
Abstract
Metacaspases (MCAs) are distant orthologues of caspases and have been proposed to play a role in programmed cell death in yeast and plants, but little is known about their function in parasitic protozoa. The MCA gene of Leishmania major (LmjMCA) is expressed in actively replicating amastigotes and procyclic promastigotes, but at a lower level in metacyclic promastigotes. LmjMCA has a punctate distribution throughout the cell in interphase cells, but becomes concentrated in the kinetoplast (mitochondrial DNA) at the time of the organelle's segregation. LmjMCA also translocates to the nucleus during mitosis, where it associates with the mitotic spindle. Overexpression of LmjMCA in promastigotes leads to a severe growth retardation and changes in ploidy, due to defects in kinetoplast segregation and nuclear division and an impairment of cytokinesis. LmjMCA null mutants could not be generated and following genetic manipulation to express LmjMCA from an episome, the only mutants that were viable were those expressing LmjMCA at physiological levels. Together these data suggest that in L. major active LmjMCA is essential for the correct segregation of the nucleus and kinetoplast, functions that could be independent of programmed cell death, and that the amount of LmjMCA is crucial. The absence of MCAs from mammals makes the enzyme a potential drug target against protozoan parasites.
Similar content being viewed by others

Main
Metacaspases (MCAs) are evolutionary distant orthologues of metazoan caspases and appear to be restricted to plants, fungi and protozoa.1 They are cysteine peptidases of Clan CD, family C14, with a conserved catalytic cysteine and histidine dyad considered essential for enzyme activity. MCAs have little sequence identity with caspases, but secondary structure predictions indicate that MCAs possess folds similar to caspase-3 and caspase-1.1 However, MCAs differ from caspases in activity and apparently function. Thus while caspases have an aspartic acid-directed substrate specificity at P1 position, some plant MCAs have been found to possess a strict arginine/lysine substrate specificity,2, 3, 4 and recently an arginine-directed specificity has also been shown for the Leishmania major MCA expressed in yeast.5 A potential role for MCAs in programmed cell death (PCD) has been claimed. The yeast MCA has been reported to play a crucial role in the induction of PCD in response to various stresses, ageing and impairment in some biological functions.3, 6, 7 Evidence has also been presented for a role for MCAs in PCD in plants. The Norway spruce MCA mcII-Pa plays a crucial role in PCD associated with plant embryogenesis8 and the tomato MCA gene is upregulated during pathogen-induced PCD.9 Moreover, heterologous expression of type I and II Arabidopsis thaliana and L. major MCAs in yeast complemented the PCD-inducing function of the yeast MCA.3, 5
Although PCD was initially characterised in multicellular organisms as a genetically programmed process of cell suicide, several forms of cell death have now been described in various single-celled eukaryotes. These include the kinetoplastid protozoa Trypanosoma cruzi, T. brucei and Leishmania.10, 11, 12, 13, 14, 15 One of the proposed functions of a PCD pathway in unicellular organisms is to control the cell population by (i) selecting for the fitter cells within the population, (ii) optimally regulating the cell number to adapt to the environmental constraints and (iii) tightly controlling the cell cycle and cell differentiation. Potentially, PCD could also be activated to maintain clonality within the population.16
The part played by MCAs in a cell death pathway in protozoa is less clear than for plants or yeast. It has recently been suggested that the two T. cruzi MCAs might be involved in human serum-induced PCD.15 In contrast, prostaglandin D2-induced cell death, the only form of PCD characterised so far in T. brucei bloodstream forms,14 still occurred in mutants deficient in three of the five MCAs present in the organism.17 These three T. brucei MCAs colocalised with RAB11, yet the proteins appeared to have a role independent of the known recycling functions of RAB11-positive endosomes.17 The finding that RNAi of the three MCAs together resulted in a rapid arrest of growth with a delay in kinetoplast segregation and a cytokinesis block17 suggested that the enzymes could play some role during key steps of the cell cycle.
The Leishmania parasite possesses a diphasic life cycle alternating between flagellated extracellular promastigotes, residing inside the gut of the sandfly, and aflagellated amastigotes, present within mammalian mononuclear phagocytes. The procyclic promastigote form multiplies and differentiates into the metacyclic promastigotes, which is the form best able to establish an infection in a mammalian host. It has been reported that only a small percentage of promastigotes differentiate into the metacyclic form and it has been proposed that the remaining procyclic forms will undergo PCD to limit the utilisation of the restricted supplies of nutrients in the insect gut and in that way allow the survival of the form more able to transmit the disease. Furthermore, in contrast to necrosis, cells dying by apoptosis-like processes are not lysed but are eliminated by phagocytosis, preventing the development of an inflammatory response. Thus death of noninfective promastigotes once inside the mammalian host by an apoptotis-like process could ensure avoidance of aspects of the immune repertoire and thus play a major role in the initial establishment of the infection18 and progression to the chronic phase of the disease.19
Although there is evidence that PCD of Leishmania occurs, very little has been elucidated about the pathways and the proteins involved. Some features similar to those of mammalian caspase-dependent apoptosis have been observed. Nuclear chromatin fragmentation, internucleosome-like DNA fragmentation, phosphatidylserine exposure on the outer leaflet of the plasma membrane, mitochondrial permeabilisation and loss of transmembrane potential occurred upon various types of stimuli.10, 11, 16, 19, 20, 21 Nevertheless, genome database comparisons revealed that a vast majority of the proteins (especially from the Bcl-2/Bax pathway) involved in mammalian apoptosis are apparently not encoded in the genome of Leishmania or related protozoa.22
Thus this study on the L. major metacaspase (LmjMCA) was undertaken to provide more definitive data on the importance of the enzyme for these parasites and the role that they play in regulating cell proliferation. The results show that LmjMCA is essential for cell cycle progression of Leishmania and that tight regulation of expression levels for this cysteine peptidase is required for proliferation.
