
Abstract
2-Methoxyoestradiol (2-MeOE2) is an endogenous oestrogen metabolite that inhibits the proliferation of cancer cells in vitro, and it is also antiangiogenic. In vivo 2-MeOE2, when administered at relatively high doses, inhibits the growth of tumours derived from breast cancer cells, sarcomas and melanomas. Sulphamoylated derivatives of 2-MeOE2 are more potent inhibitors of in vitro breast cancer cell growth than 2-MeOE2. In the present study, we have compared the pharmacokinetic profiles and metabolism of 2-MeOE2 and its sulphamoylated derivative, 2-methoxyoestradiol-bis-sulphamate (2-MeOE2bisMATE), in adult female rats. Their ability to inhibit tumour growth was compared in nude mice bearing xenografts derived from MDA-MB-435 (oestrogen receptor negative) melanoma cancer cells. After a single oral 10 mg kg−1 dose of 2-MeOE2bisMATE, significant concentrations of this compound were still detectable at 24 h. In contrast, no 2-MeOE2 or metabolites were detected in plasma at any time after a 10 mg kg−1 oral dose. Thus, the bioavailability of 2-MeOE2 is very low, whereas for 2-MeOE2bisMATE it was 85%. No significant metabolites of 2-MeOE2bisMATE were detected in plasma after oral or intravenous dosing, showing that this drug is resistant to metabolism. In the tumour efficacy model, oral administration of 2-MeOE2bisMATE, at 20 mg kg−1 day−1 daily for 28 days, almost completely inhibited tumour growth. Inhibition of tumour growth was maintained for a further 28 days after the cessation of dosing. At this dose level, 2-MeOE2 did not inhibit tumour growth. The resistance to metabolism shown by 2-MeOE2bisMATE and its ability to inhibit tumour growth in vivo suggest that this compound should have considerable potential for development as a novel anticancer drug.
Similar content being viewed by others
Main
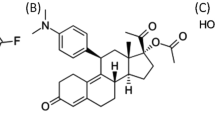
2-Methoxyoestradiol (2-MeOE2, Figure 1, 1), a metabolite of oestradiol, is generated in vivo by catechol-O-methyl transferase, an enzyme which is expressed in a plethora of mammalian tissues including the liver, kidney, brain and red blood cells (Mannisto and Kaakkola, 1999). 2-Methoxyoestradiol has been shown to inhibit the growth of ER+ and ER− breast cancer cell lines and HeLa cells (Seegers et al, 1989; Lottering et al, 1992; Raobaikady et al, 2003). It does not stimulate uterotropic growth, and has a low binding affinity for the oestrogen receptor (Ball and Knuppen, 1980; Merriam et al, 1980), and therefore it must act via an oestrogen receptor-independent mechanism (LaVallee et al, 2002). It also inhibits the in vivo growth of xenografts derived from human MDA-MB-435 melanoma cells, Meth A sarcomas, B16 melanomas and the multiple myeloma cell line KAS-6/1 in immunodeficient mice (Fotsis et al, 1994; Klauber et al, 1997; Dingli et al, 2002). However, comparatively high oral or intraperitoneal doses of 75 and 150 mg kg−1 day−1, respectively, of 2-MeOE2 were necessary to reduce the growth of melanoma or myeloma tumours (Klauber et al, 1997; Dingli et al, 2002). The relatively high doses of 2-MeOE2 required to inhibit tumour growth in vivo in the mouse may be a corollary of poor gastrointestinal absorption and/or rapid metabolic deactivation of the agent, although this remains to be experimentally determined.

Structures: compound 1, 2-MeOE2; compound 2, 2-MeOE2bisMATE; compound 3, 2-MeOE2-3S; compound 4, EMATE; compound 5, 2-MeOE1; compound 6, 2-MeOE2-3MATE; compound 7, 2-MeOE2-17MATE.
In addition to inhibiting the proliferation of cancer cells, 2-MeOE2 also possesses antiangiogenic properties (Fotsis et al, 1994). Inhibition of human umbilical vein endothelial cell (HUVEC) growth is used as a predictive assay of a drug's antiangiogenic potential. The sulphamoylated product of 2-MeOE2, 2-methoxyoestradiol-bis-sulphamate (2-MeOE2bisMATE, Figure 1, 2), was 60-fold more potent in an in vitro HUVEC growth-inhibition assay (Newman et al, 2004). 2-Methoxyoestradiol-bis-sulphamate has also been shown to inhibit neo-vascularisation in vivo in the mouse Matrigel plug model of angiogenesis (Chander et al, 2003).
