
Abstract
The Cu/Zn−superoxide dismutase (SOD1) is a ubiquitous enzyme that catalyzes the dismutation of superoxide radicals to oxygen and hydrogen peroxide. In addition to this principal reaction, the enzyme is known to catalyze, with various efficiencies, several redox side-reactions using alternative substrates, including biological thiols, all involving the catalytic copper in the enzyme’s active-site, which is relatively surface exposed. The accessibility and reactivity of the catalytic copper is known to increase upon SOD1 misfolding, structural alterations caused by a mutation or environmental stresses. These competing side-reactions can lead to the formation of particularly toxic ROS, which have been proposed to contribute to oxidative damage in amyotrophic lateral sclerosis (ALS), a neurodegenerative disease that affects motor neurons. Here, we demonstrated that metal-saturated SOD1WT (holo-SOD1WT) and a familial ALS (fALS) catalytically active SOD1 mutant, SOD1G93A, are capable, under defined metabolic circumstances, to generate cytotoxic quantities of H2O2 through cysteine (CSH)/glutathione (GSH) redox short-circuit. Such activity may drain GSH stores, therefore discharging cellular antioxidant potential. By analyzing the distribution of thiol compounds throughout the CNS, the location of potential hot-spots of ROS production can be deduced. These hot-spots may constitute the origin of oxidative damage to neurons in ALS.
Similar content being viewed by others
Introduction
The enzyme Cu/Zn-superoxide dismutase (SOD1) – a ubiquitous 32-kDa homodimeric protein – catalyzes the dismutation of superoxide radicals (O2∙−) to oxygen and hydrogen peroxide, and is critical for cell defense against reactive oxygen species (ROS)1. The Zn2+ ion plays a structural role and is required for an efficient SOD1 folding and long-term stability2, whereas active-site Cu2+ catalyzes O2∙− dismutation by alternating between the reduced and oxidized states (Reactions 1 and 2):
In the nervous tissue, SOD1 is present at exceedingly high concentrations (~1% of total protein, i.e., 100–200 μM)3 and is traditionally regarded as cytosolic protein4. However, in cell culture experiments, the SOD1 secretory pathways were shown to account for substantial quantities of extracellular SOD1, reaching ~ 20% of the intracellular SOD1 level5.
Over 150 mutations in the SOD1 gene have been described to cause amyotrophic lateral sclerosis (ALS) – a fatal neurodegenerative disease that affects the upper and lower motor neurons6,7 – in an autosomal dominant fashion, accounting for about 20% of the familial cases of ALS (fALS)8,9. The phenotypic hallmark of the SOD1-dependent ALS is the presence of amyloid aggregates of SOD1 in affected tissues, which are the ultimate outcome of SOD1 misfolding – the product of a gradual destabilization of the SOD1 structure due to the loss of metal ions and the reduction of the stabilizing intrasubunit disulfide bond10,11,12. Although fALS mutations are known to accelerate SOD1 misfolding, their presence is not obligatory; evidence exists that environmental stresses, such as oxidative stress, can trigger noxious misfolding in the wild-type SOD1 (SOD1WT)13,14,15,16,17. SOD1WT misfolding has been shown to occur in the spinal cord of sporadic (sALS) and non-SOD1 fALS patients13,14,18,19,20,21,22,23, and it was proposed that misfolded SOD1WT and mutant proteins (SOD1MUT) share a common pathogenic pathway in ALS18,23,24.
A remarkable feature of the misfolded SOD1 is its prion-like ability to self-propagate, such that a misfolded SOD1 (either SOD1WT or SOD1MUT) induces, via a yet-unknown mechanism, the misfolding of other, intact SOD1 molecules15,25. In ALS, the misfolding signal is believed to be transmitted via SOD1 secretory pathways in a cell-to-cell manner, thereby causing the disease to spread from the focal point of initiation throughout the spinal axis5,22,25,26,27,28,29,30. However, to date, the structural identity and the mechanism of neurotoxicity of the noxious SOD1 species remain obscure.
The fALS SOD1 mutants are categorized into wild-type-like (WTL), such as A4V, L38V, G37R, G41S, G72S, D76Y, D90A and G93A, that bind metals tightly and retain catalytic activity31, and those with impaired metal binding and reduced catalytic activity, such as H46R, H48Q, G85R, D124V, D125H, G127X and S134N32. The propensity to aggregate is one of the proposed noxious gain-of-functions of misfolded SOD1, although the relationships between aggregation and toxicity remain unclear. For instance, it was shown that the overexpression of fALS SOD1G93A mutant and CCS1 Cu-chaperone in mice exacerbated neuronal damage with no detectable SOD1 aggregation33. The heterogeneity of the structural/functional properties of fALS SOD1 mutants prompts the quest of a common denominator, a factor whose presence may play role in the disease etiology. Because of the dominant character of SOD1-dependent ALS, SOD1WT is present in the majority of fALS cases. Misfolding induced in the SOD1WT by interaction with a fALS SOD1MUT may confer the former toxic properties. An impaired redox chemistry catalyzed by the enzyme could be among such properties. Indeed, it has previously been demonstrated that in the SOD1WT the loss of structural Zn2+, a common attribute of SOD1 misfolding34, increases accessibility of the active site to substrates other than superoxide and enhances oxidizing ability of the catalytic copper, thus facilitating ROS production35,36. Oxidative stress is a common characteristic of ALS pathogenesis37,38,39,40.
In spite of being generally regarded as antioxidants, many biological thiol compounds, including cysteine (CSH) and homocysteine (Hcy), are toxic to various cell types, in particular to neurons, both in vitro and in vivo41,42,43,44. The cytotoxic effect of thiols is often biphasic, as they are not toxic at either low (<0.1 mM) or high (>1 mM) concentrations45. The mechanism underlying thiol cytotoxicity is not entirely understood, but their oxidation in the presence of free Cu2+ ions to produce toxic ROS was proposed as a plausible possibility45,46,47,48,49,50. In this scenario, thiol compounds act as pro-oxidants. The active site of SOD1 is relatively solvent-exposed2 and the accessibility of the catalytic Cu2+ increases upon SOD1 misfolding35,51,52. Consequently, the SOD1 active-site copper has been demonstrated to catalyze oxidation of thiol compounds53.
Here we show that a metal-saturated SOD1WT (holo-SOD1WT) and a fALS WTL mutant SOD1G93A catalyze thiol oxidation, with CSH as a preferable substrate, to produce cytotoxic quantities of H2O2. GSH, the major cellular antioxidant contributing to both non-enzymatic and enzyme-dependent defense against ROS37,38, is an inefficient substrate for the SOD1-catalyzed H2O2 formation. However, in the presence of small quantities of CSH (or cystine), GSH becomes potent pro-oxidant that exacerbates the effect of CSH by donating reducing equivalents to regenerate free CSH. The resulting redox short-circuit, which is capable of draining GSH stores, requires sub-physiological GSH concentrations to operate, and may potentially be triggered by a combination of chronic conditions, such as aging, and an acute oxidative stress. Analyzing the distribution of the thiol compounds across the CNS indicated that, in astrocytes, the unique combination of GSH and CSH levels renders these cells a potential hot spot of ROS formation, from which, under certain pathophysiologic circumstances, neuronal damage may arise.
Results
Holo-SOD1WT and WTL SOD1G93A are cytotoxic in the presence of thiol compounds
We tested the effect of CSH and other thiol compounds (namely, Hcy and GSH) on the viability of human neuroblastoma SH-SY5Y cells in the presence of extracellular holo-SOD1WT. When applied separately, neither holo-SOD1WT nor thiol compounds were cytotoxic at any of the tested concentrations (Fig. 1A,B). Conversely, their simultaneous application resulted in cytotoxicity, whose extent depended on the concentration of both holo-SOD1WT and the thiol compounds. Thiol cytotoxicity was not observed in the presence of metal-depleted SOD1WT (apo-SOD1WT) (Fig. 1B).

