
Abstract
Poikiloderma with Neutropenia (PN) is an autosomal recessive genodermatosis characterized by early-onset poikiloderma, pachyonychia, hyperkeratosis, bone anomalies and neutropenia, predisposing to myelodysplasia. The causative C16orf57/USB1 gene encodes a conserved phosphodiesterase that regulates the stability of spliceosomal U6-RNA. The involvement of USB1 in splicing has not yet allowed to unveil the pathogenesis of PN and how the gene defects impact on skin and bone tissues besides than on the haematological compartment. We established a zebrafish model of PN using a morpholino-knockdown approach with two different splicing morpholinos. Both usb1-depleted embryos displayed developmental abnormalities recapitulating the signs of the human syndrome. Besides the pigmentation and osteochondral defects, usb1-knockdown caused defects in circulation, manifested by a reduced number of circulating cells. The overall morphant phenotype was also obtained by co-injecting sub-phenotypic dosages of the two morpholinos and could be rescued by human USB1 RNA. Integrated in situ and real-time expression analyses of stage-specific markers highlighted defects of primitive haematopoiesis and traced back the dramatic reduction in neutrophil myeloperoxidase to the myeloid progenitors showing down-regulated pu.1 expression. Our vertebrate model of PN demonstrates the intrinsic requirement of usb1 in haematopoiesis and highlights PN as a disorder of myeloid progenitors associated with bone marrow dysfunction.
Similar content being viewed by others

Introduction
Poikiloderma with Neutropenia, Clericuzio-type (PN; OMIM#604173) is a rare autosomal recessive genodermatosis affecting the skin and the haematopoietic system, in particular the myeloid lineage. It is characterized by early onset poikiloderma, a cutaneous lesion with a pattern of reticulated hypo-hyper-pigmentation, mild atrophy and telangiectasias, pachyonychia, palmo-plantar hyperkeratosis, bone alterations and non-cyclic neutropenia, usually revealed by recurrent infections during infancy and childhood1,2,3,4,5. Discovery of the causative C16orf57 gene by autozygosity mapping and next-generation sequencing of the linkage region on chromosome 166, confirmed that PN is genetically distinct from clinically overlapping entities, in particular Rothmund-Thomson syndrome (OMIM#268400) and allowed the introduction of the genetic test to validate diagnosis and provide patients with the appropriate oncological surveillance7,8,9,10,11. Neutropenia, the distinctive PN hallmark, confers on patients a high risk to develop myelodysplasia, often evolving into acute myeloid leukaemia in the second decade of life12,13.
The C16orf57 gene, renamed USB1 (U six biogenesis 1), is phylogenetically conserved and ubiquitously expressed, suggesting a housekeeping function6,12.
Computational analysis of the encoded protein shows two conserved H-X-S/T-X tetra-peptide motifs (2H), which mark the active site of a 2-fold pseudosymmetric structure5. The signature of the 2H motifs, characteristic of the 2H phosphoesterase family14,15, suggested that the USB1 protein was likely to be involved in RNA processing, leading to the prediction that all the reported USB1 loss-of-function mutations cause disruption of both protein folding and catalytic site5.
Recently, it has been demonstrated that in Saccharomyces cerevisiae and human cells the USB1 gene and its yeast ortholog encode a phosphodiesterase that is essential for the biogenesis of the RNA splicing apparatus as it regulates the stability of U6 small nuclear RNA (snRNA)16,17. X-ray crystallography of the human USB1 protein has definitely confirmed that USB1 is an RNase that trims the 3′ end of the U6 transcript implicating aberrant oligoadenylation of U6 snRNA in the pathogenesis of PN18. While these insights advance our understanding of the PN pathomechanism, how USB1 mutations impact on the morphogenesis and differentiation of tissues affected in PN remains unknown.
To address this issue, we have exploited the zebrafish as a model organism. Zebrafish has a single USB1 ortholog, which is ubiquitously expressed during early development. Using a morpholino (MO) knockdown approach, we show that the morphants display developmental abnormalities that reproduce the signs of the human syndrome, namely defective pigmentation, osteochondral alterations and severe haematopoietic defects, mainly affecting the myeloid lineage.
Results
Identification and characterization of the zebrafish usb1 gene
We performed BLAST searches with the human USB1 gene and protein sequences to identify the zebrafish ortholog in the database (usb1, NM_001003460.1), which is present in only one copy in the zebrafish genome.
The genomic structure of USB1 and usb1 orthologs is similar, with 7 exons in both cases (Fig. 1a) and the USB1- and usb1-containing regions on human chromosome 16q21 and zebrafish chromosome 25 are fairly syntenic.

The human USB1 gene, the zebrafish usb1 ortholog and the encoded proteins.
(a) Schematic genomic structure of USB1 and usb1. The lengths, not to scale, of exons (boxes), UTRs and introns (interconnecting bars) are indicated below and over the scheme, respectively. The encoded proteins share 73.4% and 46% sequence similarity and identity, respectively. (b) BLAST alignment of the human (NP_078874.2) and zebrafish (NP_001003460.1) usb1 proteins. Identical amino-acid residues and residues with the same polarity can be noted in several positions. Vertical dotted lines highlight the conserved intron–exon junctions with flanking numbers denoting exon number. The two histidine-serine residues of the HLSL motifs that are a main part of the catalytic site of the protein5,16 are boxed. (c) Prediction of the secondary usb1 protein structure using the PSIPRED server (http://bioinf.cs.ucl.ac.uk/psipred/). Barrels represent the α-helices and arrows the β-strands. The two histidine-serine residues of the HLSL motifs are boxed.
The encoded human and zebrafish proteins are 265 and 276 amino acids (aa) in length, respectively, with a sequence similarity and identity of 73.4% and 46%, respectively. The difference in length between the genomic usb1 sequence (9452 bp) and that of USB1 (22072 bp) is accounted for by shorter usb1 introns (Fig. 1a).
Alignment of the entire human and zebrafish protein sequences shows a highly evolutionarily conserved amino acids sequence from the 51st to the last aa residue (Fig. 1b).