2-MeOE2bisMATE is a derivative of oestrone-3-O-sulphamate (EMATE, Figure 1, 4) which was originally developed as a steroid sulphatase (STS) inhibitor (Purohit et al, 1998). Unexpectedly, EMATE proved to be a potent oestrogen on oral application in rats (Elger et al, 1995). The observed oestrogenicity of oestrogen sulphamates, such as EMATE, is thought to be a consequence of their sequestration into red blood cells (RBCs) and slow release of the oestrogen moiety into plasma (Elger et al, 1998). It is conceivable that, similarly, the sulphamoylation of 2-MeOE2, and the subsequent uptake of the ester into erythrocytes, may reduce the first pass metabolism of the agent and thus enhance its oral bioavailability. To investigate this postulate we compared the pharmacokinetics of 2-MeOE2bisMATE and 2-MeOE2 following administration of a single oral or intravenous dose in rats.
In addition, as part of the pharmacokinetic study, we also examined the metabolism of these two compounds in vivo. As sulphamoylated steroids can act as STS inhibitors, the ability of 2-MeOE2bisMATE to inhibit the in vivo activity of this enzyme was also examined (Reed et al, 1996). To compare the abilities of 2-MeOE2 and 2-MeOE2bisMATE to inhibit tumour growth in vivo, their effects on the growth of xenografts derived from MDA-MB-435 (ER−) human melanoma cells were also examined. This cell line was chosen for in vivo studies, as it had previously been used to test the efficacy of 2-MeOE2 (Klauber et al, 1997). Since the present study was initiated, it has been revealed that the MDA-MB-435 cell line is not derived from a breast carcinoma, but probably originates from a melanoma (Ellison et al, 2002). However, as this cell line is highly metastatic and ER−, it remains a good model for testing agents which should be active against a wide range of hormone-independent cancers.
Materials and methods
Chemicals and reagents
The following reagents were purchased from the suppliers listed: high-performance liquid chromatography (HPLC) grade methanol, diethyl ether (Fisher Scientific UK Limited, Loughborough, Leicestershire, UK); Halothane Astra Zeneca, Cheshire, UK); propylene glycol, ammonium sulphate, sodium azide, tetrahydrofuran (THF) (Sigma-Aldrich Comp. Ltd, Poole, Dorset, UK); 17α-epitestosterone (Steraloids, Newport, RI, USA); [6,7-3H] oestrone sulphate (43-50 Ci mmol−1, Perkin-Elmer, Boston, MA, USA). 2-methoxyoestrone (2-MeOE1, Figure 1, 5), 2-MeOE2, 2-MeOE2bisMATE, 2-MeOE2-3S (Figure 1, 3), 2-methoxyoestradiol-3-sulphamate (2-MeOE2-3MATE, Figure 1, 6) and 2-methoxyoestradiol-17-sulphamate (2-MeOE2-17MATE, Figure 1, 7) were synthesised from oestrone. All new compounds exhibited spectroscopic and analytical data in accordance with their structure. Full details of their synthesis will be reported elsewhere.
In vivo pharmacokinetic and metabolism studies
Female Wistar (155–165 g) rats were purchased from Charles River UK Ltd (Margate, Kent, UK) and housed in a dedicated animal facility. Rats received RM1 rodent maintenance diet (SDS, Kent, UK), water ad libitum, and were maintained in positive pressure isolators under a 12 h light–dark cycle. These experiments were carried out under conditions that complied with institutional requirements. Rats received 2-MeOE2 or 2-MeOE2bisMATE (10 mg kg−1, oral or intravenous), with control animals receiving vehicle only (propylene glycol: THF, 9 : 1 v v−1). There were two reasons for selecting 10 mg kg−1 for both oral and intravenous dosing. Firstly, a single dose of EMATE administered by these routes has been shown to inhibit rat liver sulphatase by at least 99% (Purohit et al, 1995). Secondly, administration of this dose was found to elicit sufficiently high levels of the agents for their detection in plasma. Rats were subjected to terminal anaesthesia (Halothane) and blood removed by cardiac puncture at 5, 15 and 30 min and 1, 3, 8 and 24 h after intravenous administration, and 15 and 30 min and 1, 3, 8, 24 and 48h following oral administration of 2-MeOE2 or 2-MeOE2bisMATE. Plasma was prepared from whole blood by centrifugation (2800 g, 4°C, 15 min). Plasma (0.5 ml) was extracted with diethyl ether (4 ml) and frozen in a methanol : solid carbon dioxide mixture. 17α-Epitestosterone (28 μg ml−1) was used as an internal standard after purification by HPLC. The organic phase was decanted to a fresh tube and evaporated to dryness under a stream of air at room temperature. The extraction efficiencies for 2-MeOE2 and 2-MeOE2bisMATE from plasma were 71±5 and 75±4% (n=6), respectively. The residues were stored at −20°C until analysis by HPLC.