Thiol compounds induce holo-SOD1WT cytotoxicity. (A) The viability of SH-SY5Y cells was determined after 6 h of incubation in the absence or presence of 50 μM (monomer-based) holo-SOD1WT and the indicated concentrations of thiol compounds. (B) The viability of SH-SY5Y cells was determined after 6 h of incubation in the absence or presence of 0.5 mM Hcy and the indicated concentrations of holo-SOD1WT, apo-SOD1WT or CuCl2. (C) Time course of Hcy-induced cytotoxicity. The viability of SH-SY5Y cells was determined at the indicated time points in the absence or presence of 0.5 mM Hcy and either holo-SOD1WT (50 μM) or CuCl2 (5 μM). The viability of untreated cells was set to 100% (control). Results represent normalized means ± SD and are representative of at least three independent experiments performed in triplicates.
The fast kinetics of holo-SOD1WT-induced cell death in the presence of thiol compounds, as demonstrated by Hcy (Fig. 1C), suggest that the origin of cytotoxicity is extracellular, rather than due to the internalization of holo-SOD1WT by the cells54,55. Moreover, separating holo-SOD1WT from the cells using a 3-kDa cutoff membrane did not abolish cytotoxicity, although the kinetics of cell death were significantly delayed (Supplementary Fig. S1), demonstrating that cytotoxicity was facilitated by a diffusible low-molecular weight substance. The divalent metal ion chelator N,N,N’,N’-Tetrakis(2-pyridylmethyl)ethylenediamine (TPEN) abolished Hcy-induced holo-SOD1WT cytotoxicity in a dose-dependent manner (Fig. 2A), implying that metal ions are involved in the mechanism responsible for this cytotoxicity.

Thiol-dependent holo-SOD1WT cytotoxicity is irreversible and can be prevented by Cu2+ chelator or by catalase. (A) The viability of SH-SY5Y cells was determined after 6 h of incubation in the presence of 10 μM (monomer-based) holo-SOD1WT, 0.5 mM Hcy, and the indicated concentrations of TPEN. (B) The viability of SH-SY5Y cells was determined after 6 h of incubation with or without 500 U/ml catalase in the presence of 0.5 mM Hcy and the indicated concentrations of holo-SOD1WT. (C) SH-SY5Y cells were incubated with 50 μM holo-SOD1WT in the presence of 0.5 mM Hcy for the indicated periods of time, followed by further incubation in a fresh medium (without SOD1 or Hcy), and the viability was determined after 12 h of incubation in total. The viability of untreated cells was set to 100%. Results represent normalized means ± SD and are representative of at least three independent experiments performed in triplicates.
We also tested thiol-induced cytotoxicity in the presence of two fALS SOD1 mutants: WTL SOD1G93A and metal binding-impaired SOD1G85R, which were purified and reconstituted with metals using the same procedure as the holo-SOD1WT (see Methods). The SOD1G93A demonstrated a reduced cytotoxic potency as compared to the holo-SOD1WT, while metal-deficient SOD1G85R was essentially non-toxic at any of the tested concentrations of either Hcy or the protein (Fig. 3A,B).

Wild-type-like SOD1G93A but not metal-deficient SOD1G85R mutant is cytotoxic in the presence of thiol compounds. (A) The viability of SH-SY5Y cells was determined after 6 h of incubation in the absence (control) or presence of 50 μM (monomer-based) metal reconstituted holo-SOD1WT, SOD1G93A or SOD1G85R and the indicated concentrations of Hcy. (B) The viability of SH-SY5Y cells was determined after 6 h of incubation in the absence (control) or presence of 0.5 mM Hcy and the indicated concentrations of holo-SOD1WT, SOD1G93A or SOD1G85R. The viability of control cells was set to 100%. Results represent normalized means ± SD and are representative of at least three independent experiments performed in triplicates.
H2O2 is produced during the SOD1-catalyzed oxidation of thiol compounds
The toxicity of CSH and Hcy toward various cell types in the presence of free Cu2+ (as CuCl2) has been previously demonstrated and ascribed to the toxicity of H2O2 and/or of hydroxyl radicals produced during Cu2+-catalyzed thiols oxidation46,47,48,49,50. Although free Cu2+ was 5-fold more potent than holo-SOD1WT in inducing cell death (Fig. 1B), the kinetics of the Hcy-dependent cell death were almost identical in both cases (Fig. 1C). Moreover, the cytotoxicity of Hcy was completely abolished by adding catalase at all examined concentrations of holo-SOD1WT (Fig. 2B), indicating that this cytotoxicity was mediated by H2O2, and Cu2+ ions are involved in the mechanism of its formation. The luck of cytotoxicity in apo-SOD1WT and metal-deficient SOD1G85R is in line with this conclusion.
Extracellular H2O2 is an effective and potent neurotoxin, which can induce either apoptosis or necrosis in neurons56,57,58. Such cell death is associated with a burst-like increase in intracellular Ca2+ after a brief (several minutes) exposure to H2O2, as a result of the influx of Ca2+ from the extracellular space via redox-sensitive Ca2+ channels, such as TRPM2, which are expressed in neurons59. In agreement with this mechanism of neuronal death, a brief (<15 min) exposure of SH-SY5Y cells to extracellular holo-SOD1WT in the presence of Hcy irreversibly damaged the cells (Fig. 2C).
Since the plasma membrane is permeable to H2O260, extracellularly produced H2O2 may also aggravate existing oxidative stress conditions. Cells with an impaired ability to scavenge ROS are expected to be less resistant to an oxidative insult, including one of extracellular origin. Consistent with this idea, the cytotoxicity of holo-SOD1WT in the presence of Hcy was significantly potentiated in SH-SY5Y cells pre-treated with buthionine sulfoximine (BSO), an inhibitor of glutathione synthesis61 (Fig. 4).

Cells pretreated with BSO are sensitized to holo-SOD1WT cytotoxicity. The SH-SY5Y cells were pretreated for 18 h with or without (control) 1 mM BSO, followed by the addition of 0.3 mM Hcy and the indicated concentrations of holo-SOD1WT (monomer based). The cells were incubated for additional 6 h and viability was determined as described in the Methods. Results represent normalized means ± SD and are representative of at least three independent experiments performed in triplicates.
We then directly quantified the formation of H2O2 in the mixtures of SOD1 and thiol compounds using a phenol red/HRP H2O2 assay62. In cell experiments, all tested thiol compounds were toxic in the presence of holo-SOD1WT, with GSH being the most potent (Fig. 1A); in contrast, in a reconstituted reaction mixture, different rates of H2O2 formation were observed for different thiols (Fig. 5A and Supplementary Fig. S2). The fastest rates of oxidation were obtained with CSH, whereas Hcy was inferior and GSH failed to produce any significant amount of H2O2. Similar (but not identical) patterns of H2O2 production were observed in the presence of free Cu2+ (CuCl2) as a catalyst (Supplementary Fig. S2). In particular, the oxidation of Hcy in the presence of free Cu2+ exhibited a lag phase followed by a sigmoidal increase in the rate of H2O2 formation, whereas a monotonic asymptotic increase without lag phase was obtained in the presence of holo-SOD1WT (Supplementary Fig. S2C,D).

Metallated SOD1 catalyzes oxidation of thiol compounds to produce hydrogen peroxide. (A) Holo-SOD1WT (20 μM) was incubated in 10 mM glycyl-glycine buffer, pH 7.5, 50 mM NaCl at 37 °C in the presence of 0.3 mM thiol compounds, and H2O2 concentration was measured at the indicated time points. (B) As in (A), but with the addition of 200 μM cystine. (C) Metal reconstituted holo-SOD1WT, SOD1G93A or SOD1G85R (20 μM) were incubated in the presence of 0.3 mM CSH, and H2O2 concentration was measured at the indicated time points. The maximal concentration of H2O2 produced by holo-SOD1WT was set to 100%. Results represent means ± SD and are representative of at least three independent experiments performed in triplicates.
The apparent discrepancy between the cytotoxic potencies of the thiol compounds and their respective rates of H2O2 formation in the reconstituted reaction mixture (in the presence of catalytically active SOD1) was reconciled when thiol oxidation was performed in the presence of cystine (oxidized cysteine dimer, CS-SC, 200 μM) – a component of the standard DMEM cell growth medium used in our cytotoxicity experiments. In the presence of cystine, all thiol compounds, including GSH, demonstrated similar rates of holo-SOD1WT-catalyzed H2O2 production (Fig. 5B), which were consistent with their cytotoxic potencies (Fig. 1A). Comparable rates of H2O2 formation were also observed when the measurements were performed directly in the DMEM cell growth medium (Supplementary Fig. S3).