The zebrafish protein shares with the human protein the distinctive series of secondary structure elements (α-helices and β-strands) and has the two HLSL motifs (boxed in Fig. 1b,c), which are the main part of the catalytic site in the human protein5,16,17,18.
Expression profile of usb1 during zebrafish development
Analysis of usb1 expression during zebrafish embryonic development by reverse transcription-PCR (RT-PCR) showed that usb1 transcript was both maternally and zygotically expressed (Fig. 2a). In adult animals, usb1 expression was recorded in all analysed tissues (brain, eyes, liver, gills, heart, muscle, ovary and testis) (Fig. 2a).

Expression profiling of usb1 in zebrafish.
(a) Temporal and spatial usb1 expression during embryogenesis and among adult tissues. Total RNA was extracted from embryos at the indicated time-points and from the specified adult tissues and subjected to RT-PCR with primers specific for usb1 (upper panel) or β-actin (lower panel). The sizes of the obtained PCR fragments are indicated. (b) WISH analysis of usb1. The signal obtained with usb1 antisense probe was ubiquitous until the 5-somite stage. It showed a higher intensity in the cephalic region at the 15 somites stage and displayed decreasing expression from the head to the caudal region (20 somites and 26 hpf). At 36 hpf, the signal only marked the head and the pharyngeal arches-derived structures. hpf: hour post fertilization; dpf: day post fertilization. Images showing the results obtained with sense probe are provided at each developmental stage. Scale bars: 200 μm.
Whole-mount in situ hybridization analysis (WISH) with an antisense probe targeting the full-length transcript (1,126 bp) was also used to characterize usb1 expression patterns during embryo development (Fig. 2b). At early developmental stages, from two cells up to the 5 somites stage, usb1 expression was ubiquitous and consistent with the expression found by RT-PCR (Fig. 2a). By the 15 somites stage until 26 hours post fertilization (hpf), expression of usb1 was higher in the anterior head region than in the posterior tail (Fig. 2b). From 26 hpf, usb1 transcripts were detected in the cephalic region and in specific trunk regions that at 36 hpf were recognized as the pharyngeal arches, the budding liver and the pectoral fins, while no expression was observed in the caudal region (Fig. 2b). At 36 hpf the expression persisted only in the anterior head and trunk regions (Fig. 2b). No signal was observed in embryos at the same developmental stage when stained with usb1 sense probe (Fig. 2b).
Design and validation of splice-blocking morpholinos
To investigate the role of usb1 during zebrafish embryogenesis, we performed loss-of-function experiments using MO-mediated gene knockdown.
We injected two different splice-blocking MOs, SMO-A and SMO-B, targeting the usb1 sequence between the IVS2 splice acceptor site and exon 3 and between the IVS4 splice acceptor site and exon 5, respectively (Fig. 3a).

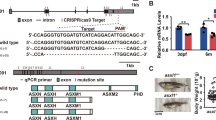
Efficiency of usb1 knockdown by splice-blocking morpholino SMO-A, targeting IVS2 acceptor splice site and SMO-B, blocking IVS4 acceptor splice site.
(a) Schematic diagram of zebrafish usb1 gene organization and position of SMO-A (blue) and SMO-B (red). Blue and red arrows indicate the positions of primer pairs used to detect the presence of aberrantly spliced usb1 transcripts obtained with SMO-A (primers F2-R3, Supplementary Table S1) and SMO-B (primers F-R2, Supplementary Table S1), respectively. (b) RT-PCR analysis of usb1 transcripts from embryos injected with SMO-A, SMO-B or Std-MO (each at 0.6 pmol/embryo). SMO-A morphants exhibited aberrantly spliced products (363 bp) as compared to Std-MO-injected embryos (547 bp) due to skipping of exon 3 (184 bp), frameshift and exposure of a stop codon 24 nucleotides downstream. SMO-B morphants exhibited aberrantly spliced products (1020 bp) as compared to Std-MO-injected embryos (1126 bp) due to skipping of exon 5 (106 bp) following the block of the IVS4 acceptor splice site. MW, molecular weight markers. (c) Schematic representation of SMO-A- and SMO-B-mediated misspliced transcripts inferred by sequencing. Aberrant proteins are predicted in both cases as the 107 aa SMO-A-translated protein should lack both HLSL motifs, while the 184 aa SMO-B-translated protein should lack the second HLSL motif.
Specific effects of SMO-A and SMO-B on usb1 mRNA splicing were confirmed by RT-PCR, which showed a severe reduction in the amount of normal splice products and the formation of aberrant transcripts (Fig. 3b).
Sequence analysis revealed that the SMO-A misspliced amplicon of 363 bp was characterized by the out-of-frame skipping of exon 3. The corresponding aberrant transcript should result in a truncated protein (107 aa instead of 276 aa) lacking both HLSL motifs and therefore likely to be non-functional as the HLSL motifs are essential for the enzymatic function of the protein (Fig. 3c). The SMO-B aberrant transcript skipped exon 5, resulting in a protein that was missing the second of the two functionally relevant HLSL motifs (Fig. 3c).
Overall phenotype of usb1-morphants
Following injection of SMO-A or SMO-B (each at a dosage of 0.6 pmol/embryo) into 1–2-cell embryos, specific defects could be observed in a consistent percentage of both morphant embryos at two (Fig. 4a) and five days post-fertilization (dpf; Fig. 4b) as compared to control embryos injected with the control MO (Std-MO) (Table 1).

Overall phenotype of usb1 morphants.