HPLC analysis
2-MeOE2bisMATE was separated from its putative metabolites, 2-MeOE2, 2-MeOE2-3MATE and 2-MeOE2-17MATE, using a modified version of a reversed-phase HPLC method described previously (Hildago Aragones et al, 1996). An Agilent 1100 (Cheshire, UK) autosampler, photodiode array detector and solvent delivery system were used. The agents were separated from endogenous plasma components by an isocratic mobile phase consisting of 58% methanol in 0.02 M ammonium sulphate. Sodium azide (1 mM) was added to the mobile phase in order to decrease microbial growth. Extracted samples were reconstituted in mobile phase and aliquots of 100 μl were injected on to a C3-phenyl column (250 × 5 mm, 5 μm) purchased from Phenomenex (Cheshire, UK). 2-Methoxyoestradiol-bis-sulphamate, 2-MeOE2 and their metabolites were analysed with a photodiode array detector with detection at 285 nm. The method was validated by spiking plasma with 2-MeOE2bisMATE (960 ng ml−1). The inter-day and intra-day coefficients of variation were 8.2% (n=6) and 3.8% (n=6), respectively. Quantification was achieved by spiking plasma with 2-MeOE2bisMATE and the internal standard 17α-epitestosterone, extraction with diethyl ether and subsequent analysis by HPLC. Plasma calibration curves were found to be linear from 40 to 9000 ng ml−1. The limits of detection (LODs) and quantification of 2-MeOE2bisMATE in plasma were 11 and 40 ng ml−1, respectively.
Pharmacokinetic analysis
Pharmacokinetic parameters were calculated using WinNonlin software (Pharsight Corporation, Mountview, CA, USA). The area under the curve (AUC) was calculated using the linear trapezoidal method, with extrapolation of the terminal phase to infinity. Other parameters calculated were: distribution and elimination rate constants (α and β); total body clearance (Cl)=dose/AUC; volume of distribution (Vd)=Cl/β; distribution half-life (t1/2α)=0.693/α; elimination half-life (t1/2β)=0.693/β; bioavailability (% F=(AUCoral/AUCi.v) × 100).
Liver oestrone sulphatase activity
Livers were obtained from animals administered with 2-MeOE2 or 2-MeOE2bisMATE either orally or intravenously, to assess their effects on steroid sulphatase activity. Steroid sulphatase activity was measured as described previously (Purohit et al, 1995).
In vivo inhibition of tumour growth
To compare the anticancer effects of 2-MeOE2 and 2-MeOE2bisMATE, xenografts derived from MDA-MB-435 (ER−) human melanoma cells were transplanted into female nude mice with eight mice per group. These studies were carried out by Anti-Cancer Inc. (San Diego, CA, USA). Treatment was initiated when tumour volumes reached 100–200 mm3. Drugs were dissolved in a minimum volume of THF, diluted with propylene glycol and administered at 20 mg kg−1, oral, daily for 28 days. The length (l) and width (w) of tumours was measured at weekly intervals, from which the tumour volumes were calculated using the formula (l × w2/2). Monitoring of tumour volumes continued for a further 28-day period after the end of drug administration. The body masses of control and treated animals were also measured at weekly intervals as an indicator of any toxicity that might be associated with the use of these drugs.
Statistics
Student's t-test was used to assess the significance of differences in tumour volumes between control and treated animals.
Results
HPLC analysis
A reproducible, robust and sensitive analytical method was developed for the detection of 2-MeOE2 and 2-MeOE2bisMATE in plasma. The method facilitated separation of these agents from the putative metabolites 2-MeOE1, 2-MeOE2-17MATE and 2-MeOE2-3MATE, the internal standard, 17α-epitestosterone, and endogenous plasma components.
