
Abstract
The epidermal growth factor receptor (EGFR) is a member of the erbB family of tyrosine kinase receptors (RTK). The EGFR is involved in cell proliferation, metastasis and angiogenesis, and is expressed in a large proportion of epithelial tumours. The two main classes of EGFR inhibitors in clinical trials are the RTK inhibitors and the monoclonal antibodies. The clinical development of EGFR inhibitors has introduced new challenges to the design of phase I, II, and III trials. Both classes of agents can be safely administered at doses sufficient to inhibit the EGFR system. Receptor tyrosine kinase inhibitors have been extensively evaluated in non-small-cell lung cancer. In this setting, gefitinib has demonstrated activity in patients who fail initial chemotherapy. Monoclonal antibodies have been developed in combination with cytotoxic chemotherapy in several tumour types, most notably colorectal and head and neck cancer. The preliminary results suggest an increase in response rate and time to progression with the combination of cetuximab and chemotherapy in both disease models. Future issues in the development of EGFR inhibitors include the identification of biologic predictors of response, combination with other targeted agents, and their utilisation in earlier stage malignancies.
Similar content being viewed by others
Main
The epidermal growth factor receptor (EGFR; erbB1) is a member of the tyrosine kinase receptor family, which includes HER2/neu (erbB2), erbB3, and erbB4 (Olayioye et al, 2000; Yarden, 2001). The ErbB receptors are present at the cell surface and share a common structure composed of an extracellular ligand-binding domain, transmembrane segment, and an intracellular tyrosine kinase domain (Yarden, 2001). In normal tissue, the ErbB receptors are activated by a variety of receptor-specific ligands. The ligands specific to the EGFR are epidermal growth factor and transforming growth factor-α (TGF-α) (Yarden, 2001). After ligand binding, the receptors form homo- or heterodimeric complexes activating the tyrosine kinase domain (Olayioye et al, 2000; Yarden, 2001). Subsequently, intracellular proteins involved in signalling pathways are phosphorylated and activated, resulting in modulation of gene transcription (Schlessinger, 2000).
The function of the ErbB receptors is dysregulated in several malignant disorders including among others lung, breast, colorectal, squamous cell cancer of the head and neck (SCCHN), and prostrate cancer (Salomon et al, 1995; Mendelsohn, 2002). Mechanisms involved in the activation of the ErbB receptors include: (1) receptor overexpression (Hirsch et al, 2003, 2) mutant receptors resulting in ligand-independent activation (Hirsch et al, 2003; Moscatello et al, 1995, 3) autocrine activation by overproduction of ligand (Prenzel et al, 1999) or (4) ligand-independent activation through other receptor systems such as the urokinase plasminogen receptor (Liu et al, 2002). Activation of the EGFR is involved in malignant transformation and tumour growth through the inhibition of apoptosis, cellular proliferation, promotion of angiogenesis, and metastasis.
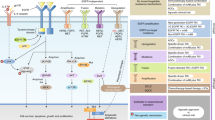
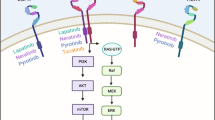
At the cellular level, three major signalling pathways mediate the downstream effects of EGFR activation (Figure 1). The first pathway involves the Ras-Raf-MAP kinase pathway (Lewis et al, 1998). The second pathway involves phosphatidylinositol 3-kinase (PI-3 K) and Akt (Chan et al, 1999; Vivanco and Sawyers, 2002). The third pathway involves the stress-activated protein kinase pathway, involving Jak/Stat and protein kinase C (Sato et al, 1983; Boudny and Kovarik, 2002).

The EGFR signalling pathways. After ligand activation, the EGFR phosphorylates and activates the Ras-Raf-MAP kinase, PI-3K/Akt, and Stat/Jak pathways. This in turn results in activation of transcription factors and modulation of the cell cycle, growth, apoptosis, and angiogenic processes.
Strategies targeting the EGFR pathway
Four strategies for targeting the EGFR are at different stages of development. These include: (1) monoclonal antibodies against the EGFR (Sato et al, 1983, 2) inhibition of the receptor tyrosine kinase (RTK) domain (Lichtner et al, 2001, 3) inhibition of receptor trafficking to the cell membrane (Yamazaki et al, 1998), and (4) inhibition of EGFR synthesis through antisense oligonucleotides (Ciardiello et al, 2001b). Only the monoclonal antibody and RTK inhibitor class of agents have been evaluated through phase III trials.
Monoclonal antibodies
Monoclonal antibodies bind to the extracellular domain of the EGFR and inhibit ligand binding to the receptor (Sato et al, 1983). After binding to the EGFR, the monoclonal antibodies induce receptor dimerisation and downregulation. Cetuximab (IMC-C225, Erbitux ImClone Systems Inc, New York, NY, USA), ABX-EGF (Abgenics, San Francisco, CA, USA), and EMD 72000 are monoclonal antibodies directed against the EGFR that are currently in clinical trials. Another class of monoclonal antibodies consists of bispecific antibodies that can bind the EGFR and an immunologic effector cell (Negri et al, 1995; Tosi et al, 1995; Curnow, 1997). Examples of this class of agents include M26.1, MDX-447, and H22-EGF. These agents have shown promising activity in early clinical trials (Negri et al, 1995; Tosi et al, 1995; Curnow, 1997).