Representative live-microscopy-4X magnification-pictures of 2 dpf (a) and 5 dpf (b) embryos injected with the same dosage (0.6 pmol/embryo) of Std (top), SMO-A (central) or SMO-B (bottom) morpholinos. The morphants display a smaller size, with defective and irregular pigmentation of the skin, as compared to the continuous stripes of the control embryos and pericardial oedema. (c) Massive skeletal defects revealed by Alcian blue staining in usb1 morphants at 5 dpf. From left to right: diagram illustrating the normal pharyngeal arches-derived bone architecture19; ventral view of a control embryo (left panel) and representative pictures of the mild (left) and severe (right) phenotypes of SMO-A and SMO-B morphants. The Meckel’s (yellow in the colour-coded diagram), the palatoquadrate (green) and ceratohyal (purple) structures appear misshaped in both mildly- and severely-affected embryos; moreover, the ceratobranchial structures (light blue) appear disorganized or missing in half of SMO-A and in a third of SMO-B severely-affected embryos. (d) Reduction of gata1-positive cells (red fluorescence) (middle panels) and a regular vasculature pattern (green fluorescence) (lower panels) in 2 dpf tg(gata1:dsRED;flk1:GFP) embryos injected with SMO-A (0.7 pmol/embryo) (right) as compared to control (left). (e) Myeloid lineage defects evinced by a reduction in mpx-positive cells (green fluorescence) (lower panels) in 2 dpf tg(mpx:GFP;lyzC:dsRED) embryos injected with SMO-A (0.7 pmol/embryo) (right) as compared to control (left). Scale bars: (a–e) 200 μm.
At 2 dpf, most of SMO-A morphants appeared smaller than the controls, with smaller head and 80% had pericardial oedema. Interestingly, 58% of the SMO-A-injected embryos (grown without 1-phenyl-2-thiourea) displayed a defective and scattered pattern of spotted pigment in the skin and eyes as compared to the Std-MO embryos (Fig. 4a and Table 1). At 2 dpf the phenotype of SMO-B morphants was remarkably similar to that of SMO-A morphants as they showed reduced body size, pericardial oedema (59%) and pigmentation defects (12%) (Fig. 4a and Table 1).
Furthermore, in vivo analysis at 2 dpf showed that about half of both SMO-A and SMO-B morphants had severe blood circulation defects ranging from a complete absence to reduced numbers of circulating cells (Table 1), despite a beating heart. WISH analysis of the cardiac myosin light chain 2 (cmlc2) expression did not show differences in the 2 dpf SMO-A and SMO-B morphants as compared to the controls, thus excluding heart morphology defects (Supplementary Fig. S1).
At 5 dpf, the majority of usb1-morphants never gained circulation and developed severe oedema extending to both the trunk and the yolk (Fig. 4b).
As the cardiac oedema and pigmentation defects observed in the usb1-morphants can be ascribed to an abnormal patterning of derivatives of the neural crest cells19, we explored the extent of usb1 loss-of-function on another neural crest-specified process, chondrogenesis, by staining the cartilage of the skull and pharyngeal skeleton of 5 dpf embryos with Alcian Blue.
As shown in Fig. 4c and detailed in Table 1, 89% of Std-MO-injected larvae displayed a normal cartilages architecture. By contrast, 5 dpf embryos injected with either SMO-A or SMO-B (Fig. 4c, central and right panels) displayed either mild or severe craniofacial defects (Table 1). We consider the “mild” phenotype to be mainly characterized by abnormalities of the first two pharyngeal arches P1 (misshaped Meckel’s cartilage/lower jaw and misjoined to the palatoquadrate) and P2 (kinked and displaced ceratohyal cartilage), with grossly preserved posterior pharyngeal arches P3-P7. The “severe” phenotype was defined as reduced or absent P3-P7 bilateral branchial arches associated with the P1 and P2 defects mentioned above.
Among the SMO-A morphants, cartilage alterations were observed in 81% of the examined embryos with 31% in the mild and 50% in the severe phenotype subgroups (Table 1). Of the SMO-B morphants, 59% displayed the cartilage alterations, 27% with the mild and 32% with the severe phenotype (Table 1).
Haematopoietic defects of usb1-morphants
To investigate the lineage-specific effect of usb1-knockdown on embryonic haematovascular morphogenesis and for further analysis of the circulation defects observed in usb1-morphants, we used the tg(gata1:dsRED;flk1:GFP) zebrafish line, which is transgenic for the red cell progenitor marker gata1 and the vascular marker flk1. At 2 dpf, 62% of tg(gata1:dsRED;flk1:GFP) SMO-A morphants showed a reduction in gata1-positive cells in the context of a grossly regular vasculature pattern (Fig. 4d, Table 1).
Another transgenic line, tg(mpx:GFP;lyzC:dsRED), in which the promoter of the myeloid-specific marker myeloperoxidase (mpx) drives the expression of GFP protein, was used to investigate neutrophils. At 2 dpf, a reduction in mpx-expressing cells was observed in 70% of tg(mpx:GFP;lyzC:dsRED) SMO-A-injected embryos (Fig. 4e, Table 1).
These data prompted us to focus on the effects of usb1-knockdown on the primitive haematopoietic cascade in the SMO-A morphants using WISH and real-time PCR analysis. The real-time PCR analysis evidenced a substantial decrease in expression (40%) of the myeloid progenitor marker pu.1 in 10 somites SMO-A morphants, whereas expression of the erythroid progenitor marker gata1 was not perturbed (Fig. 5a). At 22 hpf, WISH and real-time PCR analysis showed only slightly decreased gata1 levels (20%) (Fig. 5a,b), but at 30 hpf, gata1-expression levels in SMO-A and Std-MO embryos were similar (Fig. 5a).

Effect of usb1 knockdown on haematopoiesis.
(a) Real-time PCR analysis of pu.1, gata1 and mpx expression at the indicated developmental stages of embryos injected with SMO-A or Std-MO (each at 0.6 pmol/embryo). Samples were run in triplicate and data are expressed as the mean ± standard deviation. Asterisks (*) indicate statistically significant differences (t- test; p < 0.05). (b) WISH of gata1 and mpx in SMO-A and control embryos at the indicated developmental stages. (c) O-dianisidine staining for haemoglobin in embryos injected with Std-MO (top), SMO-A (central) and SMO-B (bottom panels) (each at 0.6 pmol/embryo). The left and the right images are representative of the slight and severe erythropoiesis defects observed at the indicated percentages in SMO-A- and SMO-B-injected embryos. The # points to the caudal region of the morphant where the accumulation of blood is observed. Scale bars: (b,c) 200 μm.