Pharmacokinetics of 2-MeOE2 and 2-MeOE2bisMATE
In order to compare the pharmacokinetics of 2-MeOE2 and 2-MeOE2bisMATE, rats were administered with either a single intravenous or oral dose of the agents. Using the HPLC analytical method developed, it was possible to detect both compounds in plasma after intravenous administration (Figure 2B, C), but only 2-MeOE2bisMATE was detected after oral administration (Figure 2E, F). The identities of these agents were corroborated by electrospray ionisation liquid chromatography-mass spectrometry in the selected ion mode (results not shown). When 2-MeOE2 was given to rats as an intravenous bolus, the agent was found to be rapidly removed from the plasma (Figure 3A) and was below the LOD (11 ng ml−1) 1 h after administration of the dose. Consequently, it was not possible to determine the pharmacokinetic parameters for 2-MeOE2. In contrast, when 2-MeOE2bisMATE was administered as an intravenous bolus, the parent compound was quantifiable in plasma 8 h after administration of the agent (1.52±0.10 μg ml−1), although it was not detectable by 24 h. The decrease in plasma concentration of 2-MeOE2bisMATE followed a biexponential pattern with an initial distribution half-life (t1/2α) of 0.22±0.10 h and a terminal half-life (t1/2β) of 8.50±1.16 h. The area under the plasma concentration–time curve (AUC) was 513.99±153.88 h μg ml−1. Pharmacokinetic parameters after oral administration were calculated using an extravascular noncompartmental model. Maximum levels of 2-MeOE2bisMATE in plasma (Cmax=3.90±0.25 μg ml−1) were reached 3 h after oral administration of the agent (Figure 3B and Table 1) with 0.53±0.04 μg ml−1 still detectable after 24 h. A terminal plasma half-life of 6.51±1.09 h with an AUC of 388.84±28.84 h μg ml−1 was achieved. The bioavailability for 2-MeOE2bisMATE based on AUC after intravenous or oral administration was 85.19±16.09%. The putative metabolites of 2-MeOE2bisMATE, 2-MeOE2-17MATE, 2-MeOE2-3MATE and 2-MeOE2 were not above the LOD in plasma.

HPLCs of diethyl ether plasma extracts. Plasma was prepared from whole blood following administration of intravenous (A) propylene glycol, (B) 2-MeOE2, (C) 2-MeOE2bisMATE or oral, (D) propylene glycol, (E) 2-MeOE2 and (F) 2-MeOE2bisMATE. The blood was removed from animals 0.25 and 3 h after intravenous and p.o. dosing, respectively. The identities of 2-MeOE2, 2-MeOE2bisMATE and 17α-epitestosterone (internal standard) as being peaks 1, 2 and 3, respectively, were corroborated by co-elution with authentic standards and liquid chromatography-mass spectrometry. For details of extraction and HPLC analysis, see ‘Materials and Methods’. The peak eluting at 3.9 min was an endogenous plasma component.

Concentrations of 2-MeOE2 and 2-MeOE2bisMATE in rat plasma following a single intravenous (A) or oral (B) bolus of drug (10 mg kg−1) in THF/propylene glycol. The values shown are the means ±s.e.m. (n=3). 2-Methoxyoestradiol was below the LOD (11 ng ml−1) in plasma after oral administration. Where no error bars are shown, the coefficient of variation is <10%.
Inhibition of liver sulphatase
Hepatic sulphatase activity was inhibited by 99.5±0.5% compared with the activity measured in control animals, following administration of a single oral or intravenous dose of 2-MeOE2bisMATE. This level of inhibition of sulphatase activity was maintained for 48 h following administration of 2-MeOE2bisMATE. 2-Methoxyoestradiol did not affect rat liver sulphatase activity (data not shown).
In vivo inhibition of tumour growth
To compare the effects of 2-MeOE2 and 2-MeOE2bisMATE on in vivo tumour growth in nude mice that had been inoculated with xenografts derived from MDA-MB-435 human melanoma cells, animals were treated with drugs (20 mg kg−1, oral) daily for 28 days (Figure 4). At this dose, 2-MeOE2 had no significant effect on tumour growth during the treatment or post-dosing periods. In contrast, by week 2 of dosing, the tumour volumes of mice receiving 2-MeOE2bisMATE were significantly smaller (P<0.05) than those of the control group. Tumour volumes for mice treated with 2-MeOE2bisMATE then remained significantly smaller (P<0.01) for the remaining period of the study. At 4 weeks, at the end of the dosing period, the mean tumour volumes in mice receiving 2-MeOE2bisMATE were 14% of that of tumour volume in the control animals. This difference (86%) in tumour volumes between the two groups was maintained for a further 4 weeks after the cessation of drug administration. No significant differences in the change in weights between the control and treated animals were detected during the treatment or post-dosing periods (Figure 5).