Results
Expression of LmjMCA during the L. major life cycle
L. major metacaspase (LmjMCA, LmjF35.1580) is a syntenic orthologue of T. brucei metacaspase 5 (TbMCA5) and has a predicted N-terminal signal sequence, possesses a proline-, glutamine- and tyrosine-rich C-terminal extension of unknown function, and has several potential N-glycosylation sites. Affinity-purified antibodies raised against a peptide comprising the C-terminal 15 amino acids of LmjMCA recognised three major proteins (∼50, ∼40 and ∼30 kDa) in procyclic promastigote extracts (Figure 1a, lane L*). Antibodies previously raised against an 80-mer peptide covering the active site region of TbMCA2 and TbMCA317 gave a similar but not identical pattern, but with a more sensitive detection threshold and less background (Figure 1a, lane L; compare with lane L*). The ∼50 kDa protein detected by both antibodies is close to the predicted size for the full-length LmjMCA (47.2 kDa), the other bands detected are likely to be processing products of the full-length polypeptide. LmjMCA protein was also detected in amastigotes freshly isolated from a mouse lesion, again with some processed forms being apparent (Figure 1b). LmjMCA had a higher level of expression and processing in actively dividing logarithmic phase promastigotes (Figure 1a, lane L) than in stationary phase promastigotes (Figure 1a, lane S; note the lack of ∼40 and ∼30 kDa bands).

LmjMCA expression in L. major. LmjMCA expression in L. major promastigotes (a) and lesion amastigotes (b). Promastigotes in logarithmic phase (L, 5 × 106 cells/ml) and stationary phase (S, 2.2 × 107 cells/ml) of culture were analysed by western blot, as were amastigotes extracted from mouse lesions. The equivalent of 107 (L, S) or 108 (L*, amastigotes) cells were loaded per lane of 12% (w/v) SDS-PAGE gels before electrophoresis and transfer to PVDF membranes. The blots were then incubated with either affinity-purified rabbit anti-TbMCA2/3 or anti-LmjMCA antibodies. L. major cysteine synthase (CS) was used as a loading control
LmjMCA localisation within Leishmania changes during the cell cycle
Different phases of the cell cycle were determined by visualisation of the number and position of the nucleus (N), kinetoplast (K) and flagellum (F), as well as staining of the mitotic spindle with anti-β-tubulin antibodies (positive cells were designated N*M). Cells with one of each of these organelles (1N1K1F) enter S phase and duplicate their DNA content (1N*1K*1F cells, where * denotes duplicated DNA). Initiation of the growth of the second flagellum then begins (1N*1K*2F cells), whereupon segregation of the two daughter kinetoplasts is initiated. Nuclear mitosis starts before kinetoplast segregation reaches completion (1N*M1K*2F cells). Kinetoplast segregation then concludes (1N*M2K2F cells) followed by nuclear segregation (2N2K2F cells), whereupon cytokinesis begins (Figure 2A, Ambit A, Woods K, Coombs GH and Mottram JC, unpublished data).

LmjMCA localisation during the cell cycle. (A) Cell cycle progression in L. major. Schematic representation of the various cellular configurations observed during cell cycle progression of L. major promastigotes. (B, C). Immunofluorescence was performed on wild-type promastigotes. Fixed cells were labelled with anti-TbMCA2/3 antibodies and Alexa Fluor 488 (green)- or Alexa Fluor 548 (red)-conjugated anti-rabbit antibodies. Anti-β-tubulin antibodies were used together with Alexa Fluor 488-conjugated anti-mouse antibodies (green). DAPI staining of the nucleus and kinetoplast is shown in blue. Differential Interference Contrast (DIC) images are shown in the inset panels. The black arrowheads (in Bc, Bd and Ca) indicate the presence of a second growing flagellum. Merged pictures are magnified. The scale bars apply to all pictures and equal 10 μm. (B) Immunolocalisation of LmjMCA in logarithmic phase (a) and stationary phase (b) promastigotes. Examples of fluorescent staining observed for cells in G1 (a and b) and G2/M (c and d) are shown. (C) LmjMCA associated with the mitotic spindle during mitosis. Colocalisation of LmjMCA (red) with β-tubulin (β-tub, green) was observed by immunofluorescence (yellow in merged image)
By analysing LmjMCA subcellular location and cell cycle status in a population of unsynchronised cells with anti-TbMCA2/3 antibody, it was found that LmjMCA associated with different organelles at different stages of the cell cycle (Figure 2B). In interphase cells, LmjMCA had a punctate distribution throughout the cell body; with more puncta detected in logarithmic phase promastigotes (Figure 2Ba) than stationary phase promastigotes (Figure 2Bb). The LmjMCA-associated punctate structures were not labelled by markers of organelles that occur in Leishmania; glycosomes (using antibodies against phosphofrutokinase23) and acidocalcisomes (using antibodies against a vacuolar-type H+-pyrophosphatase24) (Supplementary Figure S1). In 1N*M1K*2F and 1N*M2K*2F cells, LmjMCA was in the nucleus (Figure 2Bc and d; 2Cb). Immunocolocalisation using anti-β-tubulin antibodies (Figure 2C) revealed LmjMCA to be associated with the mitotic spindle within the nucleus.25 Upon the disassembly of the mitotic spindle and the start of cytokinesis, LmjMCA was found to have a random punctate distribution throughout the cytoplasm with no apparent location in the nucleus (data not shown).
L. major MCA associates with microtubules
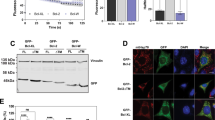
The association of LmjMCA with the mitotic spindle raised the question of whether the protein might also associate with other microtubules in the parasite. Using established methods for preparing cytoskeleton fractions in trypanosomes,26, 27 LmjMCA was found by western blot analysis to cofractionate, in part, with β-tubulin (Figure 3A). RAB11, a protein found on the membrane of endosomes28 but not associated with the cytoskeleton, was absent from the cytoskeleton fraction, demonstrating the effectiveness of the detergent extraction. Interestingly, elongation factor 1 alpha (EF1α) was also detected in the cytoskeleton fraction (Figure 3A). In mammalian cells, EF1α has been shown to stabilise microtubules in a calcium/calmodulin-dependent manner in vitro and thus is recognised as a microtubule-associated protein (MAP).29 EF1α in T. brucei similarly binds to calmodulin in vitro.30 Little is known about the physical properties of Leishmania EF1α, although it has been reported to occur in a Triton-X 100 insoluble fraction.31 These findings now suggest that L. major EF1α may also be a MAP.