In agreement with the data obtained with the tg(mpx:GFP;lyzC:dsRED) morphants, the terminally-differentiated neutrophil marker mpx was shown to be significantly reduced in the SMO-A-injected embryos: real-time PCR analysis showed 60% and 40% decreased expression levels of mpx in 22 hpf (p = 0.002) and 30 hpf morphants (p = 0.0096), respectively (Fig. 5a), fairly matching WISH results (Fig. 5b).
To determine whether red cell maturation was affected by usb1-knockdown, O-dianisidine staining for haemoglobin was performed. At 2 dpf, a reduction of the mature red blood cells was observed in 77% of SMO-A and 61% of SMO-B morphants, compared with a reduction in only 13% of Std-MO-injected embryos (Fig. 5c). Two subgroups could be identified: one with a slight and one with a severe reduction in haemoglobinized erythrocytes. The slightly reduced red cell phenotype was 43% for SMO-A and 27% for SMO-B morphants. A similar percentage of SMO-A (33%) and SMO-B (34%) embryos showed the severe phenotype.
Ruling out off-target effects and validating the phenotype of usb1 morphants
Co-injection of SMO-A and p53-MO, to check for potential off-target effects of the usb1-MO, resulted in a phenotype similar to that of only usb1-depleted embryos (Supplementary Fig. S2). In vivo analysis at 3 dpf showed that pericardial oedema, abnormal pigmentation and reduced circulating blood cells were all maintained (Supplementary Fig. S2).
The specificity of the usb1 morphant phenotype was further corroborated by the co-injection of SMO-A and SMO-B at sub-phenotypic doses (each at 0.3 pmol/embryo) (Fig. 6a). When each morpholino was injected alone at this low dose, no phenotypic defects were observed. Conversely, the co-injection of both morpholinos at sub-critical doses induced an abnormal phenotype that was comparable to the one observed in the morphants injected with 0.6 pmol/embryo of SMO-A or SMO-B alone. In vivo analysis at 2 dpf showed that 56% of double-injected morphants displayed a paucity of circulating cells and 70% had oedema (Fig. 6a) and Alcian blue staining at 5 dpf revealed that 71% of double-injected morphants had a defective architecture of pharyngeal arches respect to controls and to embryos injected with SMO-A or SMO-B at sub-phenotypic doses (Fig. 6b).

Phenotypes of morphants co-injected with SMO-A and SMO-B at subphenotypic dosage and rescue of morpholino-induced phenotypes with human USB1 RNA.
(a) Representative pictures of zebrafish embryos injected with Std-MO (0.6 pmol/embryo), SMO-A (0.3 pmol/embryo), SMO-B (0.3 pmol/embryo) and co-injected with SMO-A and SMO-B (each at 0.3 pmol/embryo). (b) Alcian blue staining at 5 dpf highlights the regular morphologic architecture of the pharyngeal arch cartilages in embryos injected with Std-MO, SMO-A and SMO-B at sub-phenotypic dosages and the aberrant cartilaginous structures in embryos co-injected with SMO-A and SMO-B (each at 0.3 pmol/embryo). (c) In the left panels, lateral views of 2 dpf embryos injected with Std-MO, SMO-A (0.6 pmol/embryo) and SMO-A and human USB1 RNA (300 pg/embryo). In the right panels, fluorescent images of tg(mpx:GFP;lyzC:dsRED) embryos at 2 dpf. The green signals, representing mpx-expressing cells, are reduced in embryos injected with SMO-A (0.6 pmol/embryo) as compared to the controls, but are enhanced in embryos co-injected with human USB1 RNA (300 pg/embryo). The fractions of embryos exhibiting the investigated phenotypes out of the total number of those examined are indicated. All images are at the same magnification. Scale bars: 200 μm.
Rescue experiments were performed on the tg(mpx:GFP;lyzC:dsRED) line to further confirm the specificity of the usb1 MO-induced phenotype. Co-injection of human wild-type USB1 mRNA (300 pg/embryo) together with SMO-A (0.6 pmol/embryo) rescued the usb1 morphant phenotype (Fig. 6c). At 2 dpf, 58% of co-injected embryos were similar to the controls and had a normal circulation, at difference of SMO-A injected embryos, of which only a small proportion (21%) was without defects in body structure and circulation (Fig. 6c, left panels).
An increase of mpx-positive cells could also be appreciated in 55% of embryos co-injected with USB1 RNA (300 pg/embryo) and SMO-A (0.6 pmol/embryo) (Fig. 6c, right panels), indicating recovery of the number of mature neutrophils.
Discussion
In this study we showed that the developmental abnormalities observed in patients affected by Poikiloderma with Neutropenia syndrome can be effectively recapitulated in zebrafish through the knockdown of the single usb1 gene. The two exploited splice-blocking MOs were both capable of giving rise to the signs of the human clinical spectrum, although SMO-A showed a higher penetrance of the abnormal phenotypic traits than SMO-B.
PN has two hallmarks: “poikiloderma” and mild to severe “neutropenia”. The latter predisposes to myelodysplasia and acute myeloid leukaemia3,4,5,6,12,13 thus featuring PN as a disorder of the haematopoietic cascade, mainly of the myeloid lineage. Biochemical studies on yeast and lymphoblastoid cells from PN patients have shown that USB1 plays a crucial role in the stability and recycling of U6 snRNA, a component of the RNA splicing machinery16,17,18. As mutations affecting spliceosomal genes that result in defective splicing delineate a new leukaemogenic pathway20,21, the defective function of USB1 could take part in this pathomechanism. However, splicing defects could be observed only in the yeast model, not in PN lymphoblastoid cell lines, leaving the pathogenesis of PN incompletely understood22.
In line with the substantial evidence that the zebrafish is an excellent system for the study of haematopoiesis during development23, our model, together with that recently generated by Patil et al.24, demonstrate that the usb1 gene has a key role in normal haematopoiesis and, when dysfunctional, leads to significant reduction of mature mpx-neutrophils, making it a candidate for a leukaemia-causing gene.