In vivo effect of 2-MeOE2 or 2-MeOE2bisMATE on the growth of MDA-MB-435 (ER−) xenografts in nude mice. Vehicle (THF/propylene glycol) or compounds were administered daily (20 mg kg−1, oral) for 28 days. Tumour volumes were monitored at weekly intervals for the duration of drug administration and for a further 4-week period after the end of drug administration (means±s.e.m., n=8). Results are expressed as percentage change in tumour volumes detected at weekly intervals. At this dose, 2-MeOE2 had no significant effect on tumour growth during the treatment or post-dosing periods. For 2-MeOE2bisMATE-treated animals, tumour volumes were significantly smaller than those in control animals by week 2 (P<0.05) and remained significantly smaller (P<0.01) for the rest of the study period.

Changes in weights of control animals and those administered 2-MeOE2 or 2-MeOE2bisMATE. Animals were weighed at weekly intervals and the results are expressed as the percentage change compared with their pre-dosing weights (means±s.e.m., n=8).
Discussion
The sulphamoylated oestrogen derivative 2-MeOE2bisMATE has previously been shown to be a more potent inhibitor of cancer cell proliferation than its parent compound, 2-MeOE2 (Raobaikady et al, 2003). Furthermore, in in vitro models of angiogenesis, 2-MeOE2bisMATE proved to be considerably more potent as an inhibitor of angiogenesis than 2-MeOE2 (Newman et al, 2004). The main finding to emerge from the present in vivo study goes some way to offer an explanation for the enhanced potency of 2-MeOE2bisMATE. In this study, the plasma concentrations of both drugs were determined after oral or intravenous dosing, together with an examination of the duration for which the compounds and putative metabolites were detectable in plasma. After intravenous administration of 2-MeOE2, the compound was rapidly cleared from the plasma, suggesting metabolic removal. 2-Methoxyoestradiol eluded detection in plasma after its oral administration and, by 1 h after intravenous dosing, levels were below the LOD for the assay. In contrast, significant concentrations of 2-MeOE2bisMATE were detectable in plasma after its oral or intravenous administration with 450 ng ml−1 still being detectable at 24 h after oral dosing. Recently, a daily oral dose of 1000 mg 2-MeOE2 was given to 24 patients with advanced metastatic breast cancer (Sledge et al, 2002; Lakhani et al, 2003). It was shown that 2-MeOE2 was metabolised to 2-MeOE1 and 2-MeOE2/2-MeOE1 glucuronide and sulphate conjugates. Demethylated 2-MeOE1 (2-OHE1) has also been measured in the urine of humans administered with [3H]2-MeOE2, although this metabolite could not be detected in plasma (Longcope et al, 1980).
After intravenous administration of 2-MeOE2, its half-life in plasma was approximately 14 min. Pharmacokinetic studies in rats indicated that 2-MeOE2bisMATE was detectable in plasma for up to at least 24 h after oral administration. The calculated bioavailability for 2MeOE2bisMATE from the AUC was 85.2%. A possible explanation for this finding may be that, initially, there is sequestration of 2-MeOE2bisMATE into blood components, possibly RBCs (unpublished data), or binding to plasma proteins, resulting in high AUC orally. This may, in part, account for the slow clearance rates and the high volume of distribution achieved by both intravenous and oral administration. Nevertheless, it is apparent that 2-MeOE2bisMATE has a higher level of bioavailability after oral administration, compared with that of the non-sulphamoylated 2-MeOE2.
Further evidence of the enhanced potency of 2-MeOE2bisMATE compared with that of 2-MeOE2 was obtained from the in vivo study carried out in nude mice bearing xenografts of MDA-MB-435 tumours. 2-Methoxyoestradiol had little effect on the growth of these tumours. In contrast, for animals receiving 2-MeOE2bisMATE, tumour growth was almost completely inhibited for the 4-week treatment period, and its growth-inhibiting effects were maintained for 4 weeks after the cessation of dosing. It is evident from the present study that 2-MeOE2bisMATE can inhibit in vivo tumour growth. Further studies will be required to ascertain whether it is acting by inhibition of angiogenesis or induction of apoptosis or, as most likely, a combination of both mechanisms. No decreases in body weights were detected in animals receiving either 2-MeOE2 or 2-MeOE2bisMATE, indicating that, at the dose tested, these compounds have little or no toxicity.