We also monitored the kinetics of CSH substrate consumption in the course of H2O2 formation by using Ellman’s reagent (DTNB). The oxidation of CSH went to a completion in the presence of the holo-SOD1WT, while no significant thiol oxidation was observed with the metal-depleted apo-SOD1WT, Fig. 6A. As expected, the rate of CSH oxidation decreased under the conditions of limiting oxygen, and the reaction came to a halt when the remaining O2 was consumed.

Cysteine-dependent redox short-circuit depletes GSH in the presence of holo-SOD1WT. (A) CSH (100 μM) was incubated at 37 °C in 20 mM Tris-HCl buffer, pH 7.5, 100 mM NaCl, in the absence (control) or presence of 10 μM holo-SOD1WT or apo-SOD1WT. In a separate experiment the reaction mixture was purged with N2 for 1 h before holo-SOD1WT addition. (B) The mixtures of CSH (100 μM) and the indicated concentrations of GSH were incubated in the presence of 10 μM holo-SOD1WT. The free thiol group concentration was measured at the indicated time points using DTNB reagent as described in the Methods. Results represent means ± SD and are representative of at least three independent experiments performed in triplicates.
In agreement with their respective cytotoxic potencies (Fig. 3), the WTL SOD1G93A was by 40% less efficient in catalyzing H2O2 formation in the presence of CSH as compared to the holo-SOD1WT, whereas metal-deficient SOD1G85R failed to produce any significant amounts of H2O2 (Fig. 5C). We suggest that the efficiency of thiol oxidation catalyzed by SOD1 is a function of its Cu2+ content, which in turn reflected the efficiency, with which Cu2+ was incorporation into the tested SOD1 variants upon the reconstitution (see Methods).
Although GSH cannot be oxidized directly by SOD1 to produce H2O2, it is apparently capable of reducing cystine to regenerate free CSH: 2GSH + CS-SC → GS-SG + 2CSH53,63. The CSH that is thereby produced enters another cycle of oxidation. Similarly to GSH, Hcy is capable of regenerating CSH from cystine53,63 (Fig. 5A,B). The total amount of H2O2 produced by a given amount of CSH or cystine was proportional to the amount of GSH present in the system (Fig. 7). The dependence of H2O2 accumulation on GSH concentrations, however, was bell-shaped, demonstrating that H2O2 buildup was inhibited at high GSH concentrations (Fig. 7C).

Glutathione promotes holo-SOD1WT-catalyzed H2O2 production via a cysteine-dependent redox short-circuit. Holo-SOD1WT (20 μM) was incubated at 37 °C in 10 mM glycyl-glycine buffer, pH 7.5, 50 mM NaCl in the presence of 50 μM CSH (A) or cystine (B) and the indicated concentrations of GSH. The H2O2 concentration was measured at the indicated time points. In the absence of GSH, cystine does not produce H2O2. Results represent means ± SD and are representative of at least three independent experiments performed in triplicates. (C) The area under the curve of the time courses of H2O2 formation were calculated from (A) and (B) (between 0 and 120 min) using Prism 6 software (GraphPad) and plotted against GSH concentration. The shaded area represents the region, in which oxidative stress may occur. Results represent means ± SE calculated from two independent experiments performed in triplicates.
To distinguish between the possibilities that the inhibitory effect of high GSH concentrations was caused by the ability of the thiol compounds (including GSH) to effectively scavenge H2O2 in a non-enzymatic Cu2+-independent process (2GSH + H2O2 → GS-SG + 2H2O)64, or alternatively, by the inhibition of H2O2 formation, we monitored the kinetics of free thiol oxidation in the CSH/GSH mixtures in the presence of holo-SOD1WT, Fig. 6B. Although GSH alone was resistant − at any of the tested concentrations − to the holo-SOD1WT-catalyzed oxidation (not shown), in the presence of small quantities of CSH (100 μM), GSH was rapidly and completely oxidized, demonstrating a marked ability of the redox short-circuit thus established to deplete GSH stores. At the GSH concentrations higher than 2 mM, however, no significant GSH oxidation was observed, consistent with the lack of H2O2 formation at high GSH concentrations (Fig. 7). The mechanism responsible for the inhibitory effect of high GSH concentrations on the rate of H2O2 formation is unclear, but it may involve a competitive inhibition of CSH binding to the SOD1 active-site by GSH65; this competition, however does not prevent much smaller substrates, such as superoxide, from accessing the catalytic site, therefore enabling the enzyme’s action at the physiological GSH concentrations.
Thiol compounds increase the accessibility of Cu2+ to external chelators
As high-affinity binding of metals to SOD1 is characterized by fast association–dissociation dynamics, strong chelators may interfere with the equilibrium2. To test whether thiol compounds increase the accessibility of holo-SOD1WT metals to external chelators, we used as chelator the colorimetric divalent metal sensor 4-(2-pyridylazo)resorcinol (PAR)36. The incubation of holo-SOD1WT with PAR in the presence of thiol compounds resulted in an increased formation of PAR-copper complexes (Fig. 8), while virtually no effect of thiol compounds was observed on the rate of PAR-Zn2+ complex formation (not shown).

Thiol compounds increase the accessibility of SOD1 copper to exogenous chelator. Holo-SOD1WT (50 μM) was incubated for 20 h at 37 °C with 100 μM PAR in the presence of the indicated concentrations of thiol compounds. PAR absorbance was measured at 490 nm and 520 nm, and the concentrations of released (PAR-complexed) metals were calculated as described in the Methods. The results were normalized to the amount of metals released in the absence of thiol compounds (fold change, mean ± SD) and are representative of at least three independent experiments performed in triplicates.
The mechanism by which the thiol compounds increase the accessibility of PAR to SOD1 copper is unknown. It is possible that thiol compounds reduce Cu2+ to Cu+ directly in the active site of SOD153, thus changing its coordination geometry to non-tetrahedral and facilitating ligand replacement (PAR binding)52. In addition, the formation of a binary thiol ̶ Cu+ complex could increase copper accessibility to PAR. The latter possibility, however, appears unlikely, since in line with the assumption that the thiol oxidation is catalyzed by the enzyme-bound Cu2+, CSH (similarly to other thiols) was incapable of extracting metal ions (Cu2+ or Zn2+) from the active site of holo-SOD1WT (Fig. 9). The filtrate of the reaction mixture containing 100 μM holo-SOD1WT and 0.5 mM CSH was analyzed for the metal presence using PAR. No labile metal ions were detected in the filtrate after the complete oxidation of the remaining thiols, indicating that, during the catalysis, no soluble binary complexes between the thiol compound and SOD1-derived Cu2+ were formed.

Thiol compounds form no binary complexes with SOD1WT-derived copper. Holo-SOD1WT (100 μM) was incubated in the absence or presence of 0.5 mM CSH at 37 °C in 10 mM glycyl-glycine buffer, pH 7.5, 50 mM NaCl for 15 min (corresponding to the peak of H2O2 formation, Fig. 3). The reaction mixture was separated from the protein by ultrafiltration, and the filtrate was analyzed for the presence of labile metal ions after the complete oxidation of the remaining thiols (10 h at 37 °C) by adding 100 μM PAR. Metal standards (0–10 μM) were added to separate reaction mixtures to calibrate the system. The concentration of metals was determined as described in the Methods.