Association of LmjMCA with the cytoskeleton. (A) Cofractionation of LmjMCA with the cytoskeleton. Promastigotes were extracted with 0.5% NP40 in PEME buffer. The total (T), soluble (S) and insoluble (I) fractions were separated by SDS-PAGE, transferred to PVDF membrane and immunoblotted with rabbit anti-TbMCA2/3 antibodies. Anti-β-tubulin, anti-TbRAB11 and anti-TbEF1α antibodies were used as controls. (B) Schematic representation of LmjMCA and various GFP fusion proteins. The 47.2 kDa LmjMCA contains a putative N-terminal signal peptide (orange) with a predicted cleavage site between residues 28 and 29 (black arrowhead). The predicted active site histidine (H147) and cysteine (C202) residues are indicated. The C-terminal extension of LmjMCA starts at residue 333 (yellow). A GFP fusion (green) of full-length LmjMCAH147A is 74.3 kDa. An N-terminal region of LmjMCA ending at residue E101 was fused to GFP, yielding a 41.1 kDa protein. The scale bar represents 50 amino acids. (C) LmjMCAH147A-GFP partially associates with the cytoskeleton while Nterm-GFP does so only weakly. Cell lines were analysed as in (A) and were probed with mouse anti-GFP antibodies. Anti-β-tubulin, anti-TbRAB11 and anti-TbEF1α antibodies were used as controls. (D) LmjMCAH147A-GFP localisation is consistent with an association with microtubules. Nuclear and kinetoplast DNA of live GFP-expressing cells was stained with DAPI for 2 min at 25°C. The cells were then washed in ice-cold PBS and observed. While most of the detected signal was cytosolic, punctate signals were observed mainly at the posterior end of the cell (a and b, white arrowhead) but also in the flagellum (a, white arrow) and a tubular structure along the cell body (a, grey arrow). (a) interphase cell (b) cell in G2/M (1N*1K*2F) (the presence of a second growing flagellum is indicated by a black arrowhead on the DIC picture). (E) The N-terminal domain of LmjMCA targets GFP to the mitochondrion. Live WT [N-term-GFP] promastigotes were labelled with either MitoTracker Red CMXRos (a) or MitoTracker Green FM (b) and were observed directly (a) or after immunofluorescence staining with anti-TbMCA2/3 antibodies (b). Images were taken with an exposure time of 100 ms (a) or 300 ms (b). Merged images of the different signals are shown. Areas of colocalisation appear in yellow. (D–E) Scale bars represent 10 μm. (F) Immunolocalisation of LmjMCA in logarithmic phase L. major in G2/M. The black arrowhead indicates the presence of a second growing flagellum. Inset shows intense staining close to the kinetoplast (K). The fixed cell was labelled with anti-TbMCA2/3 antibodies and Alexa Fluor 488 (green)-conjugated anti-rabbit antibodies. DAPI staining of the nucleus and kinetoplast is shown in blue. A DIC image is shown. Merged picture is magnified. The scale bar is 10 μm
While most of the methods for identifying MAPs rely on their ability to bind microtubules in vitro, association with microtubules in vivo can be performed by immunofluorescence analysis.32 Immunofluorescence using anti-MCA antibodies allowed the visualisation of LmjMCA interactions with the microtubules (notably the mitotic spindle, Figure 2A). However, basic proteins have been reported to bind to β-tubulin in a nonspecific manner27 and LmjMCA has a predicted pI of 8.3. To investigate whether the association of LmjMCA with microtubules is physiological, we generated an L. major line that expressed LmjMCA-green fluorescent protein (GFP). The level of expression of LmjMCA-GFP was unfortunately too low for the GFP-fusion to be visualised by direct fluorescence, most likely due to the toxicity of active protein (see below). When expressed in yeast, the L. major MCAH147A mutant, in which the catalytic histidine residue of LmjMCA had been changed to alanine, had significantly reduced enzyme activity.5 We therefore generated an LmjMCAH147A-GFP, which we hypothesised would be overexpressed readily as the protein would be enzymatically compromised. Despite the fact that overexpression of LmjMCAH147A-GFP was toxic, mutant cells would grow – but only slowly and only to low cell density. Nevertheless, LmjMCAH147A-GFP could be visualised fluorescing in WT [MCAH147A-GFP] cells (Figure 3D) and was also detectable by western blot analysis. In the latter, LmjMCAH147A-GFP partly associated, as expected, with the cytoskeleton fraction (Figure 3C). In live cells analysed using fluorescent microscopy, LmjMCAH147A-GFP associated with punctate structures at the tip of the cell (Figure 3Da). These punctate structures were observed moving along the cell body and the flagellum. In some cases, a tubular structure was also labelled (Figure 3Da, grey arrow). This tubular structure was not the multi-vesicular tubule-lysosome (MVT-lysosome) as it did not label with FM4-64 (N-(3-triethylammoniumpropyl)-4-(6-(4-(diethylamino)phenyl)hexatrienyl) pyridinium dibromide). In G2/M (1N*1K*2F) cells, an accumulation of LmjMCAH147A-GFP was observed in punctate structures at the posterior end of the cell (Figure 3Db, arrowheads).
The role of the N-terminal domain as a signal sequence
To investigate if the N-terminal region of LmjMCA predicted in silico to be a signal sequence was indeed involved in targeting LmjMCA to any of the observed subcellular locations, an N-terminal domain-GFP fusion (designated Nterm-GFP), comprising the first 101 amino acids of LmjMCA linked to GFP, was expressed in L. major (Figure 3B). Detergent extractions showed that Nterm-GFP only weakly associated with the cytoskeleton (Figure 3C). Live cell imaging showed that Nterm-GFP was targeted throughout the mitochondrion, as it colocalised with MitoTracker (Figure 3Ea). In contrast, native LmjMCA was not apparently located throughout the matrix of the mitochondrion as it did not colocalise with MitoTracker (Figure 3Eb). Rather, LmjMCA was found to be associated with the kinetoplast in 1N*1K*2F cells (Figure 3F), consistent with an involvement in segregation of this organelle.