Both our SMO-A and SMO-B morphants engendered haematological defects, as shown by the significantly reduced expression of mpx in the tg(mpx:GFP;lyzC:dsRED) morphants. Furthermore, by injecting SMO-A into the tg(gata1:dsRED;flk1:GFP) line, it was possible to clarify cell lineage relationships and establish that the observed reduction in blood flow was due to haematopoietic deficiency rather than a vascular defect. Parallel analysis of haematopoiesis-specific markers by WISH and real-time PCR demonstrated that the usb1 deficit impacted on the myeloid precursors traced by the pu.1 marker in 10 somites embryos and on the downstream mature neutrophils, as revealed by the dramatic down-regulation of mpx expression in 22 hpf and 30 hpf morphants. This finding attests that the myeloid is the most severely affected lineage by usb1 knockdown during primitive haematopoiesis. There is a parallel between the neutropenia seen in the human syndrome, which clinically is revealed by recurrent infections and the reduction in mpx-positive neutrophils in zebrafish cells that have been shown to have anti-inflammatory properties25. Furthermore, the effect of usb1 deficiency also affected the erythroid lineage, as mature red cells were reduced in number in 2 dpf SMO-A and SMO-B morphants. The expression levels of the erythroid progenitor marker gata1 were variable over time: a decreased expression was observed by WISH and real-time PCR analysis in the 22 hpf morphants and was documented in vivo in 2 dpf tg(gata1:dsRED;flk1:GFP) SMO-A morphants. Conversely, real-time PCR analysis of gata1 at 10 somites and 30 hpf showed levels of expression comparable to those in control embryos.
Taken together, our haematological results indicate the involvement of usb1 early enough in the haematopoietic cascade to affect erythro-myeloid precursors which are present prior to the emergence of multi-lineage haematopoietic stem cells26, although its preeminent effect is exerted downstream of the critical bifurcation point between erythroid and myeloid lineage.
Interestingly, although almost all PN patients show involvement of the myeloid lineage, a few have been reported with multi-lineage deficiency3,4,6,12,27,28.
Other clinical signs of PN, such as poikiloderma and skeletal defects, were fully reproduced by both SMO-A and SMO-B morphants. The impact of usb1 depletion on the zebrafish pigmentation and developing skeleton suggests that it is essential for the correct migration of ectoderm-derived neural crest-cells that differentiate in a wide spectrum of cell types, including melanocytes and craniofacial skeleton. Consistent with the physiological role of USB1 in the morphogenesis and homeostasis of developing skin and skin annexes, our SMO-A morphants, including those generated in the two transgenic lines, displayed stripes of pigment much thinner than those in the Std-MO-injected embryos. Reduction in the clusters of spotted pigment was also observed in SMO-B morphants, in a smaller percentage and to a less degree.
Skeletal defects, including osteopenia and delayed skeletal maturation, are suggested to be more frequent in PN patients than reported, as they are mostly detectable only by X-ray5. The usb1 morphants had overt and massive defects of the pharyngeal arches-derived bones, particularly those involving the Meckel’s, palatoquadrate and ceratohyal structures. The severe disruption of a few branchial arches and associated cartilages accounts for the small head of the morphants and their craniofacial defects. The leaner and smaller body of the morphants in comparison to Std-MO-injected embryos mirrors the small stature of PN patients. Interestingly, the zebrafish model of Shwachman-Bodian-Diamond syndrome (SBDS; OMIM#260400), a well characterized defect of ribosome biogenesis associated with haematopoietic dysfunction and increased cancer risk, due to mutations of the SBDS gene, is also characterized by skeletal defects in addition to chronic neutropenia29.
Moreover, the concurrence of skeletal defects, disorganized distribution of melanocytes and cardiac oedema has been observed in zebrafish with knockdown of the pinin (pnn) gene, which encodes a nuclear and desmosome-associated protein that modulates alternative splicing of a specific subset of target genes30.
The observation that SMO-A, SMO-B and co-injection of both morpholinos at sub-phenotypic doses caused a similar alteration of the overall phenotype (body size, pericardial oedema, dyspigmentation and osteocartilagineous defects) and of haematopoietic stage-specific markers (mpx and O-dianisidine) confirms the specificity and efficiency of the two MO-mediated usb1-knockdowns. Moreover, the developmental defects observed in SMO-A morphants, as the reduced body size and the paucity of neutrophils, could be almost completely rescued by co-injection with human USB1 mRNA. All together these evidences indicate that the overall morphological and neutrophil abnormalities were caused by loss of usb1 and support the specific effect of SMO-A and SMO-B.
The overall phenotype of the morphants co-injected with p53-MO and SMO-A resembled that of usb1 morphants, suggesting that usb1-knockdown effects have not been caused by aspecific p53-activation. This result is in line with similar findings obtained with the double sbds- and p53- knockdown morphants29.
Finally we want to comment our data taking into account the work of Patil et al.24 who exploited as we did the morpholino knockdown technology for successful modelling Poikiloderma with Neutropenia in zebrafish. Despite differing in the main focus and the prioritised experimental tools, a few merging issues are highlighted by both studies, in particular the impact of usb1 deficit on the neutrophil commitment and differentiation.
Indeed, while Patil and coworkers mainly explored the neutrophil specific markers, also employing the most markedly affected ela3l (elastase) marker for the rescue experiments, linking the significantly reduced number of neutrophils to incomplete splicing of the myeloid genes, we could monitor, by real-time PCR, the effect of defective usb1 at the level of the myeloid progenitor through the alteration of pu.1 expression. Our work also provides evidence on the multiple severe morphological abnormalities characteristic of the human syndrome which are associated with loss-of-function of the constitutively expressed usb1 gene. These include pigmentation and osteochondral defects which are fairly recapitulated by SMO-A and SMO-B morphants and also by embryos co-injected with subphenotypic dosages of both morpholinos. Overall, the Japanese work and our study are well complementary and demonstrate the appropriate tool offered by the zebrafish model for further investigation of the pathogenesis of human PN syndrome.