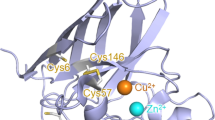
Thiol oxidation promotes the formation of the intramolecular disulfide in holo-SOD1WT
The H2O2 produced during holo-SOD1WT-catalyzed thiol oxidation would increase the oxidizing potential of the protein’s immediate environment, potentially affecting redox-sensitive surface-exposed groups of the protein. The SOD1 contains a highly conserved intramolecular disulfide bond (Cys57-Cys146), which is required for the long-term stability and full catalytic activity of SOD166. The formation of this bond in newly synthesized SOD1 is facilitated by CCS1 Cu-chaperone and is coupled to copper insertion66,67. The process is initiated by a copper-mediated (and CCS1-independent) oxidative step, in which one of the SOD1 cysteine residues becomes sulfenylated. The sulfenylation is subsequently resolved, in the presence of CCS1, to form a stable disulfide bond66. This disulfide, which is surface-exposed, is rather unusual feature for the protein found predominantly in the highly reducing cytosolic environment66,67,68. We tested the effect of holo-SOD1WT-catalyzed CSH oxidation on the redox status of the SOD1 intramolecular disulfide bond (Fig. 10). The holo-SOD1WT or apo-SOD1WT were initially reduced by a high concentration of DTT (5 mM) and then exposed to the increasing concentrations of CSH (0–1 mM). After blocking free cysteine groups with iodoacetamide to prevent disulfide bond scrambling, the disulfide status of the proteins was analyzed using a non-reducing SDS-PAGE69,70. Counterintuitively, considering the cysteine’s status as cellular reductant, increasing concentrations of CSH resulted in a progressively increasing proportion of holo-SOD1WT, but not apo-SOD1WT, containing oxidized disulfide bond (Fig. 10). We therefore concluded that the disulfide bond formation in the metallated SOD1 was facilitated, in the absence of CCS1, by ROS generated via the active-site Cu2+-catalyzed CSH oxidation. Understanding the physiological significance of such ability of metallated SOD1WT to ‘self-repair’ its disulfide bond in the presence of ubiquitous biological thiols, and how this ability is affected by fALS mutations, requires further investigation. It is tempting to speculate, however, that the ROS-producing reaction may contribute to the long-term stability of SOD1 by preserving the oxidized status of its conserved disulfide in a highly reducing atmosphere of cellular interior.

Cysteine promotes disulfide bond formation in metallated SOD1WT. Holo-SOD1WT or apo- SOD1WT (50 μM) were fully reduced by DTT (5 mM) and then exposed to the indicated concentrations of CSH for 30 min at 37 °C. After blocking free cysteine groups with iodoacetamide, the protein was separated by a non-reducing 12% SDS-PAGE. Data are representative of three independent experiments. Full-length gels are presented in Supplementary Figure S4.
Discussion
The active-site Cu2+ of holo-SOD1WT and fALS WTL SOD1G93A mutant is capable of catalyzing the oxidation of various thiol compounds, including CSH and Hcy, with a concomitant production of H2O2. Conversely, the ubiquitous cellular thiol antioxidant GSH is resistant to the SOD1-catalyzed oxidation. In the presence of small quantities of CSH or cystine, however, GSH becomes potent pro-oxidant that fuels the CSH-dependent H2O2 formation by reducing cystine back to CSH. The GSH/CSH mixtures, therefore, may constitute a potent redox short-circuit that, under certain pathophysiological metabolic circumstances, could drain − in the presence of metallated SOD1 − GSH stores and, thereby, discharge the antioxidant potential of the cell.
Further investigation is required to elucidate the significance of the SOD1-catalyzed thiol oxidation with the concomitant H2O2 production to ALS pathogenesis. The reaction involves catalytic copper and as such it characterizes both holo-SOD1WT and WTL fALS SOD1 mutants. Conversely, metal-deficient fALS mutants lack this ability, which raises the possibility that the described reaction is a peculiar catalytic ability of metallated SOD1 and it may not be related to ALS. Alternatively, one may hypothesize that ALS is facilitated by a synergistic interplay between a fALS SOD1 mutant and SOD1WT. According to this scenario, a catalytically inactive SOD1 mutant may act as a prion-like transmitter of the misfolding signal to efficiently spread the pathology, whereas the neurotoxic effect per se is caused by the ubiquitous SOD1WT, the structural and catalytic properties of which compromised by its interaction with the misfolded SOD1 mutant.
Under the assumption that the described mechanism of H2O2 production by thiol oxidation contributes to ALS pathogenesis, two important questions need to be addressed. The first one is whether the metabolic conditions required for the CSH-dependent short-circuit of GSH oxidation exist in CNS. A significant fraction of the cellular SOD1 may reach the extracellular space5. Although the cytotoxicity of extracellular SOD1 may represent a plausible mechanism of pathogenesis in disorders characterized by high concentrations of extracellular thiols (e.g., in homocystinuria, where plasma Hcy may reach 500 μM71), it is unlikely the pathogenic mechanism in ALS. This is because the extracellular, as demonstrated by CSF, concentrations of both GSH and CSH (or cystine) are very low72,73,74,75,76,77. Although the intracellular concentration of GSH in neurons is high77,78,79, the CSH content is very low (under the detection limit77) and, therefore, an efficient CSH-dependent GSH oxidation is unlikely in these cells. In astrocytes, by contrast, the intracellular content of GSH is one of the highest among mammalian cells (8–10 mM)77,78,79, and the level of CSH is substantial80. Supplemented with the ubiquitously expressed SOD181, this combination renders astrocytes a possible locale for the CSH-dependent short-circuiting of GSH oxidation to produce high quantities of H2O2.
In healthy astrocytes (8–10 mM GSH) a bell-shaped dependency of H2O2 formation on GSH concentration (Fig. 7C) would effectively prevent H2O2 accumulation. However, under certain pathophysiologic circumstances, including aging, the total glutathione and/or GSH/GS-SG ratio may drop significantly38,39,40. According to the bell-shaped dependence curve, this change may result in the production of substantial quantities of H2O2 (shaded area in Fig. 7C). The increased formation of ROS may compromise the neuro-supportive (neuro-protective) potential of astrocytes resulting in neurotoxicity82,83,84,85,86,87,88,89,90,91.
The second question is as to how misfolding may affect the SOD1 catalytic properties to facilitate disease. H2O2 is considered a relatively benign ROS due to the presence of H2O2-detoxifying enzymes, such as catalase and GSH peroxidases (although the performance of the latter is compromised by low GSH concentrations). It was previously demonstrated that SOD1 can utilize H2O2 as a sole substrate to produce hydroxyl radical51,92,93, a highly potent ROS that targets lipids, sugars, DNA bases, amino acids, and organic acids38,94, and against which no enzymatic defense exists. In this process, a reduction of SOD1 Cu2+ by H2O2 (Reaction 3) is followed by a Cu+ oxidation by another H2O2 in a Fenton-type reaction to generate a hydroxyl radical (Reaction 4)51,92,93:
The hydroxyl radical formation is markedly accelerated in the fALS SOD1 mutants, as compared with the native SOD1WT, a feature attributed to structural instability of the former manifested in the increased openness and accessibility of the active-site Cu2+ to substrates other than superoxide35,51,95. However, due to the low affinity of H2O2 to the SOD1 active site, non-physiologically high concentrations of H2O2 (10–20 mM) were required to produce substantial amounts of HO∙ 92, questioning the significance of this reaction to ALS pathogenesis. It was proposed that the HO∙ formation in the presence of H2O2 could, in principle, be facilitated by a cellular reductant other than H2O2 (whose identity remains unknown) capable of activating the active-site Cu2+ 35,51, therefore decreasing the amount of H2O2 required to produce HO∙ 57,59. We speculate that ubiquitous thiol compounds may play the role of such activating substance. Moreover, during thiol oxidation, H2O2 substrate of the second step (Reaction 4) is generated by SOD1 itself, thus the effective local concentration of H2O2 near the enzyme’s active-site is high. It could be especially true when considering the abnormal tendency of misfolded SOD1 to accumulate on particular locations in the cell96,97, hence further increasing local H2O2 concentration and accelerating the reaction by mass action.
Since the thiol oxidation by SOD1 is insensitive to the presence of Mn-SOD, it was concluded that superoxide is not formed as intermediate in this reaction53. Such resistance to the Mn-SOD presence, however, might results from the differences in the kinetics of the competing reactions or because peroxide may not be released as a free intermediate35. If superoxide is generated during the catalysis, it may react with NO to produce peroxynitrite, a powerful and much more (10,000 folds) stable oxidant than the hydroxyl radical38,98,99,100. Peroxynitrite exhibits a broad range of tissue-damaging effects, including lipid peroxidation, enzyme and ion channel inactivation, and inhibition of mitochondrial respiration,98 and the decomposition of peroxynitrite produces hydroxyl radicals101. It has been demonstrated that, in the absence of structural Zn2+, the coordination of Cu2+ in the active site of SOD1 changes, rendering Cu2+ a much more potent oxidant35,36. The Zn2+-deficient/Cu2+-SOD1WT and fALS WTL SOD1 mutants were shown to efficiently oxidize ascorbate, with a subsequent reoxidation of SOD1-Cu+ by oxygen to produce O2∙−, which combines with NO afterward to produce peroxynitrite35. We speculate that the peroxynitrite production by misfolded metallated SOD1 may benefit, similarly to the HO∙ formation, by the ability of SOD1 enzyme to accept ubiquitous biological thiols as an alternative substrate for cooper activation.