Overexpression of L. major MCA leads to aneuploid cells with a severe growth defect
LmjMCA was expressed in L. major promastigotes from an episome (to give a line designated as WT[MCA]). Two types of transfectants expressing LmjMCA were observed; type 1 had similar growth characteristics as wild-type L. major, while type 2 had a severe growth defect (Figure 4A). Western blot analysis revealed that the WT[MCA] type 2 transfectants had a higher level of LmjMCA expression than type 1 transfectants (Figure 4B; one representative set of transfectants shown from five analysed). Fluorescence-activated cell sorting (FACS) analysis of the DNA content of the transfectants revealed that WT[MCA] type 2 transfectants had twice the DNA content of wild type (Figure 4C). 4′,6-Diamidino-2-phenylindole (DAPI) staining of WT[MCA] type 2 cells showed that they had only one nucleus and kinetoplast (a 1N*1K*1F configuration, Figure 4Eb). Comparison of the various cell configurations between WT[MCA] type 2 transfectants and wild-type parasites revealed a reduction in the number of cells in early mitosis (1N*M1K*2F, from 9.2% in wild-type populations to 0.9% in WT[MCA] populations), a moderate reduction of cells with two nuclei, two kinetoplasts and two flagella (2N2K2F, from 4.9 to 1.9%) and a large increase of cells with other configurations (from 3.3 to 20.6%) (Figure 4D, left panel). Consistent with the decrease in the number of cells undergoing mitosis, an accumulation of aberrant cells with one or several mitotic nuclei (as indicated by the presence of a mitotic spindle, Figure 4Ed) and multiple kinetoplasts and flagella were observed, indicating that mitosis was disrupted in the WT[MCA] type 2 transfectants (‘impaired mitosis’, Figure 4D, right panel). Additionally, most of the 2N2K2F cells had an aberrant positioning of the nuclei and kinetoplasts (Figure 4Ec) and only very few were found to be undergoing cytokinesis, suggesting a severe defect that caused de-regulation of cell cycle control.

Overexpression of LmjMCA is detrimental to promastigote growth. (A) Growth of wild type (WT, •), WT[MCA] type 1 (▴) and WT[MCA] type 2 promastigotes (♦). Cells were grown in 15 μg/ml G418. (B) Western blot analysis of WT, WT[MCA] type 1 (lane 1) and WT[MCA] type 2 (lane 2) promastigotes with anti-TbMCA2/3 antibodies. Antiserum against L. mexicana transketolase (anti-TK) was used as a loading control. (C) FACS analysis of WT, WT[MCA] type 1 and WT[MCA] type 2 promastigotes. The peaks at 200 and 400 arbitrary fluorescence units (AFU) correspond to cells with a diploid (2C) and tetraploid (4C) DNA content, respectively. The peak at 800 AFU corresponds to octaploid cells (8C). (D) Classification of the various cell configurations observed in the wild-type (yellow bars) and WT[MCA] type 2 (red bars) populations of promastigotes based on DAPI staining and cell morphology. Subgroups of the cells classified in ‘others’ are shown on the right panel. N: nucleus; K: kinetoplast; F: flagellum; N*M nucleus in mitosis. (E) Immunofluorescence performed on L. major WT (a) and WT[MCA] type 2 cells (b–f). Fixed cells were labelled with anti-TbMCA2/3 antibodies and Alexa Fluor 488-conjugated anti-rabbit antibodies (green). DAPI staining of the nuclear and kinetoplast DNA is shown in blue. ms, mitotic spindle. White arrowheads indicate the kinetoplast
Evidence that LmjMCA is essential for L. major
Attempts to generate an L. major MCA null mutant were unsuccessful and it was not possible to remove even a single allele of LmjMCA from wild-type parasites when well-established methods for genetic manipulation of wild-type L. major and a variety of drug selectable markers were applied. A L. major mutant cell line was generated, however, when LmjMCA was fused to a C-terminal TAP tag and expressed from an episome that was originally targeted to the β-tubulin locus. Using this mutant expressing LmjMCA-TAP, the two LmjMCA alleles were successfully deleted by successive rounds of LmjMCA gene deletion with blasticidin-resistance gene (BSD) and phleomycin-resistance gene (BLE) selectable markers (data not shown). LmjMCA was integrated into the 18S rRNA locus of wild-type L. major. Integration was confirmed using polymerase chain reaction (PCR) (Figure 5A). A single LmjMCA allele could be removed successfully from this cell line (Figure 5Ac, d, e and f), but it was not possible to remove both LmjMCA alleles without the duplication of the LmjMCA gene (Figure 5A, g and h). DNA content analyses showed that attempts to delete the second allele of the chromosomal LmjMCA resulted in the duplication of the cellular DNA content (Figure 5B, line 4). These cells also exhibited a slower growth rate than wild-type parasites (Figure 5C, compare 4 to 1), and had similar defects in cell cycle progression as observed with WT[MCA] type 2 transfectants.

Evidence that LmjMCA is essential. (A) Schematic representation of the wild-type LmjMCA locus and the plasmid constructs used to integrate LmjMCA into the 18S rRNA and to delete the wild-type LmjMCA. ORFs are shown as arrows, intergenic and flanking DNA sequences are shown as boxes. BLA, blasticidin-resistance gene; HYG, hygromycin-resistance gene. The pairs of oligonucleotides used for PCR amplifications and their predicted sizes are indicated. The PCR reactions were performed to assess the integration into the 18S rRNA locus (a, b), the integration of the BSD-resistance cassette (c, d) and the integration of the HYG-resistance cassette (e, f) in the wild-type LmjMCA locus, and the integrity of the wild-type LmjMCA locus (g, h). Integration at the 5′ flank (a, c, e, g) and the 3′flank (b, d, f, h) of the specific loci were tested. Lane 1: wild-type L. major; Lane 2: PrRNA MCA; Lane 3: PrRNA MCA Δmca∷BSD/MCA; Lane 4: PrRNA MCA Δmca∷BSD/Δmca∷HYG/MCA; (B) FACS analysis of the DNA content of the various cell lines generated. (C) Growth of promastigotes in vitro. Wild type (1), PrRNA MCA Δmca∷BSD/Δmca∷HYG/MCA (4)
Discussion
The various gene knockout experiments attempted in this study suggest that the MCA is essential to L. major, as it was not possible to generate a null mutant. Moreover, the finding that we were unable to remove even the first allele of LmjMCA from the L. major genome without expression of an LmjMCA transgene elsewhere in the parasite's genome suggests that the level of expression of LmjMCA is very tightly regulated. Similarly, even though removal of the two chromosomal copies of LmjMCA was achieved successfully in a cell line containing episomal copies of the gene, we found that the parasite modulated the number of MCA-TAP-bearing plasmids within the cell (data not shown). When an extra copy of LmjMCA was integrated in the genome, the first allele of LmjMCA locus could be disrupted, but attempts to replace the second allele led to tetraploidy. This duplication of the genome could be explained by the fact that in this cell line the extra copy of LmjMCA was integrated into the 18S rRNA locus and the regulation of the level of expression of the protein was nonphysiological and ultimately led to an accumulation of LmjMCA in the cell and a phenotype also occurring during LmjMCA overexpression (Figure 4). Overall, these data provide good evidence that LmjMCA is essential for cell survival.
