
Abstract
Convalescent plasma (CP) therapy in COVID-19 disease may improve clinical outcome in severe disease. This pilot study was undertaken to inform feasibility and safety of further definitive studies. This was a prospective, interventional and randomized open label pilot trial in patients with severe COVID-19. Twenty COVID-19 patients received two 200 ml transfusions of convalescent patient CP over 24-h compared with 20 who received standard of care. The primary outcome was the requirement for ventilation (non-invasive or mechanical ventilation). The secondary outcomes were biochemical parameters and mortality at 28 days. The CP group were a higher risk group with higher ferritin levels (p < 0.05) though respiratory indices did not differ. The primary outcome measure was required in 6 controls and 4 patients on CP (risk ratio 0.67, 95% CI 0.22–2.0, p = 0.72); mean time on ventilation (NIV or MV) did not differ. There were no differences in secondary measures at the end of the study. Two patients died in the control and one patient in the CP arm. There were no significant differences in the primary or secondary outcome measures between CP and standard therapy, although a larger definitive study is needed for confirmation. However, the study did show that CP therapy appears to be safe in hospitalized COVID-19 patients with hypoxia.
Clinical trials registration NCT04356534: 22/04/2020.
Similar content being viewed by others
Introduction
Coronavirus disease 2019 (COVID-19) is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and has developed into a pandemic with serious global public health and economic sequelae. As of 25 February 2021, more than 113 million cases have been confirmed worldwide leading to over 2.5 million deaths1. There have been a number of reports of medications, such as remdesivir, with antiviral properties that have shown efficacy against SARS-CoV-2 with shorter time to recovery2. Whilst there are now several effective vaccines emerging and being used globally, the rate of COVID-19 disease and its complications currently remain high.
Plasma therapy using Convalescent Plasma (CP) transfusion refers to a form of passive therapy, where neutralizing antibodies from a recovered donor are injected into the infected patient with the aim of altering the course of the disease3. This has been shown to be effective in severe acute respiratory syndrome4, Ebola virus infection5 and in H1N1 influenza6. More recently, there has been a report of the use of CP in the treatment of five mechanically ventilated COVID-19 patients with the suggestion of expedited recovery as the patients improved one week after the transfusion7. A randomized trial with CP therapy in severely and critically ill COVID-19 patients undertaken in China was stopped early as recruitment slowed down due to the decrease in COVID-19 cases in China8. Similarly, an open label randomized trial in the Netherlands was stopped prematurely due to the detection of baseline neutralizing antibodies in the majority of patients, with antibody titers similar to that of the donors9. An open label Expanded Access Program in the US studied the effect on CP on mortality in more than 35,000 participants with COVID-19 and concluded that CP transfusion with high antibody levels and early during admission was associated with decreased mortality10. However, no studies have been undertaken with CP therapy in hypoxic patients and therefore this pilot trial was undertaken.
Methods
This was a prospective, randomized, controlled open label pilot study involving 40 patients with severe COVID-19 disease confirmed by RT-PCR testing11. All patients gave written informed consent. This study was approved by the National COVID-19 Research Committee, and the Bahrain Defense Force Hospital Ethics committee, and was conducted in accordance with the Declaration of Helsinki and local regulations. The trial was conducted upon the approved protocol by the National Research and Ethics committee. The trial protocol is shown in Supplementary Information S1.
Participants
Patients were recruited from two medical centres. The study recruitment was from April 2020 to June 2020.
Inclusion criteria
Inclusion criteria were: (1) signed informed consent; (2) aged at least 21 years; (3) COVID-19 diagnosis based on polymerase chain reaction (PCR) testing; (5) hypoxia (oxygen saturation of less than or equal 92% on air, or PO2 < 60 mmHg arterial blood gas, or arterial partial pressure of oxygen (PaO )/fraction of inspired oxygen (FIO) of 300 or less and the patient requiring oxygen therapy; (6) pneumonia confirmed by chest imaging.
Exclusion criteria
Exclusion criteria were the following: (1) Patients with mild disease not requiring oxygen therapy; (2) Patients with a normal CXR or CT scan; (3) Patients requiring ventilatory support (non-invasive or mechanical); (4) Patients with a negative PCR test for SARS-CoV-2; (5) Patients with a history of allergy to plasma, sodium citrate or methylene blue, or those with a history of autoimmune disease or selective IGA deficiency.
