with acetate as the unique carbon source. After 96 hour of culture, the growth abilities of these three strains were assessed by the cell density measured at OD600.
Geographic division
The Yangtze River has been considered as a historically significant boundary of the Chinese population52,53,54. In addition, the previously estimated carrier rate of SLC25A13 mutations was 1/48 in the south but 1/940 in the north of this river28. Accordingly, the distribution of the mutations and genotypes in this study were compared among the north, border and south regions relevant to this boundary, based on the origin of the parents of each case. In this study, individuals in the north area referred to those from Beijing, Inner Mongolia, Shangdong, Shanxi, Shaanxi, Henan, Hebei, Liaoning, Jilin and Heilongjiang; in the border, from Shanghai, Jiangsu, Anhui, Hubei, Sichuan and Chongqing; and in the south, form Guangdong, Guangxi, Yunnan, Guizhou, Hunan, Fujian, Zhejiang, Jiangxi, Hainan and Taiwan, respectively.
Calculation of homozygosity
The theoretical homozygosity (J) at a locus in a given population is measured by J = ∑Χi2, where ∑ stands for summation over all alleles, and Χi is the frequency of the ith allele55,56. If the number of the ith allele is m, Χi is calculated to be m/N, where N is the total number of the mutant alleles being investigated. The alleles harboring obscure mutations were counted as SLC25A13 alleles different from those with detected variations, and thus, each of them was defined to have a frequency of 1/N.
Statistical analysis
The frequencies of the SLC25A13 mutations and genotypes, as well as the homozygosity values among the three geographic areas, were compared by means of Chi-square test or Fisher’s exact tests, respectively. When the Chi-square test of 3 × 2 table was significant with P < 0.05, pairwise comparisons were then performed with Bonferroni corrections of the P values. There were 3 pairwise comparisons: (1) north vs border, (2) north vs south, and (3) border vs south; accordingly, the adjusted P values of 2 × 2 Chi-square test for significance was 0.017 (0.05/3)57. The data of growth abilities of the yeast strains were analyzed by using one-way ANOVA followed by the Games-Howell test for the pairwise comparison of the non-homogeneity of variances, with P < 0.05 as the significant criteria. All statistical calculations were performed on the software SPSS17.0.
Additional Information
How to cite this article: Lin, W.-X. et al. Molecular diagnosis of pediatric patients with citrin deficiency in China: SLC25A13 mutation spectrum and the geographic distribution. Sci. Rep. 6, 29732; doi: 10.1038/srep29732 (2016).
References
Kobayashi, K. et al. The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat Genet. 22, 159–163 (1999).
Palmieri, F. Mitochondrial transporters of the SLC25 family and associated diseases: a review. J Inherit Metab Dis. 37, 565–575 (2014).
Palmieri, F. & Monné, M. Discoveries, metabolic roles and diseases of mitochondrial carriers: A review. Biochim Biophys Acta. pii: S0167-4889(16)30060-X (2016).
Thangaratnarajah, C., Ruprecht, J. J. & Kunji, E. R. Calcium-induced conformational changes of the regulatory domain of human mitochondrial aspartate/glutamate carriers. Nat Commun. 5, 5491 (2014).
Tomomasa, T. et al. Possible clinical and histologic manifestations of adult-onset type II citrullinemia in early infancy. J Pediatr. 138, 741–743 (2001).
Ohura, T. et al. Neonatal presentation of adult-onset type II citrullinemia. Hum Genet. 108, 87–90 (2001).
Tazawa, Y. et al. Infantile cholestatic jaundice associated with adult-onset type II citrullinemia. J Pediatr. 138, 735–740 (2001).
Saheki, T. & Kobayashi, K. Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD). J Hum Genet. 47, 333–341 (2002).
Saheki, T. et al. Pathogenesis and pathophysiology of citrin (a mitochondrial aspartate glutamate carrier) deficiency. Metab Brain Dis. 17, 335–346 (2002).
Yamaguchi, N. et al. Screening of SLC25A13 mutations in early and late onset patients with citrin deficiency and in the Japanese population: Identification of two novel mutations and establishment of multiple DNA diagnosis methods for nine mutations. Hum Mutat. 19, 122–130 (2002).
Song, Y. Z. et al. Failure to thrive and dyslipidemia caused by citrin deficiency: a novel clinical phenotype. Chin J Contemp Pediactr. 11, 328–332 (2009).
Avdjieva-Tzavella, D. M. et al. First Bulgarian case of citrin deficiency caused by one novel and one recurrent mutation in the SLC25A13 gene. Genet Counsel. 25, 271–276 (2014).
Moriyama, M. et al. Mechanism for increased hepatic glycerol synthesis in the citrin/mitochondrial glycerol-3-phosphate dehydrogenase double-knockout mouse: Urine glycerol and glycerol 3-phosphate as potential diagnostic markers of human citrin deficiency. Biochim Biophys Acta. 1852, 1787–1795 (2015).
Takeuchi, S. et al. An Adolescent case of Citrin Deficiency with severe Anorexia Mimicking Anorexia Nervosa. Pediatrics. 136, e530–e534 (2015).