The detection of LmjMCA in different cellular compartments at different stages of the cell cycle and in association with the dividing kinetoplast and the nuclear spindle suggests that at least one essential role of the Leishmania protein is in DNA segregation. LmjMCA is expressed at a higher level in actively dividing logarithmic phase cells than in quiescent stationary phase cells, which would be consistent with a role for the protein in cell division. Other MCAs have also been shown to have a nuclear location, including Saccharomyces cerevisiae YCA1.33 During plant embryogenesis, the Norway spruce MCA (mcII-Pa) translocates from the cytoplasm of the terminally differentiated suspensor cells to their nuclei in order to trigger PCD and thus mediate their removal.8 The MCA5 of T. cruzi also relocates to the nucleus during human serum-induced PCD.15 The main observed location of MCAs in T. brucei was in RAB11-positive endosomes, yet the finding that RNAi knock-down of the proteins resulted in a cell cycle defect, with a delay in kinetoplast segregation, and a cytokinesis defect17 may well be consistent with the findings reported here for Leishmania. The studies with T. brucei did not rule out the presence of MCAs in the nucleus. Thus a nuclear location for MCAs appears to be widespread, although current evidence for the roles of MCAs differs with organism.
LmjMCA has different domains that could be relevant to both its trafficking within the cell and also its function. A GFP-fusion protein with the first 101 amino acid of the LmjMCA (designated Nterm-GFP) localised to the mitochondrion (Figure 3Ea), suggesting that the N-terminal domain is a targeting signal sequence for this organelle. The Nterm-GFP fusion protein was detected distributed throughout the matrix of the mitochondrion, while native LmjMCA could only be found directly associated with the dividing kinetoplast within the organelle. This may simply reflect the higher level of expression of the GFP-fusion protein, with the resultant ease of detection, but perhaps also a change in distribution (as previously observed with the overexpression of a GFP-fused DNA polymerase in T. brucei34). The mitochondrial matrix localisation could also have resulted from the GFP-fusion protein lacking other domains present in the native LmjMCA, domains that could be required for interaction with proteins associated with the kinetoplast. Notably, LmjMCA possesses a proline-, glutamine- and tyrosine-rich C-terminal extension that could mediate such protein : protein interactions.
Western blot analysis using MCA-specific antibodies on L. major extracts revealed a complex pattern of proteins, both in promastigotes and amastigotes (Figure 1), indicating that LmjMCA might be subject to processing. This would be similar to caspases, which are processed as part of their activation. Comparable processing was observed when LmjMCA was expressed in yeast with a C-terminal tag,5 with the results suggesting that LmjMCA processing occurs close to the N- and C-termini of the protein. The catalytic domain of LmjMCA was more active than the full-length protein when expressed and purified from yeast suggesting processing is required for optimal activity.5 The data presented here do not allow judgement on whether this processing is required for activation of the cysteine peptidase activity of LmjMCA in Leishmania or whether the processing is required to direct the LmjMCA to different compartments of the cell at different phases of the cell cycle. Indeed we have recently showed that recombinant T. brucei MCA2 does not require processing for cysteine peptidase activity (CX Moss, GH Coombs and JC Mottram, manuscript in preparation).
We were able to demonstrate that LmjMCA associates with cytoskeletal components extracted with detergent (Figure 3A). Overexpression of LmjMCA led to a rapid growth arrest, and defects in kinetoplast segregation, mitosis and cytokinesis. In T. brucei, kinetoplast segregation and cytokinesis are mediated by subpellicular microtubules.35 T. brucei procyclic forms overexpressing the subpellicular proteins CAP15 and CAP17 duplicate but fail to segregate their kinetoplast and lose their kinetoplast duplication/cytokinesis coordination.36 By analogy, an association of LmjMCA with filaments of the cytoskeleton could be the explanation for the severe impairment of cell division when LmjMCA is overexpressed.
L. major MCA has been shown to complement the PCD-induction function in the yeast MCA mutant,5 yet no role for LmjMCA in L. major PCD has been identified. Moreover, evidence for an involvement of MCAs of T. brucei in PCD is lacking.17 It is now becoming clear that caspases do not only function in PCD, but they also have functions in cell proliferation and differentiation.37 Our finding that LmjMCA plays an essential role in the Leishmania cell cycle could thus be somewhat analogous to the roles of some caspases.37, 38, 39 In mammals, cell proliferation and cell death are essential yet opposing cellular processes. Although the occurrence of PCD in parasitic protozoa remains controversial, it is interesting to postulate that MCAs in these cells might mediate crosstalk between the cell cycle and PCD and so promote a balance between proliferation and death. Irrespective of the precise function of LmjMCA in Leishmania, as LmjMCA is expressed in both life stages of the parasite, it is crucial for cell cycle progression and has no mammalian homologue, thus its biochemical characterisation, identification of its natural substrates and, crucially, obtaining specific inhibitors should provide excellent opportunities for developing novel drugs to treat leishmaniaisis.
Materials and Methods
Parasites and transfections
L. major (MHOM/JL/80/Friedlin) promastigotes were grown in HOMEM medium (modified Eagle's medium) with 10% (v/v) heat-inactivated foetal calf serum (FCS) at 25°C. In total, 25 μg of linear or plasmid DNA were used to transfect 4 × 107 logarithmic phase promastigotes in 400 μl of electroporation buffer (20 mM HEPES, pH 7.5, 135 mM NaCl, 5 mM KCl, 1 mM sodium phosphate buffer, pH 7.35, 0.12% glucose) in a 0.2 cm pulse-cuvette at 450 V and 500 μF. After 24 h recovery in 5 ml of HOMEM with 10% (v/v) FCS, the transfectants were cloned by limiting dilution with the appropriate antibiotics: G418 (Life Technologies) from 15–50 μg/ml hygromycin B (Calbiochem) at 50 μg/ml phleomycin (Cayla) at 10 μg/ml and blasticidin S (hydrochloride, Calbiochem) at 10 μg/ml.