Exploiting the zebrafish as a vertebrate model of PN, with its single highly-conserved usb1 gene, could provide a suitable evolutionary intermediate between yeast and human to carry out further biochemichal studies aimed at clarifying the pathomechanism of PN. Targeted analysis of usb1-deficient zebrafish might lend insights suitable to be translated into therapies for the human disorder, particularly concerning the predisposition to myelodysplasia and acute myeloid leukaemia.
Methods
Zebrafish lines and maintenance
Zebrafish (Danio rerio) embryos were raised and maintained under standard conditions and national guidelines (Italian decree 4th March 2014, n.26). All experimental procedures were approved by IACUC (Institutional Animal Care and Use Committee).
Zebrafish AB strains obtained from the Wilson lab, University College London, London, United Kingdom and the transgenic lines tg(gata1:dsRED;flk1:GFP) kindly provided by the Santoro lab, Molecular biotechnology centre Università di Torino, Torino, Italia31 and tg(mpx:GFP;lyzC:dsRED) kindly provided by dr. Deflorian (IFOM Istituto FIRC di Oncologia Molecolare)32,33.
Embryos were staged according to morphological criteria34 and embryonic ages are expressed in somites (s), hour post fertilization (hpf) and day post fertilization (dpf). Since 24 hpf embryos were cultured in fish water containing 0,01% methylene blue to prevent fungal growth and 0,003% 1-phenyl-2-thiourea (Sigma-Aldrich, Saint Louis, Missouri, USA) to prevent pigmentation in all experiments, except those aimed at evaluating the skin phenotype.
Embryos were washed, dechorionated and anaesthetized, with 0.016% tricaine (Ethyl 3-aminobenzoate methanesulfonate salt; Sigma-Aldrich), before observations and picture acquisitions. Embryos were fixed overnight in 4% paraformaldehyde (Sigma-Aldrich) in PBS at 4 °C, then dehydrated stepwise to methanol and stored at −20 °C.
usb1 identification
The human USB1 amino-acid sequence was used as a query to identify in-silico the zebrafish usb1 gene. NCBI (http://www.ncbi.nlm.nih.gov/BLAST/), ClustalW (http://www.ebi.ac.uk/Tools/clustalw/) and SMART (http://smart.embl-heidelberg.de/) tools were used for basic handling and analyses of the nucleotide and protein sequences.
Analysis of synteny was performed with Genomicus software, version 57.01 (http://www.dyogen.ens.fr/genomicus-69.01/cgi-bin/search.pl).
Expression analysis
Total RNAs were isolated from embryos at different developmental stages and from different adult organs using the “SV Total RNA Isolation System” (Promega, Madison, Wisconsin, USA).
After treatment with DNase I RNase-free (Roche, Basel, Switzerland) to avoid possible genomic contamination, 1 μg of RNA was reverse-transcribed using the “ImProm-II™ Reverse Transcription System” (Promega) and random primers according to manufacturer’s instructions.
According to the sequence information gained from bioinformatic analysis, we amplified a fragment and the full coding sequence of usb1 (primers F-R and F-R2, respectively; Supplementary Table S1) using GoTaq polymerase (Promega) following the manufacturer’s instructions. Specific β-actin primers35 were also used to check cDNA quality and possible genomic contamination. Reaction products were analysed by 1% agarose-gel electrophoresis. Amplicons were sequenced using Big Dye Terminator v.3.1 Cycle Sequencing Kit according to the manufacturer’s protocol on the ABI PRISM 3130 sequencer (Applied Biosystems, Foster City, California, USA). Electropherograms were analyzed with ChromasPro software 1.42 (Technelysium Pty Ltd, Tewantin QLD, Australia) using NM_001003460 as reference.
The full coding region of usb1 was cloned into pCR™4-TOPO® TA vector using “TA Topocloning for sequencing” (Invitrogen, Paisley, UK).
Sense and antisense RNA probes were respectively transcribed using T7 and T3 RNA polymerase (Roche) on templates linearized with SpeI or NotI (New England Biolabs Inc, Ipswich, Massachusetts, USA). Probes were labelled with digoxigenin using the “DIG-RNA Labelling Kit” (Roche).
WISH was performed as described36 with BM-Purple (Roche) as substrate. Controls with sense riboprobes were performed in parallel with antisense analyses.
Images of stained embryos were taken on a Leica MZFLIII epifluorescence stereomicroscope equipped with a DFC 480 digital camera and LAS Leica imaging software (Leica, Wetzlar, Germany).
Morpholinos-mediated knockdown, phenotype analysis and rescue experiments
Antisense morpholinos (MOs; GeneTools, Philomath, Oregon, USA) were designed against the acceptor splice site of the usb1 IVS2 (SMO-A, 5′–GGATCATCTGAAATTTAGGCAGGAA-3′) and IVS4 (SMO-B, 5′–CCAAGAAAAGTCCTGCGTCAACAAT-3′). A standard control oligo (Std-MO) with no target in zebrafish embryos, to check for nonspecific effects due to the injection procedure and p53-MO were also used according to previous reports37,38.
All morpholinos were diluted in Danieau solution39 and pressure-injected into 1- to 2-cell-stage embryos using Eppendorf FemtoJet Micromanipulator 5171. Rhodamine dextran (Molecular Probes, Life technology) was usually co-injected as dye tracer. The optimal dose for each MO was selected based on phenotypic effect.
RT-PCR analysis to determine efficacy of SMO splicing inhibition was carried out on RNA isolated from 2 dpf embryos using Go Taq polymerase (Promega) and F2-R3 and F-R2 usb1 specific primers (Supplementary Table S1). Amplicons were sequenced as above mentioned.
Alcian blue and O-dianisidine staining were performed as described40,41.
Probes for pu.142, mpx25, gata143 and clmc244 were synthesized and used for WISH on at least 2 batches of control and morphant embryos at each developmental stage.