Receptor tyrosine kinase inhibitors
Receptor tyrosine kinase inhibitors compete with ATP for the intracellular catalytic site of the EGFR. In contrast to the monoclonal antibodies, this class of agents does not downregulate the EGFR. Receptor tyrosine kinase inhibitors differ with respect to reversibility of inhibition and specificity to the EGFR vs the other ErbB receptors. Based on these differences, four different classes of RTK inhibitors can be identified and these include: (1) reversible EGFR inhibitors (e.g. gefitinib, erlonitib), (2) irreversible EGFR inhibitors (e.g. EKB-569), (3) reversible dual-ErbB inhibitors (e.g. GW2016), and (4) irreversible pan-ErbB inhibitors (e.g. CI-1033) (Mendelsohn and Baselga, 2003).
Comparison of the monoclonal antibody and RTK compounds
Both these classes of agents result in downregulation of the MAPK, PI3K/Akt, and Jak/Stat signal transduction pathways (Bruns et al, 2000; Albanell et al, 2001). Monoclonal antibodies also downregulate EGFR expression, while RTKs inhibit receptor phosphorylation without affecting expression. At the cellular level, EGFR inhibitors result in cell cycle arrest at the G1 phase (Wu et al, 1995; Busse et al, 2000), decrease tumour neovascularisation by downregulating expression of angiogenic mediators such as vascular endothelial growth factor (VEGF) (Perrotte et al, 1999; Ciardiello et al, 2001a), and promote apoptosis (Moyer et al, 1997; Liu et al, 2000). While monoclonal antibodies require an intact EGFR ligand-binding domain to be active, the RTK inhibitors are active against mutated forms of the EGFR.
At the clinical level, several differences between RTK inhibitors and monoclonal antibodies exist. The RTK compounds are orally administered while the monoclonal antibodies require intravenous administration. While both classes of agents are associated with acenform rash (Baselga et al, 2000; 2002), only RTK inhibitors have been associated with gastrointestinal toxicity (Baselga et al, 2002; Herbst et al, 2002). The preliminary results of clinical trials also suggest different disease-specific activity for each class of agents. For example, cetuximab (Saltz et al, 2001a; 2002) and EMD 72000 (Tewes et al, 2002) are both active in colorectal cancer, in contrast to erlonitib (Townsley et al, 2002) and gefitinib (Seymour et al, 2002), which have failed to demonstrate activity against this tumour type, but have shown activity against non-small-cell lung cancer (NSCLC).
Results of the clinical trials evaluating the RTK inhibitors
Gefitinib
In the initial phase I clinical trials, patients were treated with escalating doses of gefitinib (50–925 mg day−1) for 14 days of a 28-day cycle (Ranson et al, 2002; Nakagawa et al, 2003). In these trials, the maximal tolerated dose (MTD) was 700 mg day−1. The dose-limiting toxicities were diarrhoea and aceniform rash. Objective responses were observed across all doses starting at the 225 mg day−1 dose, raising the possibility that inhibition of the EGFR may be achieved at doses lower than the MTD. In order to determine the optimal biologic dose for gefitinib, two identical multicentre Phase I pharmacodynamic (PD) trials were performed in patients with five tumour types known to express EGFR (NSCLC, SCCHN, ovarian, colorectal, or prostate cancer) (Baselga et al, 2002; Herbst et al, 2002). Secondary objectives were to determine the pharmacokinetic (PK) profile, to investigate the feasibility and sensitivity of the Functional Assessment of Cancer Therapy (FACT) questionnaire and the seven-item Lung Cancer Subscale (LCS) of FACT in assessing improvements in quality of life and disease-related symptoms, respectively. Dose escalation proceeded until the MTD (800 mg day−1) was determined. Common adverse events were mild dose-related skin toxicity and diarrhoea. Biologically relevant plasma concentrations were maintained at doses ⩾150 mg day−1, and skin biopsies demonstrated EGFR inhibition at the same dose as well as inhibition of the downstream signalling pathways involving MAPK, p27, and keratinocyte proliferation index (Albanell et al, 2002). Both the LCS and FACT questionnaires were found to be feasible and sensitive tools with which to assess improvements in these areas. Patients with NSCLC who had stable disease for ⩾6 months also had improvements or stabilisation in disease-related symptoms (LCS scores), while those patients with disease progression had worsened LCS scores (LoRusso et al, 2003). These trials reported the utility of alternative end points in early clinical trials of novel, targeted, anticancer agents.
Based on the phase I trials, two dose levels were selected for Phase II/III studies: 250 and 500 mg day−1. The former is above the lowest dose shown to produce biologic and antitumour activity, thereby ensuring adequate gefitinib drug exposure. Pharmacokinetics from phase I trials also identified plasma levels greater than the targeted cell line IC90 values (100 ng ml−1) in 100% of patients treated at this dose. The 500 mg dose was the highest dose tolerated by most patients on a chronic daily dosing schedule. It also provided greater exposure than the 250 mg dose.