Generation of cell lines expressing LmjMCA
For episomal expression, the LmjMCA gene (LmjF35.1580) was amplified by PCR from 100 ng of L. major genomic DNA using the Taq Plus Precision polymerase mixture (Stratagene) with the primer pair OL1399 (XbaI) and OL1189 (BamHI) and cloned into the pTEX vector to generate pGL1128. For episomal expression of green fluorescence protein (GFP) fusion of LmjMCA (MCA-GFP), LmjMCA was amplified with OL1396 (NdeI) and OL1398 (KpnI) and cloned into the NdeI/KpnI-digested pNUS-GFPcN to give pGL985. MCAH147A was created as described previously.5 The cassette containing the H147A mutation was digested by NruI/BstEII and cloned into pGL1128 to give pGL1406. The MCAH147A fragment was then freed from pGL1406 by NruI/MluI digestion and cloned into pGL985 to give pGL1402. For episomal expression of the first 101 N-terminal amino acids of LmjMCA fused to the GFP (Nterm-GFP), the 5′ end of LmjMCA was amplified with OL1396 (NdeI) and OL2006 (NdeI) and cloned in the NdeI-digested pNUS-GFPnN to give, after confirmation of correct orientation, pGL1403. After transfection of wild-type L. major with episomal plasmids, populations of transfectants were obtained under 15 μg/ml G418 selection. For integration, LmjMCA was obtained by PCR with OL1188 (XhoI) and OL1189 (BamHI) and cloned into XhoI/BglII-digested pRIB plasmid, yielding plasmid pGL1399. The integration cassette was finally excised by digestion with PacI and PmeI before transfection of L. major. For the LmjMCA knock-out constructs, 582 and 566 bp DNA fragments from the 5′ and 3′ UTR of LmjMCA were amplified with the primers pairs OL1190 (HindIII)/OL1191 (SalI) and OL1192 (XmaI)/OL1193 (BglII), respectively, and cloned in pGL792 yielding pGL842. The blasticidin-resistance cassette of pGL842 was then replaced, by SpeI/BamHI digestion, by the hygromycin-resistance cassette of pGL720 or by the bleomycin-resistance cassette of pGL726, yielding pGL849 and pGL1033, respectively. The sequences of the primers used for this study are given in Table 1.
Antisera and immunoblotting
A peptide corresponding to the C-terminal 15 amino acids of the predicted LmjMCA sequence (QPPPAQYTFSPLPPG) was synthesised (Seqlon) and used to immunise a rabbit (Seqlon). Synthetic peptide (1 mg) was then immobilised on an AminoLink Coupling Gel (Pierce) and used to affinity purify the antibodies. The affinity-purified rabbit anti-LmjMCA antibodies were used at 1 : 50 dilution. Affinity-purified rabbit anti-TbMCA2/3 antibodies were previously produced in the laboratory17 and were found to optimally recognise LmjMCA at a 1 : 10 000 dilution. L. major cysteine synthase-specific rabbit antiserum (Williams RAM and Coombs GH, unpublished data) was used at 1 : 1000.
Cells were lysed in 2% SDS with a cocktail of peptidase inhibitors (0.1 mg/ml leupeptin, 1 mM phenantroline, 0.5 mg/ml Pefabloc SC, 5 μg/ml Pepstatin A and 0.1 mg/ml PMSF). After BCA protein quantification (Pierce), Laemmli buffer was added. Whole-cell lysates were then resolved with a 12% (w/v) SDS-PAGE, transferred to a polyvinylidene fluoride (PVDF) membrane using a semidry blotter (Biorad) and immunoblotted with the appropriate primary antibodies diluted in PBS with 5% (w/v) skimmed milk and 0.05% Tween-20. Anti-rabbit or anti-mouse HRP-conjugated secondary antibodies and the SuperSignal West Pico chemoluminescent system (Pierce) were then used to visualise the antigens.
L. major cytoskeleton preparations
The protocol was based on methods previously used for the extraction of the T. brucei cytoskeleton.26, 27 Promastigotes were lysed in 0.5% Nodinet P40 in PEME buffer (100 mM Pipes, pH 6.9, 2 mM EGTA, 1 mM MgSO4, 0.1 mM EDTA40) on ice for 5 min to give a total extract (T). The sample was centrifuged at 250 × g for 30 min at 4°C. The soluble fraction (S) was collected and the insoluble pellet was washed twice in PEME before being finally resuspended in 0.5% NP40-PEME (to give I, the insoluble fraction). The T, S and I fractions were separated by SDS-PAGE in a 12% (w/v) acrylamide gel. The quantity of protein within the T, S or P fraction groups was normalised after BCA protein quantification (Pierce) and Coomassie Blue staining. The samples were then processed for immunoblotting.
Immunofluorescence
For immunolocalisation of LmjMCA and colocalisation with β-tubulin, promastigotes were washed in PBS and fixed in 200 μl of 1% (v/v) formaldehyde in PBS (0.15 M NaCl, 5 mM potassium phosphate, pH 7.4) for 30 min at room temperature. The cells were then permeabilised by addition of 0.1% Triton-X 100/PBS for 10 min and the free aldehyde groups were neutralised with 0.1 M glycine/PBS for 10 min at room temperature. The treated cells were then settled on poly-lysine-treated slides for 15 min. Affinity-purified rabbit anti-TbMCA2/3 and monoclonal anti-β-tubulin (KMX, kind gift from Keith Gull) antibodies diluted in 0.1% (v/v) Triton-X 100, 0.1% (w/v) bovine serum albumin (BSA), PBS (TB) at 1 : 1000 were then applied, alone or at the same time, to the slides for 2 h at room temperature. After a 10 ml wash with PBS, anti-rabbit Alexa Fluor 488- and anti-mouse IgG2b Alexa Fluor 548-conjugated antibodies (green and red, respectively, Molecular Probes), at 1 : 1000 in TB, were added to the appropriate slides, for 1 h, in the dark and at room temperature. The cellular DNA was then stained with 0.5 μg/ml of DAPI (Sigma) for 1 min before proceeding to a final 10 ml PBS wash. A mounting solution (2.5% 1,4-diazabicyclo[2.2.2]octane (DABCO) in 50% glycerol) was then applied to the slide. Cells were viewed with a Zeiss UV microscope, and images were captured by an Orca-ER camera (Hammamatsu) and Openlab software version beta. Images were prepared for presentation using Adobe Photoshop CS. Pictures were taken with an exposure time of 400 ms unless stated otherwise.