Randomization
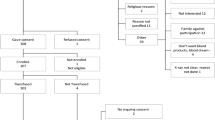
Following informed consent and screening, the patients were block randomised (in blocks of 4) by computer-generated random numbering to either the standard therapy or CP arms (Fig. 1).

Flow chart of study.
Patients and clinicians were not blinded to the treatment given.
Convalescent plasma donors
Patients who had recovered from COVID-19 and had been discharged from hospital for more than 2 weeks were approached to be volunteer donors. The criteria for donors included (1) ability to give informed consent; (2) men or nulliparous women (all women had a pregnancy test except for postmenopausal women); (3) PCR COVID-19 negative from respiratory tract; (4) patients were symptom free; (5) patients above the ages of 21; (6) body weight more than 50 kg; (7) met all donor selection criteria employed for routine plasma collection and plasmapheresis procedures at the collection centre. Convalescent plasma collection was performed followed by plasma extraction as detailed.
COVID-19 CP collection and handling was undertaken at Bahrain Defence Force Hospital Blood Bank. History and demographics were obtained from each of the plasma donors who met all donor selection criteria employed for routine plasma collection and plasmapheresis procedures at the collection center. Approximately 450 ml of whole blood was venesected together with testing for anti-SARS-CoV-2 antibody titres.
Whole blood units were separated into packed red blood cells and plasma; the plasma was removed and stored at less than − 18 °C. Laboratory tests including blood group (ABO/Rh), antibody screening, blood phenotype and transfusion-transmissible infection screening (which includes hepatitis B virus, hepatitis C virus, HIV and syphilis). Convalescent compatible plasma was thawed and stored at 2–6 °C for 24-h. Antibody levels were measured using Lansionbio COVID-19 IgM/IgG Test kit (Lansion Biotechnology Co., Ltd, China).
Convalescent plasma transfusion
ABO matched CP units were selected for transfusion and transfused into the COVID-19 patients using standard clinical transfusion procedures.
The dosage of CP was 400 ml, given as 200 ml over 2hrs over 2 successive days; the infusion rate was monitored and amended if there was a risk of fluid overload.
Patients prior to CP therapy were on standard supportive treatment including control of fever (paracetamol) and possible therapy including antiviral medications, Tocilizumab and antibacterial medication.
Standard supportive treatment
The standard supportive treatment included control of fever (paracetamol) and possible therapy including antiviral medications, Tocilizumab and antibacterial medication.
Primary outcome measure
The primary end points were the requirement for non-invasive ventilation (NIV) or mechanical ventilation (MV), and for those patients who required ventilation, the duration of ventilation.
Secondary outcome measures
Secondary outcome measures included C reactive protein, procalcitonin, lactate dehydrogenase, troponin, ferritin, D-Dimer, brain natriuretic peptide, lactate changes and 28-day mortality rate.
Laboratory measurements
White blood cell count was measured by flow cytometry, lactate dehydrogenase (LDH) was measured using a kinetic method, C-reactive protein (CRP) and D-Dimer were measured by an immuno-turbidimetric assay, whilst troponin, ferritin and procalcitonin were measured by an electrochemiluminescence immunoassay according to the manufacturers’ instructions.
Secondary analysis
Measurement of the effect of early CP transfusion (less than 3 days from admission) compared to late transfusion for the primary outcome (requirement of NIV or MV) was undertaken. Further analysis was done to compare the mean antibody level of the CP on the patients who developed the primary outcome to those who did not.
Statistical analysis
Power and sample size for pilot studies was based on Birkett and Day12. They concluded that a minimum of 20 degrees-of-freedom was required to estimate effect size and variability for normally distributed variables and hence 20 patients per group were recruited in this study; no allowance was made for dropouts, who are unlikely to be an issue in a study of this sort.
Baseline continuously distributed data are presented as means and standard deviations unless the data are skewed in which case the data median (25th/75th centiles) are presented; categorical data are shown by number and percent. Intention to treat analysis was used throughout.
For all statistical analyses, a two-tailed p < 0.05 is considered to indicate statistical significance. Time to event analysis and logistic regression are used to analyse event-related outcomes. Effect size for secondary outcomes is shown as the Hodges-Lehmann median difference and its associated confidence interval, calculated using the community-contributed Stata command cendiff13. It should be noted that this is the median of all pairwise differences between the groups and does not correspond to the difference between the medians of the two groups.