Two large, dose-randomised, double-blind, parallel-group, multicentre Phase II trials (IDEAL 1 and 2, Iressa™ Dose Evaluation in Advanced Lung cancer) independently evaluated the activity of 250 and 500 mg day−1 gefitinib in a combined total of 425 patients with advanced NSCLC who failed prior chemotherapy (Fukuoka et al, 2003; Kris et al, 2003). In both trials, fewer and less severe side effects were observed using 250 mg day−1 compared with 500 mg day−1, while no differences in efficacy end points (response rate, disease control rate, overall survival, and symptom improvement) were seen between the two doses. Response rates ranged from 9 to 19% and, overall, approximately 40% of patients experienced disease control and symptom improvement. These two trials resulted in the recommendation of the 250 mg dose for use in further clinical trials.
Two randomised trials (INTACT 1 and 2, Iressa NSCLC Trial Assessing Combination Treatment) evaluated the effect of combining gefitinib and chemotherapy as first-line therapy for NSCLC. In the first trial, 1250 patients were randomised to receive gemcitabine and cisplatin with either placebo or gefitinib at either 250 or 500 mg day−1 (Giaccone et al, 2002). In the second trial, 1037 patients were randomised to receive carboplatin and paclitaxel with either placebo, gefitinib 250 or 500 mg day−1 (Herbst et al, 2003). In both trials, no difference in survival, progression-free survival or symptom control was observed between the gefitinib/chemotherapy and the chemotherapy alone groups. One possible interpretation for the lack of synergy between gefitinib and cytotoxic agents is related to the G1 arrest of cells continuously exposed to gefitinib. Human cancer xenograft models comparing pulsatile to continuous administration of gefitinib in combination with paclitaxel demonstrated superior tumour kill with the pulsatile schedule (Solit et al, 2003). Based on these preclinical data, trials designed to evaluate pulsatile administration of gefitinib in combination with cytotoxic agents in NSCLC are being conducted.
Cohen et al (2002) reported the results of gefitinib (500 mg day−1) in 52 patients with recurrent SCCHN. Of the 40 response-evaluable patients, eight patients had an objective response and 14 patients had stable disease. Phase II trials of gefitinib in prostate (Moore et al, 2002), breast, colorectal (Seymour et al, 2002), and gastric cancer have been reported or are ongoing. Table 1 summarises the results of these trials.
Erlonitib
Based on the phase I trial, the MTD of erlonitib is 150 mg day−1 (Hidalgo et al, 2001). Erlotinib was evaluated in a phase II trial in 57 patients with non-small-cell lung cancer, who had failed first-line chemotherapy (Perez-Soler et al, 2001). The overall response rate was 12% and the 1-year survival was 40%. Currently, erlonitib vs placebo is being evaluated in a phase III trial in patients with refractory NSCLC and in first-line setting with combination chemotherapy.
Erlonitib has also been evaluated in phase II trials in ovarian (Finkler et al, 2001), SCCHN (Senzer et al, 2001), and hepatocellular carcinoma (Philip, 2004). A summary of the results and design of the above studies is provided in Table 1.
Results of clinical trials evaluating monoclonal antibodies against EGFR
Cetuximab
Phase I trials have established the optimal biologic dose range of cetuximab to be 200–400 mg m−2 (Baselga et al, 2000). At this dose range, cetuximab downregulates EGFR and inhibits downstream signalling. The major toxicity was aceniform rash. Allergic or anaphylactic reactions were observed in 2% of the patients. Cetuximab has been evaluated in colorectal, SCCHN, NSCLC, and pancreatic cancer.
In contrast to the development of RTK inhibitors, early clinical trials with cetuximab have focused on combination therapy with cytotoxic agents. This was based on the nonoverlapping toxicity as well as the experiments in cell culture and human xenograft models demonstrating the potentiation of the effects of cytotoxic agents by cetuximab. Table 2 summarises the results of recent trials involving monoclonal antibodies against the EGFR. In a phase II trial, 120 patients with colorectal cancer who had progressed on irinotecan were treated with cetuximab and irinotecan. The observed response rate was 22.5% (Saltz et al, 2001b). A subsequent phase II trial demonstrated that the response rate to cetuximab in a similar group of patients was 11%, suggesting that cetuximab can modulate the mechanism of irinotecan resistance (Saltz et al, 2002). Cunningham et al (2003) reported on a phase III trial randomising patients with colorectal cancer, who had progressed on irinotecan to cetuximab with or without irinotecan. A total of 329 patients were enrolled. Response rate (cetuximab/irinotecan 23 vs cetuximab 11%, P=0.074) and time to progression (cetuximab/irinotecan 4.1 months vs cetuximab 1.5 months, P<0.001) were significantly improved by the combination. No significant difference in survival was observed. A phase II trial evaluating cetuximab in patients with advanced SCCHN refractory to platinum-based regimens has recently been reported (Baselga et al, 2003). In all, 75 patients were enrolled in the study. The observed response rate was 11%. A phase III trial compared cisplatin and cetuximab to cisplatin and placebo in patients with recurrent SCCHN previously treated with cisplatin (Burtness et al, 2003). A total of 118 patients were enrolled in the study. The response rates were significantly higher in the group of patients on the combination arm (25.7 vs 10.2%, P=0.048). There was no significant difference with respect to median progression-free survival and overall survival between the two arms of the study.