For colocalisation of Nterm-GFP with the MitoTracker in live cells, 107 promastigotes were labelled with 1 nM of MitoTracker Red CMXRos (Molecular probes, M7512, in dimethyl sulphoxide (DMSO)) for 2 min at 25°C. Simultaneously, DNA was stained by addition of 1 μg/ml of DAPI. The cells were then centrifuged at 1000 × g for 30 s, washed twice with ice-cold PBS and resuspended in 20 μl of PBS before observation under a UV light microscope. For the immunolocalisation of LmjMCA with MitoTracker, 107 promastigotes were labelled with 1 μM of MitoTracker Green FM (Molecular probes, M7514, in DMSO) for 5 min at 25°C. After two washes in ice-cold PBS, the stained cells were processed as for immunofluorescence staining.
FACS
107 mid-log phase promastigotes were washed with PBS and fixed in 1 ml of 70% (v/v) methanol/PBS for at least an hour at 4°C. The fixed cells were then washed once with PBS and resuspended in 1 ml of PBS supplemented with 10 μg/ml propidium iodide and 10 μg/ml RNAse A in PBS before incubation at 37°C for 45 min. FACS analysis was performed with a Becton Dickinson FACSCalibur using the FL2-A (detecting fluorescence emission between 585 and 642 nm), the forward scatter (FSC, relative cell size) and the side scatter detectors (SSC, cell granulometry or internal complexity). Sample loading was automated with a FACS Loader (Worklist manager and Loader manager softwares). For each sample, 10 000 cells were analysed. Data were interpreted using the CellQuestPro software. Histogram plots showing the cell count relative to the FL2 fluorescence (in arbitrary fluorescence units) are shown.
Abbreviations
- BLE :
-
phleomycin-resistance gene
- BSA:
-
bovine serum albumin
- BSD :
-
blasticidin-resistance gene
- DABCO:
-
1,4-diazabicyclo[2.2.2]octane
- DAPI:
-
4′,6-diamidino-2-phenylindole
- DMSO:
-
dimethyl sulphoxide
- FM4-64:
-
N-(3-triethylammoniumpropyl)-4-(6-(4-(diethylamino)phenyl)hexatrienyl) pyridinium dibromide
- GFP:
-
green fluorescent protein
- MCA:
-
metacaspase
- MAP:
-
microtubule-associated protein
- MVT-lysosome:
-
multi-vesicular tubule-lysosome
- PCD:
-
programmed cell death
- PCR:
-
polymerase chain reaction
- PVDF:
-
polyvinylidene fluoride
- WT:
-
wild type
References
Uren AG, O’Rourke K, Aravind LA, Pisabarro MT, Seshagiri S, Koonin EV et al. Identification of paracaspases and metacaspases: two ancient families of caspase-like proteins, one of which plays a key role in MALT lymphoma. Mol Cell 2000; 6: 961–967.
Vercammen D, Van De CB, De Jaeger G, Eeckhout D, Casteels P, Vandepoele K et al. Type-II metacaspases Atmc4 and Atmc9 of Arabidopsis thaliana cleave substrates after arginine and lysine. J Biol Chem 2004; 279: 45329–45336.
Watanabe N, Lam E . Two Arabidopsis metacaspases AtMCP1b and AtMCP2b are arginine/lysine-specific cysteine proteases and activate apoptosis-like cell death in yeast. J Biol Chem 2005; 280: 14691–14699.
Vercammen D, Belenghi B, Van De CB, Beunens T, Gavigan JA, De Rycke R et al. Serpin1 of Arabidopsis thaliana is a suicide inhibitor for metacaspase 9. J Mol Biol 2006; 364: 625–636.
Gonzalez IJ, Desponds C, Schaff C, Mottram JC, Fasel N . Leishmania major metacaspase can replace yeast metacaspase in programmed cell death and has arginine-specific cysteine peptidase activity. Int J Parasitol 2007; 37: 161–172.
Buttner S, Eisenberg T, Herker E, Carmona-Gutierrez D, Kroemer G, Madeo F . Why yeast cells can undergo apoptosis: death in times of peace, love, and war. J Cell Biol 2006; 175: 521–525.
Madeo F, Herker E, Maldener C, Wissing S, Lachelt S, Herlan M et al. A caspase-related protease regulates apoptosis in yeast. Mol Cell 2002; 9: 911–917.
Bozhkov PV, Suarez MF, Filonova LH, Daniel G, Zamyatnin Jr AA, Rodriguez-Nieto S et al. Cysteine protease mcII-Pa executes programmed cell death during plant embryogenesis. Proc Natl Acad Sci USA 2005; 102: 14463–14468.
Hoeberichts FA, ten Have A, Woltering EJ . A tomato metacaspase gene is upregulated during programmed cell death in Botrytis cinerea-infected leaves. Planta 2003; 217: 517–522.
Moreira ME, Del Portillo HA, Milder RV, Balanco JM, Barcinski MA . Heat shock induction of apoptosis in promastigotes of the unicellular organism Leishmania (Leishmania) amazonensis. J Cell Physiol 1996; 167: 305–313.
Debrabant A, Lee N, Bertholet S, Duncan R, Nakhasi HL . Programmed cell death in trypanosomatids and other unicellular organisms. Int J Parasitol 2003; 33: 257–267.
Das M, Mukherjee SB, Shaha C . Hydrogen peroxide induces apoptosis-like death in Leishmania donovani promastigotes. J Cell Sci 2001; 114: 2461–2469.
Mukherjee SB, Das M, Sudhandiran G, Shaha C . Increase in cytosolic Ca2+ levels through the activation of non-selective cation channels induced by oxidative stress causes mitochondrial depolarization leading to apoptosis-like death in Leishmania donovani promastigotes. J Biol Chem 2002; 277: 24717–24727.
Figarella K, Rawer M, Uzcategui NL, Kubata BK, Lauber K, Madeo F et al. Prostaglandin D2 induces programmed cell death in Trypanosoma brucei bloodstream form. Cell Death Differ 2005; 12: 335–346.
Kosec G, Alvarez VE, Aguero F, Sanchez D, Dolinar M, Turk B et al. Metacaspases of Trypanosoma cruzi: possible candidates for programmed cell death mediators. Mol Biochem Parasitol 2006; 145: 18–28.