Statistical analyses were performed using the Stata (StataCorp. 2019. Stata Statistical Software: Release 16. College Station, TX: StataCorp LLC).
Results
The participant demographic, clinical and biochemical characteristics are shown in Table 1 and medication after randomization is shown in Table 2. The two groups showed similar baseline epidemiological characteristics. The CP group showed higher D-Dimer (p < 0.001) and ferritin levels (p = 0.049); however, respiratory indices did not differ. There were more men than women, reflecting the demographics of the workforce in Bahrain.
The use of medications following randomisation did not differ between groups (Table 2).
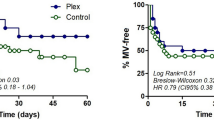
A total of 6 controls (30%) and 4 CP patients (20%) developed the primary outcome and were ventilated with either NIV or MV (risk ratio 0.67, 95% CI 0.22–2.0, p = 0.72). Primary outcome measure, time to ventilation was not different between the two groups (p = 0.52, logrank test; Fig. 2). Time on ventilation did not differ between the two groups (10.5 ± 2.9 days for control; 8.25 ± 4.42 days for CP (exact p = 0.809)). Length of stay for survivors was not different between the two groups; the mean length of stay in the control group was 18.05 ± 2.22 days compared to 14.1 ± 1.24 days in the CP group (p = 0.12). Table 3 summarizes the outcome in both groups. Steroids were used in 3 control patients and none of the CP patients, with no difference between groups (p = 0·342). To detect a difference at 90% power, alpha 0.05, with a risk ratio of 0.7 and risk in control 20%, a sample size of 822 in each group would be needed.

The plot shows cumulative ventilation rates for each group.
Table 4 shows the medians of secondary outcomes at discharge in patients who were discharged alive (18 control and 19 CP). The table also shows the Hodges Lehman median differences between the groups, with their associated confidence intervals. Significance levels are based on the Wilcoxon Mann–Whitney rank sum test.
The CP antibody level was available for 13 participants (mean 63.8 AU/ml ± 46.8 (SD), median 54.5). The patients who achieved the primary outcome and received CP had a mean antibody level of 84.95 AU/mL (SD 13.72, SE 7.9, N = 3), whilst those that did not require NIV or MV had a mean of 57.48 AU/mL (SD 51.8, SE 16.4, N = 10; p = 0.24).
30% of patients who received early CP (less than 3 days from admission) developed the primary outcome. None of the patients who received CP after 3 days from admission developed the primary outcome. Patients who received early CP had a mean antibody level of 82 AU/ml (SD 23, SE 9.5, N = 6). Those who received CP after 3 days had a mean titre 49 AU/ml (SD 58, SE 22, N = 7) (p = 0.06, rank sum test).
Two patients died in the control and one patient in the CP arm and their secondary outcome data were not included in Table 4.
Adverse events
Two patients treated with plasma reported adverse events during the study that were not considered to be related to therapy: one with diarrhoea and vomiting that settled spontaneously; and one desaturated transiently after the infusion.
Discussion
In this pilot study, the CP group appeared to have a higher systemic inflammatory response shown by the increased ferritin and D-dimer levels and more patients with chronic lung disease were included, a known risk factor for more severe disease14; however, respiratory indices did not differ between the groups. The higher ferritin and D-dimer levels shown in the CP group have been reported to predict poorer outcomes for those patients, indicating that the CP group were at a higher baseline risk15,16. These baseline differences were purely a result of chance following randomisation. While the CP therapy group had fewer patients deteriorating to NIV or MV therapy (primary outcome), and the duration of ventilation was less, this did not differ significantly from the standard therapy group. These results are in accord with a larger study done in China by Ling Li et al., that was stopped prematurely due to the decrease in COVID 19 cases8; however, as shown in this pilot, it is likely that trial was underpowered as an estimated sample size of over 822 in each group may be required to show a difference.