Methods
Purification of recombinant SOD1WT, SOD1G93A and SOD1G85R proteins and their reconstitution with metals
The human SOD1WT, SOD1G93A and SOD1G85R were produced in E. coli BL-21 grown without Zn2+ and Cu2+ supplements and purified to homogeneity under non-denaturing conditions, as described previously102. The MALDI-TOF mass spectrometry analysis of SOD1WT (Autoflex™ speed MALDI TOF/TOF mass spectrometer, Bruker, Germany) (Supplementary Fig. S5) and the purified protein’s low enzymatic activity 3.1 ± 0.2 U/mg, determined by a superoxide dismutase assay kit (Cayman Chemical, Ann Arbor, MI), indicated that this protein is demetallated (apo-SOD1WT) or metallated only partially. As shown previously, the recombinant SOD1WT produced in E. coli and reconstituted with metals by a dialysis after purification, regains its full catalytic activity and could be regarded as holo-SOD1WT 103,104,105. Indeed, we showed that saturation with Zn2+ and Cu2+ by the dialysis increased the catalytic activity of the recombinant SOD1WT by three order of magnitude (1200 ± 40 U/mg) to approach that of the SOD1 standard provided with the SOD1 activity assay kit (3700 ± 200 U/mg). The MALDI-TOF analysis revealed that the average molecular mass difference between the metallated and apo-SOD1WT is consistent, within the instrument’s sensitivity limit, with the addition of two metal ions per SOD1 monomer: 108 Da difference for the monomer peak (expected 129 Da) and 280 Da difference for the dimer peak (expected 258 Da), Supplementary Fig. S5.
Specifically, the purified SOD1 proteins were reconstituted with metals by an overnight dialysis against buffer A (50 mM Tris-HCl, pH 7.5, 0.1 M NaCl, and 10% glycerol) supplemented with ZnSO4 and CuSO4 (1 mM each). The dialyzed proteins were incubated for 1 h at 4 °C in the presence of 3 mM EDTA to chelate unbound and weakly bound metals, and then dialyzed overnight against buffer A. The dialyzed proteins were further buffer-exchanged using five 1:4 dilution (buffer A) and ultrafiltration (10 kDa cutoff, Amicon, Millipore, Burlington, MA) steps to remove residual metals and EDTA. The protein was then centrifuged at 110,000 × g for 1 h at 4 °C using an ultracentrifuge (Sorvall M120, Discovery, Thermo Fisher Scientific, UK) and stored, at the concentration 30 mg/ml, at −20 °C under argon until used. Protein concentration was measured by the Bradford method, using bovine serum albumin (fatty acid free, Sigma-Aldrich, Israel) as standard, and spectroscopically using a monomeric molar extinction coefficient at 280 nm of 5500 M−1·cm−1 (the two methods produced similar estimations).
Cell viability assay
SH-SY5Y cells were seeded in 96-well plates at a starting density of 1.5 × 104 cells/well in a high glucose Dulbecco’s modified Eagle medium (DMEM) supplemented with 1% L-glutamine, 1% penicillin-streptomycin, and 10% (vol/vol) fetal bovine serum (FBS), and incubated for 10 h prior experiments. The experiments were performed in the medium containing 2% FBS. At the end of the experiment, the cells were washed once with Hank’s balanced salt solution (HBSS) and cell viability was assessed by adding 20 µL of the CellTiter 96 AQueous One Solution reagent (Promega, Fitchburg, WI) to 100 μl HBSS, followed by incubation for 60 min at 37 °C, and the absorbance was measured at 490 nm using the Infinite 200 PRO plate-reader (Tekan, Switzerland). We found that mycoplasma infection decreased the cells susceptibility to SOD1 cytotoxicity; therefore, the cells were treated for two weeks prior the experiments with 25 μg/ml Plasmocin (Invitrogen, Sweden). We also found that the magnitude of the cytotoxic effect of SOD1 was inversely proportional to the cell density; therefore, the cell seeding conditions were adjusted to perform all the experiments at the final confluency of 70%.
In the compartmentalization experiments, SOD1 was separated from the cells cultured in a 24-well plate using a tightly fit dialysis insert equipped with a 3.5-kDa cutoff membrane (Thermo Fisher Scientific, UK).
Colorimetric metal assay
The holo-SOD1WT (50 μM, monomer based) was incubated for 20 h at 37 °C in 50 mM Hepes buffer, 50 mM NaCl, pH 7.4, in the presence of the indicated concentrations of thiol compounds and 100 μM divalent metal dye 4-(2-pyridylazo)resorcinol (PAR), in 90 μl total volume in 384-well plates covered with a transparent adhesive film (to prevent evaporation). The absorbance was measured at 490 nm and 520 nm using the Infinite 200 PRO plate-reader (Tekan, Switzerland) and the concentrations of dye-complexed copper and zinc ions were determined using the method described by Mulligan et al.106.
To determine the concentration of metals present in solution, 100 μM holo-SOD1WT were incubated for 15 min at 37 °C in 10 mM glycyl-glycine buffer, pH 7.5, 50 mM NaCl, in the absence or presence of 0.5 mM CSH. At the end of incubation, the protein was separated from the reaction mixture by ultra-filtration (5 kDa cutoff, Vivaspin 500, Sartorius, Germany) and the filtrate was analyzed for the presence of metals after a complete oxidation of the remaining thiols (10 h at 37 °C) by adding 100 μM PAR, as described above.
H2O2 assay
H2O2 was determined according to the method described by Pick et al.107 with some modifications. Briefly, a reagent solution containing 0.56 mM phenol red (Alfa Aesar, UK) and 17 U/ml horseradish peroxidase (Type II, Sigma-Aldrich, Israel) in 10 mM glycyl-glycine buffer, pH 7.5, 50 mM NaCl, was added at a 1:1 volumetric ratio to the analyzed sample, and the mixture was incubated at room temperature for 2 min (5 min produced similar results), followed by the addition of NaOH to the final concentration of 50 mM to reach pH 12.5. The absorbance was measured at 610 nm using the Infinite 200 PRO plate-reader (Tekan, Switzerland).
Free thiol assay
The analyzed samples were incubated for 15 min at room temperature in 20 mM Tris-HCl buffer, pH 7.5, NaCl 100 mM, 1 mM EDTA, 5 % DMSO in the presence of 0.5 mM Ellman’s Reagent (DTNB, 5,5′-Dithio-bis-(2-nitrobenzoic acid), Sigma-Aldrich, Israel), and the absorbance was measured at 412 nm using the Infinite 200 PRO plate-reader (Tekan, Switzerland).
Holo-SOD1WT disulfide bond status assay
The holo-SOD1WT or apo-SOD1WT (50 μM) were incubated for 0.5 h at 37 °C in 20 mM Tris buffer, pH 7.5, 0.1 M NaCl in the presence of 5 mM DTT to reduce the protein’s disulfide bonds. The residual DDT was removed from the reaction mixture by a buffer exchange using an ultrafiltration unit (10 kDa cutoff, modified PES, VWR International, USA). The fully reduced SOD1 was then incubated for 30 min at 37 °C in 20 mM Tris buffer, pH 7.5, 0.1 M NaCl in the presence of the indicated concentrations of CSH. The reaction mixture containing 0.8 mg/ml protein was then mixed (1:3, volumetric) with a SDS gel-loading solution (8% SDS, 40% Glycerol, 160 mM Tris-HCl, pH 6.8) and heated for 20 min at 65 °C. At the end of incubation, the protein was reacted at r.t with 20 mM iodoacetamide for 1 h and then separated by a non-reducing 12% SDS-PAGE. The gel was stained with Coomassie Brilliant Blue (Supplementary Fig. S4).
All data analyses were performed using the GraphPad Prism 6.0 software.
References
Rosen, D. R. et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62 (1993).