Preliminary results of a randomised trial comparing cisplatin/vinorelbine with or without cetuximab in previously untreated patients with NSCLC have been reported (Gatzemeier et al, 2003). In contrast to the INTACT trial design, only patients with EGFR expressing tumours were enrolled in the study. Of the 73 patients screened, only 65 patients (89%) expressed the EGFR. A total of 56 patients were enrolled in the study. The overall response rate was higher in the cetuximab arm (50 vs 29%). The final results of this study are pending.
A Phase II trial evaluating cetuximab and gemcitabine in advanced chemo-naïve pancreatic cancer was designed (Abbruzzese et al, 2001). In all, 41 patients were treated with weekly cetuximab and gemcitabine. The end points were objective response and time to progression. The overall response rate was 51%, with 12% partial response and 39% stable disease. Time to progression (TTP) was 12 weeks, which is longer than the historical control with gemcitabine (median TTP 8 weeks). The Southwestern Oncology Group (SWOG) is currently comparing gemcitabine with and without cetuximab in pancreatic cancer.
Future directions
Predictors of response
The advantages of defining predictors of response include: preventing the exposure of patients to potentially harmful and/ or ineffective agents, increasing the effectiveness of therapy through selecting a group of patients with a higher likelihood of response, and identifying patient populations that require different therapies. Since response to other targeted agents such as herceptin and tamoxifen depends mainly on the level of expression of the target, several trials have focused on defining a similar association in the EGFR system. Saltz et al (2001a) found no association between EGFR expression by immunohistochemistry in colorectal cancer and response to cetuximab. Similarly, no association was found between response to cetuximab and EGFR expression in SCCHN (Baselga et al, 2003), response to gefitinib in NSCLC (Bailey et al, 2003), and breast cancer (Iacobuzio-Donahue et al, 2003).
The baseline activation of the EGFR and the dependence of the downstream signalling pathways on the EGFR are other potential predictors of response. For example, preclinical models suggest that cells with mutant PTEN phosphatase resulting in EGFR-independent activation of the Akt pathway are resistant to RTK inhibitors (Anido et al, 2003). To define the translational worth of these markers, a prospective trial should be designed to incorporate an evaluation of the EGFR and the downstream signaling pathway status pre and post treatment in order to define the predictors of response to EGFR inhibitors. These trials will require serial tumour biopsies, which raise ethical and financial issues related to subjecting patients to invasive procedures. These trials could also help in defining features present in pre-treatment biopsies that could predict for response. An example of such a trial is the recently reported phase I trial of EMD 72000 in patients with colorectal cancer. In this study, only tumours with low baseline phosphorylated Akt that was inhibited post treatment had a response to EMD 72000. These results suggest that the Akt might play a central role in the antitumour effects of EGFR inhibitors. Another approach to identify predictors of response to EGFR blockade is to utilise gene microarrays. The advantage of this design is that it allows investigators to assay the effects of the EGFR inhibitors on the expression of a large number of proteins. Such trial designs would still require serial tumour biopsies.
Combination therapy involving EGFR inhibitors
As discussed previously, several recent trials have focused on combining EGFR inhibitors with cytotoxic chemotherapy. Other combinations at different stages of development include EGFR inhibitors with other targeted agents, or with radiation therapy. Cancer cells have several dysregulated and redundant pathways; therefore, combining targeted agents may be necessary in order to achieve the desired modulation of a cellular pathway. Combining inhibitors of the EGFR with inhibitors acting on the downstream signalling pathway such as MAPK or Akt could potentially result in an improved inhibition of these pathways translating into increased antitumour effects. These combinations are currently being evaluated in preclinical models. Activation of the EGFR system results in transcription of several proteins such as VEGF and cyclooxygenase-2. Therefore, inhibiting the EGFR can downregulate the expression of these targets, facilitating their inhibition by target-specific agents. The preliminary results of a phase I/II trial evaluating bevacizumab and erlonitib in patients with NSCLC have been recently reported (Mininberg et al, 2003). The preliminary results indicate that both agents can be safely administered at full dose. A phase II trial at Wayne State University is evaluating celecoxib and gefitinib in NSCLC. Since RTK inhibitors and monoclonal antibodies inhibit the EGFR system by different mechanisms, their antitumour effects could potentially be improved by combining them. Similarly, since EGFR and ErbB2 can heterodimerise and both receptors are simultaneously overexpressed in several disease models, combining herceptin with an EGFR inhibitor might be necessary to inhibit both receptors. The results of clinical trials exploring such combinations have not yet been reported. Cetuximab was safely combined with radiation therapy in a phase II trial of SCCHN (Robert et al, 2001). In all, 13 complete and two partial responses were observed in the 16 patients enrolled in the study. Encouraged by these results, a phase III trial of radiation with or without cetuximab is ongoing.