Steroids have been shown to be effective in COVID-1917; however, at the time of the study this evidence was not known and hence steroids were used at the discretion of the physicians. Steroids were used on 3 patients on standard therapy; none of the CP patients received steroids, but the sample size was inadequate to justify a hypothesis test. Of note, in the study by Ling Li et al., 45.6% CP arm and 32.7% of control arm were given steroids that may have masked the CP effect8 and, in addition, the impact of Chinese herbal medicines (used in over 50% of patients) is unclear in COVID-198. In this study, no subject received Remdesivir, but Hydroxychloroquine, Ribavirin and Lopinavir/ritonavir were used, though to date they have not been reported to be effective in COVID-19 infection18,19,20. This pilot, in accord with others8,21,22, indicated that CP therapy was safe with only three transient adverse reactions being recorded. The safety of plasma was assessed in an observational study in 20,000 hospitalized patients in the US and it reported low incidence (< 1%) of serious adverse events22. Sanfilippo et al. highlighted that plasma transfusion can be harmful in COVID-19, as plasma contains procoagulant factors and COVID-19 represents a unique scenario as they tend to have an increased risk for thrombosis23. The prothrombotic risk with plasma transfusion was not investigated in our study nor in other studies, and it has been suggested recently that the potential harm of the non-immune components of convalescent plasma should be studied, especially the prothrombotic risk24.
Patients who received early CP (less than 3 days from admission) needed NIV or MV earlier (the primary outcome measure) than those who received CP after 3 days of admission. Patients who received CP soon following admission were sicker as they deteriorated more quickly than those receiving CP later after their admission. This may confound the association between early and late CP transfusion. There was no difference between groups for length of hospital stay.
Patients who received early CP (less than 3 days from admission) had a higher mean antibody level in the transfused plasma, than those that received CP after 3 days, though the difference was not significant. A previous study reported that patients who received early CP and with higher antibody titres showed a better clinical outcome10; however, this pilot was not adequately powered to test the effect of early plasma in comparison to late plasma administration.
The CP collected from donors showed variations in mean antibody level, some extending to very low levels. The variability in the antibody level can have an effect on the effectiveness of plasma and can underestimate the effect of CP in this trial. The FDA have given recommendations to use CP with high antibody titre only25, to prevent transfusion of CP with low antibody levels that can be ineffective.
Our trial result was also in agreement with a randomized trial conducted in India in 464 adults with COVID-19. Agarwal et al. reported that convalescent plasma was not associated with a reduction in progression to severe COVID-19 or all-cause mortality. A subgroup analysis showed no difference in the outcome even after stratifying on the presence of neutralizing antibody levels (> 1:20)26.
A randomized trial comparing convalescent plasma with standard of care therapy in patients hospitalized for COVID-19 in the Netherlands was stopped early after observing that more than 79% of patients randomized to the plasma arm had median antibody titres comparable to the plasma donors prior to receiving the plasma transfusion. There was no significant difference in the median neutralizing antibody titre between recipients and donors, despite patients being randomized within 10 days of symptom onset. This may indicate that measuring the antibody titre of patients is important in order to select those patients with low antibody titre that might benefit from CP transfusion27.
A recently published randomized trial by Simonov et al. that studied CP in COVID-19 with severe pneumonia concluded that CP did not reduce mortality or improve clinical outcomes as compared with placebo28. Moreover, the length of stay was no different between the two groups, which is also in accord with our findings.
The main limitations of this study were that it was pilot whose main purpose was to guide the feasibility and safety of studies of CP therapy and, as a consequence, it lacks the statistical power to conduct outcome hypothesis tests. Determination of the optimal antibody titre from the donors should also be undertaken that was not done in this study, as well a measurement of antibody titre in the recipients before and after the infusions. Moreover, the quantification method for the antibody levels could have been improved and using the neutralizing antibody titre would have been more appropriate; however, at the time of the study, an authorized neutralizing antibody titre test was not available. Furthermore, the antibody titres were also not measured for our patients on randomization.
In conclusion, there were no significant differences in the primary or secondary outcome measures between CP and standard therapy though fewer patients required ventilation (NIV or MV) and for a shorter period of time, although a larger definitive study is needed for confirmation. However, the study did show that CP therapy appears to be safe in hospitalized COVID-19 patients with hypoxia.
References
Coronavirus Update (Live): 46,432,714 Cases and 1,200,927 Deaths from COVID-19 Virus Pandemic - Worldometer. Worldometers.info. https://www.worldometers.info/coronavirus/.
Beigel, J. H. et al. Remdesivir for the treatment of Covid-19—Preliminary report. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2007764 (2020).