Lee N, Bertholet S, Debrabant A, Muller J, Duncan R, Nakhasi HL . Programmed cell death in the unicellular protozoan parasite Leishmania. Cell Death Differ 2002; 9: 53–64.
Helms MJ, Ambit A, Appleton P, Tetley L, Coombs GH, Mottram JC . Bloodstream form Trypanosoma brucei depend upon multiple metacaspases associated with RAB11-positive endosomes. J Cell Sci 2006; 119: 1105–1117.
van Zandbergen G, Bollinger A, Wenzel A, Kamhawi S, Voll R, Klinger M et al. Leishmania disease development depends on the presence of apoptotic promastigotes in the virulent inoculum. Proc Natl Acad Sci USA 2006; 103: 13837–13842.
Zangger H, Mottram JC, Fasel N . Cell death in Leishmania induced by stress and differentiation: programmed cell death or necrosis? Cell Death Differ 2002; 9: 1126–1139.
Arnoult D, Akarid K, Grodet A, Petit PX, Estaquier J, Ameisen JC . On the evolution of programmed cell death: apoptosis of the unicellular eukaryote Leishmania major involves cysteine proteinase activation and mitochondrion permeabilization. Cell Death Differ 2002; 9: 65–81.
Kulkarni MM, McMaster WR, Kamysz E, Kamysz W, Engman DM, McGwire BS . The major surface-metalloprotease of the parasitic protozoan, Leishmania, protects against antimicrobial peptide-induced apoptotic killing. Mol Microbiol 2006; 62: 1484–1497.
Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M et al. The genome of the kinetoplastid parasite, Leishmania major. Science 2005; 309: 436–442.
Lopez C, Chevalier N, Hannaert V, Rigden DJ, Michels PA, Ramirez JL . Leishmania donovani phosphofructokinase. Gene characterization, biochemical properties and structure-modeling studies. Eur J Biochem 2002; 269: 3978–3989.
Lemercier G, Dutoya S, Luo S, Ruiz FA, Rodrigues CO, Baltz T et al. A vacuolar-type H+-pyrophosphatase governs maintenance of functional acidocalcisomes and growth of the insect and mammalian forms of Trypanosoma brucei. J Biol Chem 2002; 277: 37369–37376.
Ersfeld K, Gull K . Partitioning of large and minichromosomes in Trypanosoma brucei. Science 1997; 276: 611–614.
Broadhead R, Dawe HR, Farr H, Griffiths S, Hart SR, Portman N et al. Flagellar motility is required for the viability of the bloodstream trypanosome. Nature 2006; 440: 224–227.
Robinson D, Beattie P, Sherwin T, Gull K . Microtubules, tubulin, and microtubule-associated proteins of trypanosomes. Methods Enzymol 1991; 196: 285–299.
Jeffries TR, Morgan GW, Field MC . A developmentally regulated rab11 homologue in Trypanosoma brucei is involved in recycling processes. J Cell Sci 2001; 114: 2617–2626.
Moore RC, Durso NA, Cyr RJ . Elongation factor-1α stabilizes microtubules in a calcium/calmodulin-dependent manner. Cell Motil Cytoskeleton 1998; 41: 168–180.
Kaur KJ, Ruben L . Protein translation elongation factor-1α from Trypanosoma brucei binds calmodulin. J Biol Chem 1994; 269: 23045–23050.
Nandan D, Cherkasov A, Sabouti R, Yi T, Reiner NE . Molecular cloning, biochemical and structural analysis of elongation factor-1α from Leishmania donovani: comparison with the mammalian homologue. Biochem Biophys Res Commun 2003; 302: 646–652.
Olmsted JB . Microtubule-associated proteins. Annu Rev Cell Biol 1986; 2: 421–457.
Szallies A, Kubata BK, Duszenko M . A metacaspase of Trypanosoma brucei causes loss of respiration competence and clonal death in the yeast Saccharomyces cerevisiae. FEBS Lett 2002; 517: 144–150.
Klingbeil MM, Motyka SA, Englund PT . Multiple mitochondrial DNA polymerases in Trypanosoma brucei. Mol Cell 2002; 10: 175–186.
Robinson DR, Sherwin T, Ploubidou A, Byard EH, Gull K . Microtubule polarity and dynamics in the control of organelle positioning, segregation, and cytokinesis in the trypanosome cell cycle. J Cell Biol 1995; 128: 1163–1172.
Vedrenne C, Giroud C, Robinson DR, Besteiro S, Bosc C, Bringaud F et al. Two related subpellicular cytoskeleton-associated proteins in Trypanosoma brucei stabilize microtubules. Mol Biol Cell 2002; 13: 1058–1070.
Lamkanfi M, Festjens N, Declercq W, Berghe TV, Vandenabeele P . Caspases in cell survival, proliferation and differentiation. Cell Death Differ 2006; 14: 44–55.
Su H, Bidere N, Zheng L, Cubre A, Sakai K, Dale J et al. Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science 2005; 307: 1465–1468.
Salmena L, Lemmers B, Hakem A, Matysiak-Zablocki E, Murakami K, Au PY et al. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev 2003; 17: 883–895.
Woods A, Baines AJ, Gull K . Evidence for a Mr 88 000 glycoprotein with a transmembrane association to a unique flagellum attachment region in Trypanosoma brucei. J Cell Sci 1989; 93: 501–508.
Acknowledgements
This work was supported by the Medical Research Council.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G Salvesen
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Supplementary information
Rights and permissions
About this article
Cite this article
Ambit, A., Fasel, N., Coombs, G. et al. An essential role for the Leishmania major metacaspase in cell cycle progression. Cell Death Differ 15, 113–122 (2008). https://doi.org/10.1038/sj.cdd.4402232
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402232
Keywords
This article is cited by
-
Requirement of Toxoplasma gondii metacaspases for IMC1 maturation, endodyogeny and virulence in mice
Parasites & Vectors (2021)
-
Toxoplasma gondii metacaspase 2 is an important factor that influences bradyzoite formation in the Pru strain
Parasitology Research (2020)
-
Leishmanicidal effects of amphotericin B in combination with selenium loaded on niosome against Leishmania tropica
Journal of Parasitic Diseases (2019)
-
Evolution of caspase-mediated cell death and differentiation: twins separated at birth
Cell Death & Differentiation (2017)
-
The plant metacaspase AtMC1 in pathogen-triggered programmed cell death and aging: functional linkage with autophagy
Cell Death & Differentiation (2014)