From these studies, it is evident that 2-MeOE2bisMATE is resistant to metabolism in vivo, and this finding goes some way to account for its enhanced potency compared with 2-MeOE2. In vivo, the C3 phenolic group and 17β hydroxyl group of 2-MeOE2 are likely to be subjected to rapid conjugation or oxidation, thus reducing the potency of this compound as an anticancer and antiangiogenic drug. Several derivatives of 2-MeOE2 have been developed, such as 2-methoxymethyloestradiol and a series of 2-methoxytetrane oestradiols, which are more potent inhibitors of in vitro cancer cell proliferation than 2-MeOE2 itself (Bruggemeier et al, 2001; Tinley et al, 2003). However, as these compounds retain the C3/17β configuration of 2-MeOE2, it is unlikely that they will prove to be more potent in vivo and will undergo the same route of rapid inactivation.
EMATE was previously found to have a half-life of 4.5 h in plasma (Hildago Aragones et al, 1996), and this compares with the much shorter half-life for unconjugated oestrogen of 20–30 min (Ruder et al, 1972). Like EMATE, 2-MeOE2bisMATE is a potent steroid sulphatase inhibitor and, in the present study, liver sulphatase was rapidly inactivated after its oral or intravenous administration. Almost complete inhibition of steroid sulphatase activity (>99%) was achieved within 5 and 15 min of intravenous and oral dosing, respectively. Inhibition of steroid sulphatase activity will prevent removal of the sulphamoyl group at the C3 position, thus greatly prolonging the plasma, and presumably tissue, concentrations of the active sulphamoylated drug. The relatively high plasma concentrations of 2-MeOE2bisMATE (up to 5 μ M) detected are within the range that has been shown to inhibit cancer cell and HUVEC proliferation in vitro (Newman et al, 2004). Thus, sulphamoylation of 2-MeOE2 yields a drug that is resistant to in vivo metabolism. In vivo, its ability to inhibit STS appears to block its de-sulphamoylation, ensuring that the biologically active drug is available to exert its effects for a prolonged period of time. The findings from in vitro studies that 2-MeOE2bisMATE is more potent than 2-MeOE2, together with the results of the in vivo studies showing no evidence of conversion to 2-MeOE2, indicate that it is not acting as a pro-drug for the formation of 2-MeOE2.
There is currently considerable interest in exploring the mechanisms by which 2-MeOE2 and its sulphamoylated derivates act to inhibit cancer cell growth and angiogenesis. 2-Methoxyoestradiol is an endogenous oestrogen metabolite, and it has been suggested that it may be the body's natural anticancer metabolite (Zhu and Conney, 1998). This contention is supported by a significant body of research carried out by Bradlow et al (1998), who have convincingly demonstrated that production of 2-MeOE2 is increased in women at low risk of breast cancer. 2-Methoxyoestradiol and its sulphamoylated derivatives are thought to bind to the colchicine-binding site of tubulin, where they act to alter microtubule dynamics, leading to activation of the intrinsic apoptotic pathway via BCL-2 phosphorylation (D'Amato et al, 1994; MacCarthy-Morrogh et al, 2000). In addition, it is possible that, like 2-MeOE2, the sulphamoylated derivatives will also act to induce apoptosis via activation of the extrinsic apoptotic pathway, which involves upregulation of death receptors, such as DR5 (LaVallee et al, 2003).