Role of EGFR inhibitors in early-stage disease
Epidermal growth factor receptor inhibitors have demonstrated significant activity in patients with metastatic NSCLC, who have failed cytotoxic chemotherapy. These results raise the possibility of a role for EGFR inhibitors in locally advanced NSCLC. Currently, SWOG is conducting a randomised trial in patients with stage III NSCLC. Patients enrolled in this study will receive definitive chemo-radiotherapy, followed by docetaxel with subsequent randomisation to either gefitinib or placebo. The low incidence of toxicity associated with the EGFR inhibitors has also raised the possibility of a potential role for these agents in the adjuvant setting. SWOG is currently conducting a phase III trial randomising patients with stage I and II NSCLC to either gefitinib or placebo after resection. The results of these trials will help define the role of targeted agents after definitive treatment of early-stage and locally advanced NSCLC.
Conclusion
The EGFR inhibitors have already demonstrated activity in several advanced stage cancers including NSCLC, colorectal, and squamous cell carcinomas of the SCCHN. The role of EGFR inhibitors in early-stage disease is currently being evaluated. The preclinical and clinical development of this class of agents has required novel trial designs that could be incorporated into future trials involving other novel targeted therapies.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Abbruzzese J, Rosenberg A, Xiong Q, LoBuglio A, Schmidt W, Wolff R, Needle M, Waksal H (2001) Phase II study of anti-epidermal growth factor receptor (EGFR) antibody cetuximab (IMC-C225) in combination with gemcitabine in patients with advanced pancreatic cancer. Proc Am Soc Clin Oncol 1: 130a
Albanell J, Codony-Servat J, Rojo F, Del Campo JM, Sauleda S, Anido J, Raspall G, Giralt J, Rosello J, Nicholson RI, Mendelsohn J, Baselga J (2001) Activated extracellular signal-regulated kinases: association with epidermal growth factor receptor/transforming growth factor alpha expression in head and neck squamous carcinoma and inhibition by anti-epidermal growth factor receptor treatments. Cancer Res 61: 6500–6510
Albanell J, Rojo F, Averbuch S, Feyereislova A, Mascaro JM, Herbst R, LoRusso P, Rischin D, Sauleda S, Gee J, Nicholson RI, Baselga J (2002) Pharmacodynamic studies of the epidermal growth factor receptor inhibitor ZD1839 in skin from cancer patients: histopathologic and molecular consequences of receptor inhibition. J Clin Oncol 20: 110–124
Anido J, Matar P, Albanell J, Guzman M, Rojo F, Arribas J, Averbuch S, Baselga J (2003) ZD1839, a specific epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, induces the formation of inactive EGFR/HER2 and EGFR/HER3 heterodimers and prevents heregulin signaling in HER2-overexpressing breast cancer cells. Clin Cancer Res 9: 1274–1283
Bailey LR, Kris M, Wolf MK, Kay AC, Averbuch S, Askaa J, Janas M, Schmidt K, Fukuoka M (2003) Tumour EGFR membrane staining is not clinically relevant for predicting response in patients receiving gefitinib montherapy for pretreated non-small cell lung cancer: IDEAL 1 and 2. AACR-NCI-EORTC International Conference. Boston, MA
Baselga J, Pfister D, Cooper MR, Cohen R, Burtness B, Bos M, D'Andrea G, Seidman A, Norton L, Gunnett K, Falcey J, Anderson V, Waksal H, Mendelsohn J (2000) Phase I studies of anti-epidermal growth factor receptor chimeric antibody C225 alone and in combination with cisplatin. J Clin Oncol 18: 904–914
Baselga J, Rischin D, Ranson M, Calvert H, Raymond E, Kieback DG, Kaye SB, Gianni L, Harris A, Bjork T, Averbuch SD, Feyereislova A, Swaisland H, Rojo F, Albanell J (2002) Phase I safety, pharmacokinetic, and pharmacodynamic trial of ZD1839, a selective oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with five selected solid tumour types. J Clin Oncol 20: 4292–4302
Baselga J, Trigo J, Bourhis J, Tortochaux J, Cortes-Funes H, Hitt R, Gascon P, Muesser M, Harstrick A, Eckardt A (2003) Cetuximab (C225) plus cisplatin/carboplatin is active in patients (pts) with recurrent/metastatic squamous cell carcinoma of the head and neck (SCCHN) progressing on a same dose and schedule platinum-based regimen. Proc Am Soc Clin Oncol 1: 226a
Boudny V, Kovarik J (2002) JAK/STAT signaling pathways and cancer. Janus kinases/signal transducers and activators of transcription. Neoplasma 49: 349–355