Kobayashi, K., Saheki, T. & Song, Y. Z. Citrin deficiency. In GeneReviews [Internet] (eds Pagon, R. A. et al.) (updated 2014).
Yasuda, T. et al. Identification of two novel mutations in the SLC25A13 gene and detection of seven mutations in 102 patients with adult-onset type II citrullinemia. Hum Genet. 107, 537–545 (2000).
Dimmock, D. et al. Citrin deficiency: a novel cause of failure to thrive that responds to a high-protein, low-carbohydrate diet. Pediatrics. 119, e773–e777 (2007).
Ohura, T. et al. Clinical pictures of 75 patients with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD). J Inherit Metab Dis. 30, 139–144 (2007).
Kobayashi, K., Ushikai, M., Song, Y. Z., Gao, H. Z. & Sheng, J. S. Overview ofcitirn deficiency: SLC25A13 mutations and the frequency. J Appl Clin Pediatr. 23, 1553–1557 (2008).
Song, Y. Z., Wen, F., Chen, F. P., Kobayashi, K. & Saheki, T. Neonatal intrahepatic cholestasis caused by citrin deficiency: Efficacy of the rapeutic formulas and update ofclinical outcomes. Jpn J Inherit Metab Dis. 26, 57–69 (2010).
Tokuhara, D. et al. Novel diagnostic approach to citrin deficiency: analysis of citrin protein in lymphocytes. Mol Genet Metab. 90, 30–36 (2007).
Chen, R. et al. Different regional distribution of SLC25A13 mutations in Chinese patients with neonatal intrahepatic cholestasis. World J Gastroenterol. 19, 4545–4551 (2013).
Zeng, H. S. et al. SLC25A13 cDNA cloning analysis using peripheral blood lymphocytes facilitates the identification of a large deletion mutation: Molecular diagnosis of an infant with neonatal intrahepatic cholestasis caused by citrin deficiency. Mol Med Rep. (In production) (2016).
Zheng Q. Q. et al. Identification of a Large SLC25A13 Deletion via Sophisticated Molecular Analyses Using Peripheral Blood Lymphocytes in an Infant with Neonatal Intrahepatic Cholestasis caused by Citrin Deficiency (NICCD): A Clinical and Molecular Study. Biomed Res Int. 2016, 4124263 (2016).
Fu, H. Y. et al. The mutation spectrum of the SLC25A13 gene in Chinese infants with intrahepatic cholestasis and aminoacidemia. J Gastroenterol. 46, 510–518 (2011).
Song, Y. Z. et al. Genotypic and phenotypic features of citrin deficiency: Five-year experience in a Chinese pediatric center. Int J Mol Med. 28, 33–40 (2011).
Song, Y. Z. et al. SLC25A13 Gene Analysis in Citrin Deficiency: Sixteen Novel Mutations in East Asian Patients, and the Mutation Distribution in a Large Pediatric Cohort in China. PLoS One. 8, e74544 (2013).
Lu, Y. B. et al. Frequency and distribution in East Asia of 12 mutations identified in the SLC25A13 gene of Japanese patients with citrin deficiency. J Hum Genet. 50, 338–346 (2005).
Wongkittichote, P. et al. Screening of SLC25A13 mutation in the Thai population. World J Gastroenterol. 19, 7735–7742 (2013).
Tong, F., Yang, J. B., Huang, X. L., Zhou, X. L. & Yang, R. L. A case of neonatal intrahepatic cholestasis caused by citrin deficiency complicated with congenital biliary atresia. Zhonghua Er Ke Za Zhi. 51, 863–865 (2013).
Wen, P. Q. et al. Clinical investigation and mutation analysis of a child with citrin deficiency complicated with purpura convulsive seizures and methioninemia. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 30, 649–653 (2013).
Ricciuto, A. & Buhas, D. A novel citrin deficiency mutation in a cholestatic infant. J Pediatr Gastroenterol Nutr. 59, e52 (2014).
Zeng, H. S. et al. Inspissated bile syndrome in an infant with citrin deficiency and congenital anomalies of the biliary tract and esophagus: identification and pathogenicity analysis of a novel SLC25A13 mutation with incomplete penetrance. Int J Mol Med. 34, 1241–1248 (2014).
Zhang, Z. H. et al. Clinical, molecular and functional investigation on an infant with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD). PLoS One. 9, e89267 (2014).
Zhang, Z. H. et al. Screening for five prevalent mutations of SLC25A13 gene in Guangdong, China: a molecular epidemiologic survey of citrin deficiency. Tohoku J Exp Med. 233, 275–281 (2014).
Bijarnia-Mahay, S. et al. Citrin deficiency: A treatable cause of acute psychosis in adults. Neurol India. 63, 220–222 (2015).
Xiong, X. L. et al. Clinical research of neonatal intrahepatic cholestasis caused by citrin deficiency in Hubei Province. Chin J Appl Clin Pediatr. 30, 1064–1068 (2015).