In vitro, 2-MeOE2 has been shown to inhibit the proliferation of a wide range of cancer cells including those derived from the breast, prostate, ovary, pancreas, lung and multiple myelomas. At relatively high doses (75–150 mg kg−1), its efficacy in mouse models against melanomas, Meth A sarcomas and multiple myelomas has been demonstrated (Klauber et al, 1997; Dingli et al, 2002). As shown in the present in vivo efficacy study at a dose of 20 mg kg−1, while 2-MeOE2 was ineffective, 2-MeOE2bisMATE almost completely blocked tumour growth. The identification of 2-MeOE2bisMATE as a potent anticancer, antiangiogenic drug that is orally available, yet resistant to metabolism, should make it a good candidate for development for cancer therapy.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ball P, Knuppen R (1980) Catecholoestrogens (2- and 4-hydroxyestrogens); chemistry, biogenesis, metabolism, occurrence and physiological significance. Acta Endocrinol 232(Suppl): 1–127
Bradlow HL, Telang NT, Sepkovic DW, Osborne MP (1998) 2-Hydroxyestrone; the ‘good’ estrogen. J Endocrinol 150: S259–S265
Bruggemeier RW, Bhat AS, Lovely CJ, Coughenour HD, Yusuf F, Burak WG (2001) 2-Methoxymethyestradiol: a new 2-methoxyoestrogen analog that exhibits antiproliferative activity and alters tubulin dynamics. J Steroid Biochem Mol Biol 78: 145–156
Chander SK, Ireson CR, Fenton J, Newman SP, Leese MP, Potter BVL, Purohit A, Reed MJ (2003) Comparison of 2-methoxyestradiol and 2-methoxyestradiol-3, 17-bis-sulfamate in the Matrigel plug angiogenesis mouse model. Proc Am Assoc Cancer Res Abstr 44: 1156
D'Amato R, Lin CM, Flynn E, Folkman J, Hamel E (1994) 2-Methoxyestradiol, an endogenous mammalian metabolite, inhibits tubulin polymerisation by interacting at the colchicine site. Proc Natl Acad Sci USA 91: 3964–3968
Dingli D, Timm M, Russell SJ, Witzing TE, Rajkumar SV (2002) Promising preclinical activity of 2-methoxyestradiol in multiple myeloma. Clin Cancer Res 8: 3948–3954
Elger W, Palme H-J, Schwarz S (1998) Novel oestrogen sulfamates: a new approach to oral hormone therapy. Exp Opin Investig Drugs 7: 575–589
Elger W, Schwarz S, Hedden A, Reddersen G, Schneider B (1995) Sulfamates of various estrogens are prodrugs with increased systemic and reduced hepatic estrogenicity at oral application. J Steroid Biochem Mol Biol 55: 395–403
Ellison G, Klinowska T, Westwood PFR, Docter E, French T, Fox JC (2002) Further evidence to support the melanocytic origin of MDA-MB-435. J Clin Pathol Mol Pathol 55: 294–299
Fotsis T, Zhang Y, Pepper MS, Adlercreutz H, Montesano R, Nawroth PP, Schweigerer L (1994) The endogenous oestrogen metabolite 2-methoxyoestradiol inhibits angiogenesis and suppresses tumour growth. Nature (Lond) 368: 237–239
Hildago Aragones MI, Purohit A, Parish D, Sahm UG, Pouton CW, Potter BVL, Reed MJ (1996) Pharmacokinetics of oestrone-3-O-sulphamate. J Steroid Biochem Mol Biol 58: 611–617
Klauber N, Parangi S, Flynn E, D'Amato RJ (1997) Inhibition of angiogenesis and breast cancer in mice by the microtubule inhibitors 2-methoxyestradiol and taxol. Cancer Res 57: 81–86
Lakhani NJ, Sarkar MA, Venitz J, Figg WD (2003) 2-Methoxyestradiol, a promising anticancer agent. Pharmacotherapy 23: 165–172
LaVallee TM, Zhan XH, Herbstritt CJ, Kough EC, Green SJ, Pribluda VS (2002) 2-Methoxyestradiol inhibits proliferation and induces apoptosis independently of oestrogen receptors α and β. Cancer Res 62: 3691–3697
LaVallee TM, Zhan XH, Johnson MS, Herbstritt CJ, Swartz G, Williams MS, Hembrough WA, Green SJ, Pribluda VS (2003) 2-Methoxyestradiol up-regulates death receptor 5 and induces apoptosis through activation of the extrinsic pathway. Cancer Res 63: 468–475
Longcope C, Flood C, Femino A, Williams KIH (1980) Metabolism of 2-methoxyestrone in normal men. J Clin Endocrinol Metab 57: 277–282
Lottering M-L, Hang M, Seegers JC (1992) Effects of 17-β-estradiol metabolites on cell cycle events in MCF-7 cells. Cancer Res 52: 5926–5932