Sahu, K. K., Jindal, V., Siddiqui, A. D., Cerny, J. & Gerber, J. M. Convalescent plasma therapy: A passive therapy for an aggressive COVID-19. J. Med. Virol. 92(11), 2251–2253. https://doi.org/10.1002/jmv.26047 (2020).
Soo, Y. O. et al. Retrospective comparison of convalescent plasma with continuing high-dose methylprednisolone treatment in SARS patients. Clin. Microbiol. Infect. 10(7), 676–678. https://doi.org/10.1111/j.1469-0691.2004.00956.x (2004).
Sahr, F. et al. Evaluation of convalescent whole blood for treating Ebola Virus Disease in Freetown, Sierra Leone. J. Infect. 74(3), 302–309. https://doi.org/10.1016/j.jinf.2016.11.009 (2017).
Hung, I. F. et al. Convalescent plasma treatment reduced mortality in patients with severe pandemic influenza A (H1N1) 2009 virus infection. Clin. Infect. Dis. 52(4), 447–456. https://doi.org/10.1093/cid/ciq106 (2011).
Shen, C. et al. Treatment of 5 critically ill patients with COVID-19 with convalescent plasma. JAMA https://doi.org/10.1001/jama.2020.4783 (2020).
Li, L. et al. Effect of convalescent plasma therapy on time to clinical improvement in patients with severe and life-threatening COVID-19: A randomized clinical trial. JAMA 324(5), 1–11. https://doi.org/10.1001/jama.2020.10044 (2020).
Gharbharan, A., et al. Convalescent plasma for COVID-19. A randomized clinical trial. medRxiv. 2020.
Joyner, M. J. et al. Effect of convalescent plasma on mortality among hospitalized patients with COVID-19: Initial three-month experience. medRxiv https://doi.org/10.1101/2020.08.12.20169359 (2020).
Corman, V. M. et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance https://doi.org/10.2807/1560-7917.Es.2020.25.3.2000045 (2020).
Birkett, M. A. & Day, S. J. Internal pilot studies for estimating sample size. Stat. Med. 13(23–24), 2455–2463 (1994).
Newson, R. Confidence intervals for rank statistics: Percentile slopes, differences, and ratios. Stand. Genom. Sci. 6(4), 497–520 (2006).
Preliminary estimates of the prevalence of selected underlying health conditions among patients with coronavirus disease 2019—United States, February 12-March 28, 2020. MMWR. 2020;69(13):382–386. https://doi.org/10.15585/mmwr.mm6913e2.
Hariyanto, T. I. et al. Inflammatory and hematologic markers as predictors of severe outcomes in COVID-19 infection: A systematic review and meta-analysis. Am. J. Emerg. Med. 41, 110–119. https://doi.org/10.1016/j.ajem.2020.12.076 (2020).
Cheng, L. et al. Ferritin in the coronavirus disease 2019 (COVID-19): A systematic review and meta-analysis. J. Clin. Lab. Anal. 34(10), e23618. https://doi.org/10.1002/jcla.23618 (2020).
Ledford, H. Coronavirus breakthrough: Dexamethasone is first drug shown to save lives. Nature 582(7813), 469. https://doi.org/10.1038/d41586-020-01824-5 (2020).
Cao, B. et al. A trial of lopinavir-ritonavir in adults hospitalized with severe Covid-19. N. Engl. J. Med. 382(19), 1787–1799. https://doi.org/10.1056/NEJMoa2001282 (2020).
Hung, I. F. et al. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: An open-label, randomised, phase 2 trial. Lancet 395(10238), 1695–1704. https://doi.org/10.1016/s0140-6736(20)31042-4 (2020).
Cavalcanti, A. B. et al. Hydroxychloroquine with or without azithromycin in mild-to-moderate Covid-19. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2019014 (2020).
Joyner, M. J. et al. Early safety indicators of COVID-19 convalescent plasma in 5000 patients. J. Clin. Investig. https://doi.org/10.1172/jci140200 (2020).
Joyner, M. J. et al. Safety update: COVID-19 convalescent plasma in 20,000 hospitalized patients. Mayo Clin. 95(9), 1888–1897. https://doi.org/10.1016/j.mayocp.2020.06.028 (2020).
Sanfilippo, F., La Rosa, V., Oliveri, F. & Astuto, M. Convalescent plasma for COVID-19: The risk of pulmonary embolism should not be underestimated!. Crit. Care 24, 1–2 (2020).