Bruns CJ, Solorzano CC, Harbison MT, Ozawa S, Tsan R, Fan D, Abbruzzese J, Traxler P, Buchdunger E, Radinsky R, Fidler IJ (2000) Blockade of the epidermal growth factor receptor signaling by a novel tyrosine kinase inhibitor leads to apoptosis of endothelial cells and therapy of human pancreatic carcinoma. Cancer Res 60: 2926–2935
Burtness B, Li Y, Flood W, Mattar BI, Forastiere A (2003) Phase III trial of cisplatin+placebo versus cisplatin+C225 a monoclonal antibody directed to the epidemal growth factor-receptor: An Eastern Cooperative Group trial. AACR-NCI-EORTC International Conference. Boston, MA
Busse D, Doughty RS, Ramsey TT, Russell WE, Price JO, Flanagan WM, Shawver LK, Arteaga CL (2000) Reversible G(1) arrest induced by inhibition of the epidermal growth factor receptor tyrosine kinase requires up-regulation of p27(KIP1) independent of MAPK activity. J Biol Chem 275: 6987–6995
Chan TO, Rittenhouse SE, Tsichlis PN (1999) AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem 68: 965–1014
Ciardiello F, Caputo R, Bianco R, Damiano V, Fontanini G, Cuccato S, De Placido S, Bianco AR, Tortora G (2001a) Inhibition of growth factor production and angiogenesis in human cancer cells by ZD1839 (Iressa), a selective epidermal growth factor receptor tyrosine kinase inhibitor. Clin Cancer Res 7: 1459–1465
Ciardiello F, Caputo R, Troiani T, Borriello G, Kandimalla ER, Agrawal S, Mendelsohn J, Bianco AR, Tortora G (2001b) Antisense oligonucleotides targeting the epidermal growth factor receptor inhibit proliferation, induce apoptosis, and cooperate with cytotoxic drugs in human cancer cell lines. Int J Cancer 93: 172–178
Cohen A, Rosen F, Dekker A, Bajda C, Stenson K, Shulman K, Lamont K, Kozloff M, Vokes K (2002) Phase II study of ZD1839 (Iressa) in recurrent or metastatic squamous cell carcinoma of the head and neck (SCCHN). Proc Am Soc Clin Oncol 1: 225a
Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg B, Santoro A, Bets D, Mueser M, Harstrick A, Van Cutsem E (2003) Cetuximab (C225) alone or in combination with irinotecan (CPT-11) in patients with epidermal growth factor receptor (EGFR)-positive, irinotecan-refractory metastatic colorectal cancer (MCRC). Proc Am Soc Clin Oncol 1: 252a
Curnow RT (1997) Clinical experience with CD64-directed immunotherapy. An overview. Cancer Immunol Immunother 45: 210–215
Finkler N, Gordon A, Crozier M, Edwards R, Figueroa J, Garcia A, Hainsworth J, Irwin D, Silberman S, Allen L, Ferrante K, Fisher D, Nadler P (2001) Phase 2 evaluation of OSI-774, a potent oral antagonist of the EGFR-TK in patients with advanced ovarian carcinoma. Proc Am Soc Clin Oncol 1: 208a
Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, Nishiwaki Y, Vansteenkiste J, Kudoh S, Rischin D, Eek R, Horai T, Noda K, Takata I, Smit E, Averbuch S, Macleod A, Feyereislova A, Dong RP, Baselga J (2003) Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer. J Clin Oncol 21: 2237–2246
Gatzemeier U, Rosell R, Ramlau R, Robinet G, Szczesna A, Quoix E, Font A, JimEnez E, Mueser M, Harstrick A (2003) Cetuximab in combination with cisplatin/vinorelbine vs cisplatin/ vinorelbine alone in the first line treatment of patients with epidermal growth factor receptor positive advanced non-small cell lung cancer. Proc Am Soc Clin Oncol 1: 642
Giaccone G, Johnson D, Manegold C (2002) A phase III trial of ZD1839 (iressa) in combination with gemcitabine and cisplatin in chemotherapy naive patients with advanced non-small cell lung cancer. Ann Oncol 5: 40
Herbst R, Giaccone G, Schiller J, Miller V, Natale R, Rennie P, Ochs J, Fandi A, Grous J, Johnson D (2003) Subset analyses of INTACT results for gefitinib (ZD1839) when combined with platinum-based chemotherapy (CT) for advanced non-small-cell lung cancer (NSCLC). ASCO Proceedings. Chicago, IL
Herbst RS, Maddox AM, Rothenberg ML, Small EJ, Rubin EH, Baselga J, Rojo F, Hong WK, Swaisland H, Averbuch SD, Ochs J, LoRusso PM (2002) Selective oral epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 is generally well-tolerated and has activity in non-small-cell lung cancer and other solid tumours: results of a phase I trial. J Clin Oncol 20: 3815–3825
Hidalgo M, Siu LL, Nemunaitis J, Rizzo J, Hammond LA, Takimoto C, Eckhardt SG, Tolcher A, Britten CD, Denis L, Ferrante K, Von Hoff DD, Silberman S, Rowinsky EK (2001) Phase I and pharmacologic study of OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in patients with advanced solid malignancies. J Clin Oncol 19: 3267–3279
Hirsch FR, Varella-Garcia M, Bunn Jr PA, Di Maria MV, Veve R, Bremmes RM, Baron AE, Zeng C, Franklin WA (2003) Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis. J Clin Oncol 21: 3798–3807