Real-time PCR was performed with Fast SYBR Green Master Mix (Applied Biosystems) using the StepOne real-time PCR System (Applied Biosystems). Actin was chosen as endogenous normalising gene and relative expression of target genes (mpx, gata1 and pu.1) was determined using the ΔΔCt method45.
Total RNA was isolated from at least three batches each of 30–40 embryos injected with usb1-SMO-A and Std-MO at the indicated developmental stages (10 s, 22 hpf and 30 hpf) using SV Total RNA Isolation System (Promega). All samples were retro-transcribed in two independent experiments, each RT product was run in triplicate and standard deviation (s.d.) was calculated.
Sequences of primers used for real-time PCR are listed in Supplementary Table 2.
Statistics comparisons between groups of data were evaluated using the t test. A p-value < of 0.05 was considered statistically significant.
Total human RNA was reverse-transcribed into cDNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) with random primers; then the full-length wild-type USB1 was PCR-amplified (primer forward 5′-cggttgaggttgctggtgg-3′; primer reverse 5′-gttcctccatctcagcctg-3′). The amplicon was subcloned into the pCS2+ poly(A) vector and then used as template for generating sense RNA, in vitro synthesized using the “mMessage mMachine kit” (Ambion, Austin, Texas, USA) according to the manufacturer’s instructions. The synthesized mRNAs, diluted in RNase-free water, was injected into one- or two-cell-stage embryos.
Additional Information
How to cite this article: Colombo, E. A. et al. A zebrafish model of Poikiloderma with Neutropenia recapitulates the human syndrome hallmarks and traces back neutropenia to the myeloid progenitor. Sci. Rep. 5, 15814; doi: 10.1038/srep15814 (2015).
References
Clericuzio, C., Hoyme, H. E. & Asse, J. M. Immune deficient poikiloderma: a new genodermatosis. Am. J. Hum. Genet. 49, A661 (1991).
Van Hove, J. L. et al. Clericuzio type poikiloderma with neutropenia is distinct from Rothmund-Thomson syndrome. Am. J. Med. Genet. A 132A, 152–158 (2005).
Mostefai, R. et al. Poikiloderma with neutropenia, Clericuzio type, in a family from Morocco. Am. J. Med. Genet. 146A, 2762–2769 (2008).
Concolino, D. et al. Clericuzio type poikiloderma with neutropenia syndrome in three sibs with mutations in the C16orf57 gene: delineation of the phenotype. Am. J. Med. Genet. A 152A, 2588–2594 (2010).
Colombo, E. A. et al. Novel C16orf57 mutations in patients with poikiloderma with neutropenia: bioinformatic analysis of the protein and predicted effects of all reported mutations. Orphanet J. Rare Dis. 7, 7 (2012).
Volpi, L. et al. Targeted next-generation sequencing appoints C16orf57 as Clericuzio-type poikiloderma with neutropenia gene. Am. J. Hum. Genet. 86, 72–76 (2010).
Tanaka, A. et al. Identification of a homozygous deletion mutation in C16orf57 in a family with Clericuzio-type poikiloderma with neutropenia. Am. J. Med. Genet. A 152A, 1347–1348 (2010).
Arnold, A. W. et al. Poikiloderma with neutropenia: a novel C16orf57 mutation and clinical diagnostic criteria. Br. J. Dermatol. 163, 866–869 (2010).
Clericuzio, C. et al. Identification of a novel C16orf57 mutation in Athabaskan patients with Poikiloderma with Neutropenia. Am. J. Med. Genet. A 155A, 337–342 (2011).
Piard, J. et al. Systematic search for neutropenia should be part of the first screening in patients with poikiloderma. Eur. J. Med. Genet. 55, 8–11 (2012).
Rodgers, W. et al. Squamous cell carcinoma in a child with Clericuzio-type poikiloderma with neutropenia. Br. J. Dermatol. 168, 665–667 (2013).
Walne, A. J., Vulliamy, T., Beswick, R., Kirwan, M. & Dokal, I. Mutations in C16orf57 and normal-length telomeres unify a subset of patients with dyskeratosis congenita, poikiloderma with neutropenia and Rothmund-Thomson syndrome. Hum. Mol. Genet. 19, 4453–4461 (2010).
Larizza, L., Negri, G., Colombo, E. A., Volpi, L. & Sznajer, Y. Clinical utility gene card for: poikiloderma with neutropenia. Eur. J. Hum. Genet. 21, 1185–1189 (2013).
Mazumder, R., Iyer, L. M., Vasudevan, S. & Aravind, L. Detection of novel members, structure-function analysis and evolutionary classification of the 2H phosphoesterase superfamily. Nucleic Acids Res. 30, 5229–5243 (2002).
Sakamoto, Y., Tanaka, N., Ichimiya, T., Kurihara, T. & Nakamura, K. T. Crystal structure of the catalytic fragment of human brain 2’,3’-cyclic-nucleotide 3’-phosphodiesterase. J. Mol. Biol. 346, 789–800 (2005).
Mroczek, S. et al. C16orf57, a gene mutated in poikiloderma with neutropenia, encodes a putative phosphodiesterase responsible for the U6 snRNA 3’ end modification. Genes Dev. 26, 1911–1925 (2012).
Shchepachev, V., Wischnewski, H., Missiaglia, E., Soneson, C. & Azzalin, C. M. Mpn1, mutated in poikiloderma with neutropenia protein 1, is a conserved 3’-to-5’ RNA exonuclease processing U6 small nuclear RNA. Cell Rep. 2, 855–865 (2012).
Hilcenko, C. et al. Aberrant 3’ oligoadenylation of spliceosomal U6 small nuclear RNA in poikiloderma with neutropenia. Blood 121, 1028–1038 (2013).
Zoeller, J. J. et al. A central role for decorin during vertebrate convergent extension. J. Biol. Chem. 284, 11728–11737 (2009).
Yoshida, K. et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69 (2011).
Makishima, H. et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 119, 3203–3210 (2012).