Leinartaitė, L., Saraboji, K., Nordlund, A., Logan, D. T. & Oliveberg, M. Folding Catalysis by Transient Coordination of Zn2+ to the Cu Ligands of the ALS-Associated Enzyme Cu/Zn Superoxide Dismutase 1. J. Am. Chem. Soc. 132, 13495–13504, https://doi.org/10.1021/ja1057136 (2010).
Pardo, C. A. et al. Superoxide dismutase is an abundant component in cell bodies, dendrites, and axons of motor neurons and in a subset of other neurons. Proc. Natl. Acad. Sci. USA 92, 954–958 (1995).
Zelko, I. N., Mariani, T. J. & Folz, R. J. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 33, 337–349 (2002).
Turner, B. J. et al. Impaired Extracellular Secretion of Mutant Superoxide Dismutase 1 Associates with Neurotoxicity in Familial Amyotrophic Lateral Sclerosis. The Journal of Neuroscience 25, 108–117, https://doi.org/10.1523/jneurosci.4253-04.2005 (2005).
Miller, R. G., Mitchell, J. D., Lyon, M. & Moore, D. H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Amyotroph. Lateral Scler. Other Motor Neuron Disord. 4, 191–206 (2003).
Han-Xiang, D. et al. Molecular dissection of ALS-associated toxicity of SOD1 in transgenic mice using an exon-fusion approach. Hum. Mol. Genet. 17, 2310–2319, https://doi.org/10.1093/hmg/ddn131 (2008).
Battistini, S. et al. SOD1 mutations in amyotrophic lateral sclerosis. J. Neurol. 252, 782–788, https://doi.org/10.1007/s00415-005-0742-y (2005).
Al-Chalabi, A. & Leigh, P. N. Recent advances in amyotrophic lateral sclerosis. Curr. Opin. Neurol. 13, 397–405 (2000).
Banci, L. et al. Metal-free superoxide dismutase forms soluble oligomers under physiological conditions: A possible general mechanism for familial ALS. Proc. Natl. Acad. Sci. USA 104, 11263–11267, https://doi.org/10.1073/pnas.0704307104 (2007).
Oztug Durer, Z. A. et al. Loss of Metal Ions, Disulfide Reduction and Mutations Related to Familial ALS Promote Formation of Amyloid-Like Aggregates from Superoxide Dismutase. PLoS One 4, e5004, https://doi.org/10.1371/journal.pone.0005004 (2009).
Ray, S. S. et al. An Intersubunit Disulfide Bond Prevents in Vitro Aggregation of a Superoxide Dismutase-1 Mutant Linked to Familial Amytrophic Lateral Sclerosis. Biochemistry 43, 4899–4905, https://doi.org/10.1021/bi030246r (2004).
Forsberg, K. et al. Novel Antibodies Reveal Inclusions Containing Non-Native SOD1 in Sporadic ALS Patients. PLoS One 5, e11552, https://doi.org/10.1371/journal.pone.0011552 (2010).
Pokrishevsky, E. et al. Aberrant Localization of FUS and TDP43 Is Associated with Misfolding of SOD1 in Amyotrophic Lateral Sclerosis. PLoS One 7, e35050, https://doi.org/10.1371/journal.pone.0035050 (2012).
Grad, L. I. et al. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc. Natl. Acad. Sci. USA 108, 16398–16403, https://doi.org/10.1073/pnas.1102645108 (2011).
Brotherton, T. E. et al. Localization of a toxic form of superoxide dismutase 1 protein to pathologically affected tissues in familial ALS. Proc. Natl. Acad. Sci. USA 109, 5505–5510, https://doi.org/10.1073/pnas.1115009109 (2012).
Graffmo, K. S. et al. Expression of wild-type human superoxide dismutase-1 in mice causes amyotrophic lateral sclerosis. Hum. Mol. Genet. 22, 51–60, https://doi.org/10.1093/hmg/dds399 (2013).
Bosco, D. A. et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 13, 1396–1403, https://doi.org/10.1038/nn.2660 (2010).
Forsberg, K., Andersen, P. M., Marklund, S. L. & Brannstrom, T. Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol. 121, 623–634, https://doi.org/10.1007/s00401-011-0805-3 (2011).
Zetterström, P., Graffmo, K. S., Andersen, P. M., Brännström, T. & Marklund, S. L. Proteins That Bind to Misfolded Mutant Superoxide Dismutase-1 in Spinal Cords from Transgenic Amyotrophic Lateral Sclerosis (ALS) Model Mice. J. Biol. Chem. 286, 20130–20136, https://doi.org/10.1074/jbc.M111.218842 (2011).
Guareschi, S. et al. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc. Natl. Acad. Sci. USA 109, 5074–5079, https://doi.org/10.1073/pnas.1115402109 (2012).
Grad, L. I. et al. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA 111, 3620–3625, https://doi.org/10.1073/pnas.1312245111 (2014).
Maier, M. et al. A human-derived antibody targets misfolded SOD1 and ameliorates motor symptoms in mouse models of amyotrophic lateral sclerosis. Sci. Transl. Med. 10, eaah3924, https://doi.org/10.1126/scitranslmed.aah3924 (2018).
Rotunno, M. S. & Bosco, D. A. An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front. Cell. Neurosci. 7, 253–253, https://doi.org/10.3389/fncel.2013.00253 (2013).
Munch, C., O’Brien, J. & Bertolotti, A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. USA 108, 3548–3553, https://doi.org/10.1073/pnas.1017275108 (2011).
Ayers, J. I., Fromholt, S. E., O’Neal, V. M., Diamond, J. H. & Borchelt, D. R. Prion-like propagation of mutant SOD1 misfolding and motor neuron disease spread along neuroanatomical pathways. Acta Neuropathol. 131, 103–114, https://doi.org/10.1007/s00401-015-1514-0 (2016).
Ravits, J. M. & La Spada, A. R. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology 73, 805–811, https://doi.org/10.1212/WNL.0b013e3181b6bbbd (2009).
Polymenidou, M. & Cleveland, Don W. The Seeds of Neurodegeneration: Prion-like Spreading in ALS. Cell 147, 498–508, https://doi.org/10.1016/j.cell.2011.10.011 (2011).
Basso, M. et al. Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J. Biol. Chem. 288, 15699–15711, https://doi.org/10.1074/jbc.M112.425066 (2013).
Urushitani, M. et al. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat. Neurosci. 9, 108–118, https://doi.org/10.1038/nn1603 (2006).
Borchelt, D. R. et al. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc. Natl. Acad. Sci. USA 91, 8292–8296 (1994).
Hayward, L. J. et al. Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 277, 15923–15931, https://doi.org/10.1074/jbc.M112087200 (2002).
Son, M. et al. Overexpression of CCS in G93A-SOD1 mice leads to accelerated neurological deficits with severe mitochondrial pathology. Proceedings of the National Academy of Sciences 104, 6072–6077, https://doi.org/10.1073/pnas.0610923104 (2007).
Nordlund, A. et al. Functional features cause misfolding of the ALS-provoking enzyme SOD1. Proceedings of the National Academy of Sciences 106, 9667–9672, https://doi.org/10.1073/pnas.0812046106 (2009).
Estevez, A. G. et al. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science 286, 2498–2500 (1999).
Crow, J. P., Sampson, J. B., Zhuang, Y., Thompson, J. A. & Beckman, J. S. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J. Neurochem. 69, 1936–1944 (1997).
Barber, S. C., Mead, R. J. & Shaw, P. J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1762, 1051–1067, https://doi.org/10.1016/j.bbadis.2006.03.008 (2006).
Aoyama, K. & Nakaki, T. Impaired Glutathione Synthesis in Neurodegeneration. Int. J. Mol. Sci. 14, 21021–21044, https://doi.org/10.3390/ijms141021021 (2013).
Chi, L., Ke, Y., Luo, C., Gozal, D. & Liu, R. Depletion of Reduced Glutathione Enhances Motor Neuron Degeneration in vitro and in vivo. Neuroscience 144, 991–1003, https://doi.org/10.1016/j.neuroscience.2006.09.064 (2007).
Vargas, M. R., Johnson, D. A. & Johnson, J. A. Decreased glutathione accelerates neurological deficit and mitochondrial pathology in familial ALS-linked hSOD1(G93A) mice model. Neurobiol. Dis. 43, 543–551, https://doi.org/10.1016/j.nbd.2011.04.025 (2011).