Iacobuzio-Donahue CA, Albain KS, Ellegde R, Gradishar W, Hayes RL, Rowinsky E, Hudis C, Pusztai L, Tripathy D, Modi S, Rubin S, Hidalgo M (2003) Determination of response to gefitinib in patients with advanced breast cancer. AACR-NCI-EORTC International Conference. Boston, MA
Kris MG, Natale RB, Herbst RS, Lynch Jr TJ, Prager D, Belani CP, Schiller JH, Kelly K, Spiridonidis H, Sandler A, Albain KS, Cella D, Wolf MK, Averbuch SD, Ochs JJ, Kay AC (2003) Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA 290: 2149–2158
Lewis TS, Shapiro PS, Ahn NG (1998) Signal transduction through MAP kinase cascades. Adv Cancer Res 74: 49–139
Lichtner RB, Menrad A, Sommer A, Klar U, Schneider MR (2001) Signaling-inactive epidermal growth factor receptor/ligand complexes in intact carcinoma cells by quinazoline tyrosine kinase inhibitors. Cancer Res 61: 5790–5795
Liu B, Fang M, Schmidt M, Lu Y, Mendelsohn J, Fan Z (2000) Induction of apoptosis and activation of the caspase cascade by anti-EGF receptor monoclonal antibodies in DiFi human colon cancer cells do not involve the c-jun N-terminal kinase activity. Br J Cancer 82: 1991–1999
Liu D, Aguirre Ghiso J, Estrada Y, Ossowski L (2002) EGFR is a transducer of the urokinase receptor initiated signal that is required for in vivo growth of a human carcinoma. Cancer Cell 1: 445–457
LoRusso PM, Herbst RS, Rischin D, Ranson M, Calvert H, Raymond E, Kieback D, Kaye S, Gianni L, Harris A, Bjork T, Maddox AM, Rothenberg ML, Small EJ, Rubin EH, Feyereislova A, Heyes A, Averbuch SD, Ochs J, Baselga J (2003) Improvements in quality of life and disease-related symptoms in phase I trials of the selective oral epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 in non-small cell lung cancer and other solid tumours. Clin Cancer Res 9: 2040–2048
Mendelsohn J (2002) Targeting the epidermal growth factor receptor for cancer therapy. J Clin Oncol 20: 1S–13S
Mendelsohn J, Baselga J (2003) Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J Clin Oncol 21: 2787–2799
Mininberg ED, Herbst RS, Henderson T, Kim E, Hong WK, Mass R, Novotny W, Garcia B, Johnson D, Sandler A (2003) Phase I/II study of the recombinant humanized monoclonal anti-VEGF antibody bevacizumab and the EGFR-TK inhibitor erlotinib in patients with recurrent non-small cell lung cancer (NSCLC). Proc Am Soc Clin Oncol 1: 627
Moore M, Winquist E, Pollack M (2002) A randomized phase II study of two doses of ZD1839 in patients with hormone refractory prostate cancer: A NCI Canada Clinical Trial Group study. Ann Oncol 5: 326
Moscatello DK, Holgado-Madruga M, Godwin AK, Ramirez G, Gunn G, Zoltick PW, Biegel JA, Hayes RL, Wong AJ (1995) Frequent expression of a mutant epidermal growth factor receptor in multiple human tumours. Cancer Res 55: 5536–5539
Moyer JD, Barbacci EG, Iwata KK, Arnold L, Boman B, Cunningham A, DiOrio C, Doty J, Morin MJ, Moyer MP, Neveu M, Pollack VA, Pustilnik LR, Reynolds MM, Sloan D, Theleman A, Miller P (1997) Induction of apoptosis and cell cycle arrest by CP-358,774, an inhibitor of epidermal growth factor receptor tyrosine kinase. Cancer Res 57: 4838–4848
Nakagawa K, Tamura T, Negoro S, Kudoh S, Yamamoto N, Takeda K, Swaisland H, Nakatani I, Hirose M, Dong RP, Fukuoka M (2003) Phase I pharmacokinetic trial of the selective oral epidermal growth factor receptor tyrosine kinase inhibitor gefitinib (‘Iressa’, ZD1839) in Japanese patients with solid malignant tumours. Ann Oncol 14: 922–930
Negri DR, Tosi E, Valota O, Ferrini S, Cambiaggi A, Sforzini S, Silvani A, Ruffini PA, Colnaghi MI, Canevari S (1995) In vitro and in vivo stability and anti-tumour efficacy of an anti-EGFR/anti-CD3 F(ab′)2 bispecific monoclonal antibody. Br J Cancer 72: 928–933
Olayioye MA, Neve RM, Lane HA, Hynes NE (2000) The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J 19: 3159–3167
Perez-Soler R, Chachoua A, Huberman M, Karp D, Rigas J, Hammond L, Rowinsky E, Ferrante K, Allen L, Nadler P, Bonomi P (2001) A phase II trial of the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor OSI-774, following platinum-based chemotherapy, in patients (pts) with advanced, EGFR-expressing, non-small cell lung cancer (NSCLC). Proc Am Soc Clin Oncol 1: 310a
Perrotte P, Matsumoto T, Inoue K, Kuniyasu H, Eve BY, Hicklin DJ, Radinsky R, Dinney CP (1999) Anti-epidermal growth factor receptor antibody C225 inhibits angiogenesis in human transitional cell carcinoma growing orthotopically in nude mice. Clin Cancer Res 5: 257–265