Wang, H., Shu, S., Chen, C., Huang, Z. & Wang, D. Novel mutations in the SLC25A13 gene in a patient with NICCD and severe manifestations. J Pediatr Endocrinol Metab. 28, 471–475 (2015).
den Dunnen, J. T. & Antonarakis, S. E. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Genet. 15, 7–12 (2000).
den Dunnen, J. T. & Antonarakis, S. E. Nomenclature for the description of human sequence variations. Hum Genet. 109, 121–124 (2001).
Wongkittichote, P., Tungpradabkul, S., Wattanasirichaigoon, D. & Jensen, L. T. Prediction of the functional effect of novel SLC25A13 variants using a S. cerevisiae model of AGC2 deficiency. J Inherit Metab Dis. 36, 821–830 (2013).
Zhang, Z. H., Lin, W. X., Deng, M., Zhao, X. J. & Song, Y. Z. Molecular analysis of SLC25A13 gene in human peripheral blood lymphocytes: Marked transcript diversity, and the feasibility of cDNA cloning as a diagnostic tool for citrin deficiency. Gene. 511, 227–234 (2012).
Song, Y. Z. et al. A difficult and complicated case study: neonatal intrahepatic cholestasis caused by citrin deficiency. Zhongguo Dang Dai Er Ke Za Zhi. 8, 125–128 (2006).
Di, D. & Sanchez-Mazas, A. HLA variation reveals genetic continuity rather than population group structure in East Asia. Immunogenetics. 66, 153–160 (2014).
Di, D., Sanchez-Mazas, A. & Currat, M. Computer simulation of human leukocyte antigen genes supports two main routes of colonization by human populations in East Asia. BMC Evol Biol. 15, 240 (2015).
Sievers, F. & Higgins, D. G. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol Biol. 1079, 105–115 (2014).
Adzhubei, I. A., Schmidt, S. & Peshkin, L. A method and server for predicting damaging missense mutations. Nat Methods. 7, 248–249 (2010).
Adzhubei, I. A., Jordan, D. M. & Sunyaev, S. R. Prediction functional effect of human missense mutations using polypen-2. Curr Protoc Hum Genet. Chapter 7, Unit 7. 20 (2013).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation predition for the deep-sequencing age. Nat Methods. 11, 361–362 (2014).
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R. & Chan, A. P. Predicting the functional effect of amino acid substitutions and indels. PLOS One. 7, e46688 (2012).
Choi, Y. & Chan, A. P. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 31, 2745–2747 (2015).
Zhao, T. M. & Lee, T. D. Gm and Km allotypes in 74 Chinese populations: a hypothesis of the origin of the Chinese nation. Hum Genet 83, 101–110 (1989).
Du, R., Xiao, C. & Cavalli-Sforza, L. L. Genetic distances between Chinese populations calculated on gene frequencies of 38 loci. Sci China C Life Sci. 40, 613–212 (1997).
Chu, J. Y. et al. Genetic relationship of populations in China. Proc Natl Acad Sci USA 95, 11763–11768 (1998).
Nei, M. Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70, 3321–3323 (1973).
Guldberg, P. et al. Phenylalanine hydroxylase gene mutations in the United States: report from the maternal PKU collaborative study. Am J Hum Genet. 59, 84–94 (1996).
MacDonald, P. L. & Gardner, R. C. Type I error rate comparisons of post hoc procedures for I×J chi-square tables. Educ Psychol Meas. 60, 735–754 (2000).
Acknowledgements
We thank all patients and their parents for their kind cooperation. This research was financially supported by the Cultivation Foundation for Scientific Research (Nos 2014208 and A2014101) approved by the First Affiliated Hospital, Jinan University, the Medical Scientific Research Foundation of Guangdong Province (A2014385), the Fundamental Research Funds for the Central Universities (No. 21615470), and the projects supported by the National Natural Science Foundation of China (Nos 81070279, 81270957 and 81570793).
Author information
Authors and Affiliations
Contributions
Y.-Z.S. and Z.-H.Z. designed the research. F.-P.C. and W.-R.W. collected most of the peripheral venous blood samples. W.-X.L., H.-S.Z., Z.-H.Z., M.M., Q.-Q.Z., S.-T.Z. and Y.C. performed the experiments. W.-X.L. analyzed the data. W.-X.L. and Y.-Z.S. wrote the main manuscript. All authors reviewed the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Lin, WX., Zeng, HS., Zhang, ZH. et al. Molecular diagnosis of pediatric patients with citrin deficiency in China: SLC25A13 mutation spectrum and the geographic distribution. Sci Rep 6, 29732 (2016). https://doi.org/10.1038/srep29732
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep29732
This article is cited by
-
The mutation spectrum of SLC25A13 gene in citrin deficiency: identification of novel mutations in Vietnamese pediatric cohort with neonatal intrahepatic cholestasis
Journal of Human Genetics (2023)
-
Analysis of clinically relevant variants from ancestrally diverse Asian genomes
Nature Communications (2022)
-
The prevalence, genetic complexity and population-specific founder effects of human autosomal recessive disorders
npj Genomic Medicine (2021)
-
Citrin deficiency mimicking mitochondrial depletion syndrome
BMC Pediatrics (2020)
-
Combined primary carnitine deficiency with neonatal intrahepatic cholestasis caused by citrin deficiency in a Chinese newborn
BMC Pediatrics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