MacCarthy-Morrogh L, Townsend PA, Purohit A, Hejaz HAM, Potter BVL, Reed MJ, Packham G (2000) Differential effects of estrone and estrone-3-O-sulfamate derivatives on mitotic arrest, apoptosis, and microtubule assembly in human breast cancer cells. Cancer Res 60: 5441–5450
Mannisto PT, Kaakkola S (1999) Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev 51: 593–628
Merriam GR, MacLusky NJ, Picard MK, Naftolin F (1980) Comparative properties of the catechol estrogens. 1. Methylation by catechol-O-methyltransferase and binding to cytosol estrogen receptors. Steroids 36: 1–11
Newman SP, Leese MP, Purohit A, James DRC, Rennie CE, Potter BVL, Reed MJ (2004) Inhibition of in vitro angiogenesis by 2-methoxy and 2-ethyl estrogen sulfamates. Int J Cancer 109: 533–540
Purohit A, Vernon KA, Wagenaar Hummelinck AE, Woo LWL, Hejaz HAM, Potter BVL, Reed MJ (1998) The development of A-ring modified analogues of oestrone-3-O-sulphamate as potent steroid sulphatase inhibitors with reduced oestrogenicity. J Steroid Biochem Mol Biol 64: 269–275
Purohit A, Williams GJ, Roberts CJ, Potter BVL, Reed MJ (1995) In vivo inhibition of oestrone sulphatase and dehydroepiandrosterone sulphatase by oestrone-3-O-sulphamate. Int J Cancer 63: 106–111
Raobaikady B, Purohit A, Chander SK, Woo LWL, Leese MP, Potter BVL, Reed MJ (2003) Inhibition of MCF-7 breast cancer cell proliferation and in vivo steroid sulphatase by 2-methoxyestradiol-bis-sulphamate. J Steroid Biochem Mol Biol 84: 351–358
Reed MJ, Purohit A, Woo LWL, Potter BVL (1996) The development of steroid sulphatase inhibitors. Endocr Relat Cancer 3: 9–23
Ruder HJ, Loriaux DL, Lispett MB (1972) Estrone sulfate: production rate and metabolism in man. J Clin Endocr Metab 51: 1020–1023
Seegers JC, Aveling M-L, Van Aswegen CH, Cross M, Koch F, Joubert WS (1989) The cytoxic effects of estradiol-17β, catecholestradiols and methoxyestradiols on dividing MCF-7 and HeLa cells. J Steroid Biochem Molec Biol 32: 797–809
Sledge GW, Miller KD, Hanye LG, Nguyen DD, Storniolo AM, Phillips E, Priblunda V, Gubish ER (2002) A phase I study of 2-methoxyestradiol in patients with refractory metastatic breast cancer. Proc Amer Soc Clin Oncol, abstract number 441
Tinley TL, Leal RM, Randall-Hlubek DA, Cessac JW, Wilkens LR, Roa PN, Mooberry SL (2003) Novel 2-methoxyestradiol analogues with antitumor activity. Cancer Res 63: 1538–1549
Zhu BT, Conney AH (1998) Is 2-methoxyestradiol an endogenous estrogen metabolite that inhibits mammary carcinogenesis? Cancer Res 58: 2269–2277
Acknowledgements
We would like to thank Mrs Janine Fenton and Mr D Bennetto for their technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Ireson, C., Chander, S., Purohit, A. et al. Pharmacokinetics and efficacy of 2-methoxyoestradiol and 2-methoxyoestradiol-bis-sulphamate in vivo in rodents. Br J Cancer 90, 932–937 (2004). https://doi.org/10.1038/sj.bjc.6601591
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6601591
Keywords
This article is cited by
-
2-Methoxyestradiol inhibits carotid artery intimal hyperplasia induced by balloon injury via inhibiting JAK/STAT axis in rats
Environmental Science and Pollution Research (2022)
-
Liposomal 2-Methoxyestradiol Nanoparticles for Treatment of Uterine Leiomyoma in a Patient-Derived Xenograft Mouse Model
Reproductive Sciences (2021)
-
Novel sulphamoylated 2-methoxy estradiol derivatives inhibit breast cancer migration by disrupting microtubule turnover and organization
Cancer Cell International (2019)
-
The in vitro effects of a novel estradiol analog on cell proliferation and morphology in human epithelial cervical carcinoma
Cellular & Molecular Biology Letters (2018)
-
A novel non-sulphamoylated 2-methoxyestradiol derivative causes detachment of breast cancer cells by rapid disassembly of focal adhesions
Cancer Cell International (2018)