Pathak, E. B. Convalescent plasma is ineffective for covid-19. BMJ 371, m4072. https://doi.org/10.1136/bmj.m4072 (2020).
Recommendations for Investigational COVID-19 Convalescent Plasma. Food and Drug Administration. Accessed September 2, 2020, https://www.fda.gov/vaccines-blood-biologics/investigational-new-drug-ind-or-device-exemption-ide-process-cber/recommendations-investigational-covid-19-convalescent-plasma.
Agarwal, A. et al. Convalescent plasma in the management of moderate covid-19 in adults in India: Open label phase II multicentre randomised controlled trial (PLACID Trial). BMJ 371, m3939. https://doi.org/10.1136/bmj.m3939 (2020).
Gharbharan, A. et al. Convalescent plasma for COVID-19. A randomized clinical trial. medRxiv https://doi.org/10.1101/2020.07.01.20139857 (2020).
Simonovich, V. A. et al. A randomized trial of convalescent plasma in Covid-19 severe pneumonia. N. Engl. J. Med. 384(7), 619–629. https://doi.org/10.1056/NEJMoa2031304 (2021).
Acknowledgements
We are very grateful for the support in this trial by our fellow colleagues from physicians and nurses who have worked hard throughout the pandemic and specially during the trial period. Dr. Bandar Alnajdi, Dr. Hajer Alahmadi, Dr. Reem Almehzae, Dr. Layla Al Mutawa, Dr. Mahmood Tooq, Nurse Shafeeqa Hasan Yousif, Nurse Abdulla Mohammed Al-Ghadban, Nurse Hasan Hameed Hasan, Nurse Hamad Yusuf Habib, Nurse Mohamed Habib Hussain, Nurse Ali Abdulhadi Ali, Dr. Abdulla Alfadhel, Dr. Dana Alsharqi, Dr. Fatema Nabeel Nedham, Dr. Rashid Al Hasan, Dr. Ahmed Al Ansari, Dr. Maha Bukamal, Dr. Manar Al Sulaiti, Dr. Alya AlDoseri, Nurse Ghaida Fareed Ahmed Hasan, Nurse Elham Alfaraj, Nurse Abdulla Aman, Nursee Mariam Burshaeed, Nurse Manar Aldoseri, Nurse Ammar AlAthamneh, Nurse AlAnood Abdulrahman, Nurse Mariam Hasan Mohammed, Nurse Fatima Basheer, Nurse Ebtisam Meshal Saleh, Nurse Tareq Hasan Abdulrahman, Nurse Mariam Saleh Barshed Nurse Shouq Nader Hamada. We would also like to extend our gratitude to all health care workers and first responders worldwide and especially in Bahrain, who work day and night to protect the people and fight the virus.
Funding
This work was supported by the Ministry of Health Bahrain and the College of Surgeons in Ireland-Bahrain.
Author information
Authors and Affiliations
Contributions
A.A., A.A. and S.L.A. analyzed the data and wrote the manuscript. S.Y.A., A.Z., A.H., P.W. and M.A. performed data collection, S.O. interpreted data and edited the manuscript. R.C. performed the statistical analysis. M.A.Q. supervised data collection, data analysis and edited the manuscript. All authors reviewed and approved the final version of the manuscript. M.A. is the guarantor of this work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
AlQahtani, M., Abdulrahman, A., Almadani, A. et al. Randomized controlled trial of convalescent plasma therapy against standard therapy in patients with severe COVID-19 disease. Sci Rep 11, 9927 (2021). https://doi.org/10.1038/s41598-021-89444-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-89444-5
This article is cited by
-
Convalescent plasma and all-cause mortality of COVID-19 patients: systematic review and meta-analysis
Scientific Reports (2023)
-
Assessment of Transfusion Practices Among Doctors During COVID-19 Pandemic Using Questionnaire-Based Survey
Indian Journal of Hematology and Blood Transfusion (2023)
-
Effect of convalescent plasma transfusion on outcomes of coronavirus disease 2019: a meta-analysis with trial sequential analysis
Journal of Anesthesia (2023)
-
Differential efficacy and safety of anti-SARS-CoV-2 antibody therapies for the management of COVID-19: a systematic review and network meta-analysis
Infection (2023)
-
Randomized controlled trial of favipiravir, hydroxychloroquine, and standard care in patients with mild/moderate COVID-19 disease
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