Munday, R. Toxicity of thiols and disulphides: involvement of free-radical species. Free Radic. Biol. Med. 7, 659–673 (1989).
Pean, A. R., Parsons, R. B., Waring, R. H., Williams, A. C. & Ramsden, D. B. Toxicity of sulphur-containing compounds to neuronal cell lines. J. Neurol. Sci. 129(Suppl), 107–108 (1995).
Janaky, R., Varga, V., Hermann, A., Saransaari, P. & Oja, S. S. Mechanisms of L-cysteine neurotoxicity. Neurochem. Res. 25, 1397–1405 (2000).
Longoni, A. et al. Homocysteine Induces Glial Reactivity in Adult Rat Astrocyte Cultures. Mol. Neurobiol. 55, 1966–1976, https://doi.org/10.1007/s12035-017-0463-0 (2018).
Kachur, A. V., Held, K. D., Koch, C. J. & Biaglow, J. E. Mechanism of production of hydroxyl radicals in the copper-catalyzed oxidation of dithiothreitol. Radiat. Res. 147, 409–415 (1997).
Wang, X. F. & Cynader, M. S. Pyruvate released by astrocytes protects neurons from copper-catalyzed cysteine neurotoxicity. J. Neurosci. 21, 3322–3331 (2001).
Hirashima, Y. et al. Homocysteine and copper induce cellular apoptosis via caspase activation and nuclear translocation of apoptosis-inducing factor in neuronal cell line SH-SY5Y. Neurosci. Res. 67, 300–306, https://doi.org/10.1016/j.neures.2010.04.013 (2010).
Endo, N. et al. Vitamin B6 suppresses apoptosis of NM-1 bovine endothelial cells induced by homocysteine and copper. Biochim. Biophys. Acta 1770, 571–577, https://doi.org/10.1016/j.bbagen.2006.11.009 (2007).
Starkebaum, G. & Harlan, J. M. Endothelial cell injury due to copper-catalyzed hydrogen peroxide generation from homocysteine. J. Clin. Invest. 77, 1370–1376 (1986).
Linnebank, M. et al. Binding of copper is a mechanism of homocysteine toxicity leading to COX deficiency and apoptosis in primary neurons, PC12 and SHSY-5Y cells. Neurobiol. Dis. 23, 725–730, https://doi.org/10.1016/j.nbd.2006.06.010 (2006).
Wiedau-Pazos, M. et al. Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science 271, 515–518 (1996).
Lyons, T. J. et al. Mutations in copper-zinc superoxide dismutase that cause amyotrophic lateral sclerosis alter the zinc binding site and the redox behavior of the protein. Proc. Natl. Acad. Sci. USA 93, 12240–12244 (1996).
Winterbourn, C. C., Peskin, A. V. & Parsons-Mair, H. N. Thiol Oxidase Activity of Copper,Zinc Superoxide Dismutase. J. Biol. Chem. 277, 1906–1911, https://doi.org/10.1074/jbc.M107256200 (2002).
Waelti, E. R. & Barton, M. Rapid endocytosis of copper-zinc superoxide dismutase into human endothelial cells: role for its vascular activity. Pharmacology 78, 198–201, https://doi.org/10.1159/000096598 (2006).
Filipe, P. et al. Cellular penetration of fluorescently labeled superoxide dismutases of various origins. Mol. Med. 5, 517–525 (1999).
Hoyt, K. R., Gallagher, A. J., Hastings, T. G. & Reynolds, I. J. Characterization of Hydrogen Peroxide Toxicity in Cultured Rat Forebrain Neurons. Neurochem. Res. 22, 333–340, https://doi.org/10.1023/A:1022403224901 (1997).
Thery, C., Chamak, B. & Mallat, M. Cytotoxic Effect of Brain Macrophages on Developing Neurons. Eur. J. Neurosci. 3, 1155–1164 (1991).
Whittemore, E. R., Loo, D. T. & Cotman, C. W. Exposure to hydrogen peroxide induces cell death via apoptosis in cultured rat cortical neurons. Neuroreport 5, 1485–1488 (1994).
Kaneko, S. et al. A Critical Role of TRPM2 in Neuronal Cell Death by Hydrogen Peroxide. J. Pharmacol. Sci. 101, 66–76, https://doi.org/10.1254/jphs.FP0060128 (2006).
Bienert, G. P., Schjoerring, J. K. & Jahn, T. P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 1758, 994–1003, https://doi.org/10.1016/j.bbamem.2006.02.015 (2006).
Maeda, H. et al. Effective treatment of advanced solid tumors by the combination of arsenic trioxide and L-buthionine-sulfoximine. Cell Death Differ. 11, 737–746, https://doi.org/10.1038/sj.cdd.4401389 (2004).
Johnston, P. A. et al. Development of a 384-Well Colorimetric Assay to Quantify Hydrogen Peroxide Generated by the Redox Cycling of Compounds in the Presence of Reducing Agents. Assay Drug Dev. Technol. 6, 505–518, https://doi.org/10.1089/adt.2008.151 (2008).
Hogg, N. The effect of cyst(e)ine on the auto-oxidation of homocysteine. Free Radical Biol. Med. 27, 28–33, https://doi.org/10.1016/S0891-5849(99)00029-5 (1999).
Hanaki, A. & Kamide, H. Participation of Cu (II) Ion in the Oxidation of Cysteine with Hydrogen Peroxide. Chem. Pharm. Bull. (Tokyo) 21, 1421–1425, https://doi.org/10.1248/cpb.21.1421 (1973).
Ciriolo, M. R., Battistoni, A., Falconi, M., Filomeni, G. & Rotilio, G. Role of the electrostatic loop of Cu,Zn superoxide dismutase in the copper uptake process. Eur. J. Biochem. 268, 737–742 (2001).
Fetherolf, M. M. et al. Copper-zinc superoxide dismutase is activated through a sulfenic acid intermediate at a copper ion entry site. J. Biol. Chem. 292, 12025–12040, https://doi.org/10.1074/jbc.M117.775981 (2017).
Furukawa, Y., Torres, A. S. & O’Halloran, T. V. Oxygen-induced maturation of SOD1: a key role for disulfide formation by the copper chaperone CCS. The EMBO Journal 23, 2872–2881, https://doi.org/10.1038/sj.emboj.7600276 (2004).
Hwang, C., Sinskey, A. J. & Lodish, H. F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 257, 1496–1502 (1992).
Karch, C. M., Prudencio, M., Winkler, D. D., Hart, P. J. & Borchelt, D. R. Role of mutant SOD1 disulfide oxidation and aggregation in the pathogenesis of familial ALS. Proceedings of the National Academy of Sciences 106, 7774–7779, https://doi.org/10.1073/pnas.0902505106 (2009).
Álvarez-Zaldiernas, C. et al. Cellular Redox Systems Impact the Aggregation of Cu,Zn Superoxide Dismutase Linked to Familial Amyotrophic Lateral Sclerosis. J. Biol. Chem. 291, 17197–17208, https://doi.org/10.1074/jbc.M115.708230 (2016).
Veeranki, S. & Tyagi, S. C. Defective Homocysteine Metabolism: Potential Implications for Skeletal Muscle Malfunction. Int. J. Mol. Sci. 14, 15074–15091, https://doi.org/10.3390/ijms140715074 (2013).
Kaiser, E. et al. Cerebrospinal Fluid Concentrations of Functionally Important Amino Acids and Metabolic Compounds in Patients with Mild Cognitive Impairment and Alzheimer’s Disease. Neurodegenerative Diseases 7, 251–259 (2010).
Jones, C. M., Smith, M. & Henderson, M. J. Reference data for cerebrospinal fluid and the utility of amino acid measurement for the diagnosis of inborn errors of metabolism. Ann. Clin. Biochem. 43, 63–66, https://doi.org/10.1258/000456306775141759 (2006).
Heiblim, D. I., Evans, H. E., Glass, L. & Agbayani, M. M. Amino acid concentrations in cerebrospinal fluid. Arch. Neurol. 35, 765–768, https://doi.org/10.1001/archneur.1978.00500350069015 (1978).
Castagna, A. et al. Cerebrospinal fluid S-adenosylmethionine (SAMe) and glutathione concentrations in HIV infection: effect of parenteral treatment with SAMe. Neurology 45, 1678–1683 (1995).