Philip PA, Mahoney M, Thomas J, Pitot H, Donehower R, Kim G, Picus J, Fitch T, Geyer S, Erlichman C (2004) Phase II trial of erlotinib (OSI-774) in patients with hepatocellular or biliary cancer. Proc Am Soc Clin Onc 1: 318
Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A (1999) EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 402: 884–888
Ranson M, Hammond LA, Ferry D, Kris M, Tullo A, Murray PI, Miller V, Averbuch S, Ochs J, Morris C, Feyereislova A, Swaisland H, Rowinsky EK (2002) ZD1839, a selective oral epidermal growth factor receptor-tyrosine kinase inhibitor, is well tolerated and active in patients with solid, malignant tumours: results of a phase I trial. J Clin Oncol 20: 2240–2250
Robert F, Ezekiel MP, Spencer SA, Meredith RF, Bonner JA, Khazaeli MB, Saleh MN, Carey D, LoBuglio AF, Wheeler RH, Cooper MR, Waksal HW (2001) Phase I study of anti-epidermal growth factor receptor antibody cetuximab in combination with radiation therapy in patients with advanced head and neck cancer. J Clin Oncol 19: 3234–3243
Salomon DS, Brandt R, Ciardiello F, Normanno N (1995) Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol 19: 183–232
Saltz L, Meropol N, Loehrer P, Waksal H, Needle M, Mayer R (2002) Single agent IMC-C225 (Erbitux™) has activity in CPT-11-refractory colorectal cancer (CRC) that expresses the epidermal growth factor receptor (EGFR). Proc Am Soc Clin Oncol.
Saltz L, Rubin M, Hochster H (2001a) Cetuximab plus irinotecan is active in CPT-11 refractory colorectal cancer that expresses epidermal growth factor. ASCO proceedings. San Francisco, CA, pp 20
Saltz L, Rubin M, Hochster H (2001b) Cetuximab plus irinotecan is active in CPT-11 refractory colorectal cancer that expresses epidermal growth factor. Proceedings of the American Society of Clinical Oncology. San Francisco, CA, pp 20
Sato JD, Kawamoto T, Le AD, Mendelsohn J, Polikoff J, Sato GH (1983) Biological effects in vitro of monoclonal antibodies to human epidermal growth factor receptors. Mol Biol Med 1: 511–529
Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103: 211–225
Senzer N, Soulieres D, Siu L, Agarwala S, Vokes E, Hidalgo M, Silberman S, Allen L, Ferrante K, Fisher D, Marsolais C, Nadler P (2001) Phase 2 evaluation of OSI-774, a potent oral antagonist of the EGFR-TK in patients with advanced squamous cell carcinoma of the head and neck. Proc Am Soc Clin Oncol 1: 2a
Seymour L, Goss G, Stewart D (2002) A translational research study of ZD1839 at a dose of 750 mg in patients with pretreated advanced or metastatic colorectal cancer: NCIC CTG IND 122. Ann Oncol 5: 264
Solit DB, She Y, Moasser M, Hudis C, Kris M, Scher H, Rosen N, Sirotnak FM (2003) Pulsatile adminstration of the EGFR inhibitor gefitinib is significantly more effective than continous dosing for sensitizing tumours to taxol. AACR-NCI-EORTC International Conference. Boston, MA
Tewes M, Schleucher N, Dirsch O, Schmid K, Rosen O, Arens H, Kovar A, Seeber S, Harstrick A, Vanhoefer U (2002) Results of a phase I trial of the humanized anti epidermal growth factor receptor (EGFR) monoclonal antibody EMD 72000 in patients with EGFR expressing solid tumours. Proc Am Soc Clin Oncol 1: 95a
Tosi E, Valota O, Negri DR, Adobati E, Mazzoni A, Meazza R, Ferrini S, Colnaghi MI, Canevari S (1995) Anti-tumour efficacy of an anti-epidermal-growth-factor-receptor monoclonal antibody and its F(ab′)2 fragment against high- and low-EGFR-expressing carcinomas in nude mice. Int J Cancer 62: 643–650
Townsley C, Jackson P, Montgomery E (2002) Phase II study of OSI-774 in patients with metastatic colorectal cancer. Eur J Cancer 38 (Suppl 7): 179
Vivanco I, Sawyers CL (2002) The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2: 489–501
Wu X, Fan Z, Masui H, Rosen N, Mendelsohn J (1995) Apoptosis induced by an anti-epidermal growth factor receptor monoclonal antibody in a human colorectal carcinoma cell line and its delay by insulin. J Clin Invest 95: 1897–1905
Yamazaki H, Kijima H, Ohnishi Y, Abe Y, Oshika Y, Tsuchida T, Tokunaga T, Tsugu A, Ueyama Y, Tamaoki N, Nakamura M (1998) Inhibition of tumour growth by ribozyme-mediated suppression of aberrant epidermal growth factor receptor gene expression. J Natl Cancer Inst 90: 581–587
Yarden Y (2001) The EGFR family and its ligands in human cancer. Signalling mechanisms and therapeutic opportunities. Eur J Cancer 37 (Suppl 4): S3–S8
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