Mason, P. J. & Bessler, M. Poikiloderma with neutropenia: beginning at the end. Blood 121, 872–874 (2013).
Carroll, K. J. & North, T. E. Oceans of opportunity: Exploring vertebrate hematopoiesis in zebrafish. Exp. Hematol. 42, 684–696 (2014).
Patil, P., Uechi, T. & Kenmochi N. Incomplete splicing of neutrophil-specific genes affects neutrophil development in a zebrafish model of poikiloderma with neutropenia. RNA Biology 12, 426–434 (2015).
Lieschke, G. J., Oates, A. C., Crowhurst, M. O., Ward, A. C. & Layton, J. E. Morphologic and functional characterization of granulocytes and macrophages in embryonic and adult zebrafish. Blood 98, 3087–3096 (2001).
Bertrand, J. Y. et al. Definitive hematopoiesis initiates through a committed erythromyeloid progenitor in the zebrafish embryo. Development 134, 4147–4156 (2007).
Pianigiani, E., De Aloe, G., Andreassi, A., Rubegni, P. & Fimiani, M. Rothmund-Thomson syndrome (Thomson-type) and myelodysplasia. Pediatr. Dermatol. 18, 422–425 (2001).
Farruggia, P. et al. Poikiloderma with neutropenia: a case report and review of the literature. J. Pediatr. Hematol. Oncol. 36, 297–300 (2014).
Provost, E. et al. Ribosomal biogenesis genes play an essential and p53-independent role in zebrafish pancreas development. Development 139, 3232–3241 (2012).
Hsu, S. Y., Cheng, Y. C., Shih, H. Y. & Ouyang, P. Dissection of the role of Pinin in the development of zebrafish posterior pharyngeal cartilages. Histochem. Cell Biol. 138, 127–140 (2012).
Santoro, M. M., Samuel, T., Mitchell, T., Reed, J. C. & Stainier, D. Y. Birc2 (cIap1) regulates endothelial cell integrity and blood vessel homeostasis. Nat. Genet. 39, 1397–1402 (2007).
Renshaw, S. A. et al. A transgenic zebrafish model of neutrophilic inflammation. Blood 108, 3976–3978 (2006).
Hall, C., Flores, M. V., Storm, T., Crosier, K. & Crosier, P. The zebrafish lysozyme C promoter drives myeloid-specific expression in transgenic fish. BMC Dev Biol. 7, 42 (2007).
Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B. & Schilling, T. F. Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310 (1995).
Argenton, F. et al. Ectopic expression and knockdown of a zebrafish sox21 reveal its role as a transcriptional repressor in early development. Mech. Dev. 121, 131–142 (2004).
Wu, H. H., Brennan, C. & Ashworth, R. Ryanodine receptors, a family of intracellular calcium ion channels, are expressed throughout early vertebrate development. BMC Res. Notes 4, 541 (2011).
Pistocchi, A. et al. Cornelia de Lange Syndrome: NIPBL haploinsufficiency downregulates canonical Wnt pathway in zebrafish embryos and patients fibroblasts. Cell Death Dis. 4, e866 (2013).
Ferrari, L. et al. FAS/FASL are dysregulated in chordoma and their loss-of-function impairs zebrafish notochord formation. Oncotarget 5, 5712–5724 (2014).
Nasevicius, A. & Ekker, S. C. Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 26, 216–220 (2000).
Spoorendonk, K. M. et al. Retinoic acid and Cyp26b1 are critical regulators of osteogenesis in the axial skeleton. Development 135, 3765–3774 (2008).
Detrich, H. W. 3rd . et al. Intraembryonic hematopoietic cell migration during vertebrate development. Proc. Natl. Acad. Sci. USA. 92, 10713–10717 (1995).
Lieschke, G. J. et al. Zebrafish spi-1 (pu.1) marks a site of myeloid development independent of primitive erythropoiesis: implications for axial patterning. Dev. Biol. 246, 274–295 (2002).
Thisse, C., Thisse, B., Schilling, T. F. & Postlethwait, J. H. Structure of the zebrafish snail1 gene and its expression in wild-type, spadetail and no tail mutant embryos. Development 119, 1203–1215 (1993).
Yelon, D., Horne, S. A. & Stainier, D. Y. R. Restricted expression of cardiac myosin genes reveals regulated aspects of heart tube assembly in zebrafish. Dev. Biol. 214, 23–37 (1999).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-Delta Delta CT Method. Methods 25, 402–408 (2001).
Acknowledgements
This work was supported by Dote di Ricerca FSE, Regione Lombardia funding, to EAC [ID project: 16304, 2011–2012] and Progetto Cariplo N.O.B.E.L. (Biological and molecular characterization of cancer stem cells, Rif.2005.201610.9251) to F.C. and to L.L. We thank dr. Ludovica Volpi (Dipartimento di Biotecnologie Mediche e Medicina Traslazionale, Università degli Studi di Milano), for helpful discussion.
Author information
Authors and Affiliations
Contributions
Study concept and design: L.L., F.C. and E.A.C. Set-up of zebrafish experiments: E.A.C. and E.B. Production, analysis and interpretation of data: E.A.C., S.C., F.C. and L.L. Real-time expression analysis: L.F. and E.A.C. Drafting of the manuscript: L.L., E.A.C., L.F., F.C. and S.C. Critical revision of the manuscript for important intellectual content: L.L., F.C., E.A.C. and S.C.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Colombo, E., Carra, S., Fontana, L. et al. A zebrafish model of Poikiloderma with Neutropenia recapitulates the human syndrome hallmarks and traces back neutropenia to the myeloid progenitor. Sci Rep 5, 15814 (2015). https://doi.org/10.1038/srep15814
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep15814
This article is cited by
-
Telomerase RNA-based aptamers restore defective myelopoiesis in congenital neutropenic syndromes
Nature Communications (2023)
-
Poikiloderma with Neutropenia, Clericuzio-Type Accompanied by Loss of Digits Due to Severe Osteomyelitis
Journal of Clinical Immunology (2020)
-
Genetics of Osteopetrosis
Current Osteoporosis Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