Katz, M. et al. Cerebrospinal fluid concentrations of N-acetylcysteine after oral administration in Parkinson’s disease. Parkinsonism Relat. Disord. 21, 500–503, https://doi.org/10.1016/j.parkreldis.2015.02.020 (2015).
Wang, X. F. & Cynader, M. S. Astrocytes provide cysteine to neurons by releasing glutathione. J. Neurochem. 74, 1434–1442 (2000).
Sagara, J. I., Miura, K. & Bannai, S. Maintenance of neuronal glutathione by glial cells. J. Neurochem. 61, 1672–1676 (1993).
Vitvitsky, V., Thomas, M., Ghorpade, A., Gendelman, H. E. & Banerjee, R. A functional transsulfuration pathway in the brain links to glutathione homeostasis. J. Biol. Chem. 281, 35785–35793, https://doi.org/10.1074/jbc.M602799200 (2006).
McBean, G. J. Cysteine, Glutathione, and Thiol Redox Balance in Astrocytes. Antioxidants (Basel, Switzerland) 6, https://doi.org/10.3390/antiox6030062 (2017).
Sun, S. et al. Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc. Natl. Acad. Sci. USA 112, E6993–E7002, https://doi.org/10.1073/pnas.1520639112 (2015).
Ferraiuolo, L. et al. Dysregulation of astrocyte–motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 134, 2627–2641, https://doi.org/10.1093/brain/awr193 (2011).
Zhao, W. et al. Extracellular Mutant SOD1 Induces Microglial-Mediated Motoneuron Injury. Glia 58, 231–243, https://doi.org/10.1002/glia.20919 (2010).
Yamanaka, K. et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11, 251–253, https://doi.org/10.1038/nn2047 (2008).
Lobsiger, C. S. & Cleveland, D. W. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat. Neurosci. 10, 1355–1360, https://doi.org/10.1038/nn1988 (2007).
Yamanaka, K. et al. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc. Natl. Acad. Sci. USA 105, 7594–7599, https://doi.org/10.1073/pnas.0802556105 (2008).
Phatnani, H. P. et al. Intricate interplay between astrocytes and motor neurons in ALS. Proceedings of the National Academy of Sciences 110, E756–E765, https://doi.org/10.1073/pnas.1222361110 (2013).
Hirrlinger, J. & Dringen, R. The cytosolic redox state of astrocytes: Maintenance, regulation and functional implications for metabolite trafficking. Brain Res. Rev. 63, 177–188, https://doi.org/10.1016/j.brainresrev.2009.10.003 (2010).
Weber, B. & Barros, L. F. The Astrocyte: Powerhouse and Recycling Center. Cold Spring Harb. Perspect. Biol. 7, https://doi.org/10.1101/cshperspect.a020396 (2015).
Haskew-Layton, R. E. et al. Controlled enzymatic production of astrocytic hydrogen peroxide protects neurons from oxidative stress via an Nrf2-independent pathway. Proceedings of the National Academy of Sciences 107, 17385–17390, https://doi.org/10.1073/pnas.1003996107 (2010).
Sagara, J., Makino, N. & Bannai, S. Glutathione efflux from cultured astrocytes. J. Neurochem. 66, 1876–1881 (1996).
Yim, M. B., Chock, P. B. & Stadtman, E. R. Copper, zinc superoxide dismutase catalyzes hydroxyl radical production from hydrogen peroxide. Proceedings of the National Academy of Sciences 87, 5006–5010 (1990).
Cabelli, D. E., Allen, D., Bielski, B. H. & Holcman, J. The interaction between Cu(I) superoxide dismutase and hydrogen peroxide. J. Biol. Chem. 264, 9967–9971 (1989).
Halliwell, B. & Gutteridge, J. M. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J 219, 1–14 (1984).
Goto, J. J., Gralla, E. B., Valentine, J. S. & Cabelli, D. E. Reactions of hydrogen peroxide with familial amyotrophic lateral sclerosis mutant human copper-zinc superoxide dismutases studied by pulse radiolysis. J. Biol. Chem. 273, 30104–30109 (1998).
Vehviläinen, P., Koistinaho, J. & Gundars, G. Mechanisms of mutant SOD1 induced mitochondrial toxicity in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 8, https://doi.org/10.3389/fncel.2014.00126 (2014).
Rakhit, R. et al. An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nat. Med. 13, 754–759, https://doi.org/10.1038/nm1559 (2007).
Virag, L., Szabo, E., Gergely, P. & Szabo, C. Peroxynitrite-induced cytotoxicity: mechanism and opportunities for intervention. Toxicol. Lett. 140–141, 113–124 (2003).
Pacher, P., Beckman, J. S. & Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 87, 315–424, https://doi.org/10.1152/physrev.00029.2006 (2007).
Beckman, J. S. Peroxynitrite versus hydroxyl radical: the role of nitric oxide in superoxide-dependent cerebral injury. Ann. N. Y. Acad. Sci. 738, 69–75 (1994).
Pou, S., Nguyen, S. Y., Gladwell, T. & Rosen, G. M. Does peroxynitrite generate hydroxyl radical? Biochim. Biophys. Acta 1244, 62–68 (1995).
Banerjee, V. et al. Superoxide Dismutase 1 (SOD1)-Derived Peptide Inhibits Amyloid Aggregation of Familial Amyotrophic Lateral Sclerosis SOD1 Mutants. ACS Chem. Neurosci. 7, 1595–1606, https://doi.org/10.1021/acschemneuro.6b00227 (2016).
Beem, K. M., Rich, W. E. & Rajagopalan, K. V. Total reconstitution of copper-zinc superoxide dismutase. J. Biol. Chem. 249, 7298–7305 (1974).
Galaleldeen, A. et al. Structural and Biophysical Properties of Metal-Free Pathogenic SOD1 Mutants A4V and G93A. Arch. Biochem. Biophys. 492, 40–47, https://doi.org/10.1016/j.abb.2009.09.020 (2009).
Goto, J. J. et al. Loss of in vitro metal ion binding specificity in mutant copper-zinc superoxide dismutases associated with familial amyotrophic lateral sclerosis. J. Biol. Chem. 275, 1007–1014 (2000).
Mulligan, V. K., Kerman, A., Ho, S. & Chakrabartty, A. Denaturational stress induces formation of zinc-deficient monomers of Cu,Zn superoxide dismutase: implications for pathogenesis in amyotrophic lateral sclerosis. J. Mol. Biol. 383, 424–436, https://doi.org/10.1016/j.jmb.2008.08.024 (2008).
Pick, E. & Keisari, Y. A simple colorimetric method for the measurement of hydrogen peroxide produced by cells in culture. J. Immunol. Methods 38, 161–170 (1980).
Acknowledgements
We thank Liana Shimshilashvili, Dr. Ehud Ohana, Dr. Daniel Gitler, Dr. Uziel Hadad, Dr. N. Gabriel Lemcoff and Prof. Dan Meyerstein for help with this study. This work was supported by 343/16 grant to S.E. from the Israel Science Foundation.
Author information
Authors and Affiliations
Contributions
Conceptualization, S.E., S.B.; Methodology, S.E., S.B.; Investigation, S.B., N.C., T.Z., R.M. and M.V.; Writing – Original Draft, S.E.; Writing – Review & Editing, S.E., A.I., Z.B., and S.B.; Resources, S.E., A.I. and Z.B.; Supervision, S.E., A.I. and Z.B.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bakavayev, S., Chetrit, N., Zvagelsky, T. et al. Cu/Zn-superoxide dismutase and wild-type like fALS SOD1 mutants produce cytotoxic quantities of H2O2 via cysteine-dependent redox short-circuit. Sci Rep 9, 10826 (2019). https://doi.org/10.1038/s41598-019-47326-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-47326-x
This article is cited by
-
MIF homolog d-dopachrome tautomerase (D-DT/MIF-2) does not inhibit accumulation and toxicity of misfolded SOD1
Scientific Reports (2022)
-
SOD1 gains pro-oxidant activity upon aberrant oligomerization: change in enzymatic activity by intramolecular disulfide bond cleavage
Scientific Reports (2022)
-
Dysregulation of IGF-1/GLP-1 signaling in the progression of ALS: potential target activators and influences on neurological dysfunctions
Neurological Sciences (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
