
Abstract
Torrubiellins A (1) and B (2), two new dimeric anthraquinones were isolated from the leafhopper pathogenic fungus Torrubiella sp. BCC 28517. The structures of the new compounds were elucidated by analyses of the NMR spectroscopic and mass spectrometry data. Torrubiellin B (2) exhibited broad range of biological activities.
Similar content being viewed by others
Introduction
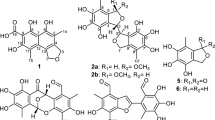
Torrubiella is a genus of arthropod-pathogenic fungi that attacks spiders, scale-insects and hoppers.1 Recently, a reclassification of the genus Torrubiella (family Clavicipitaceae) based on a multi-gene phylogeny was reported.1 Three scale insect pathogens T. luteorostrata, T. tenuis and T. petcii were assigned to the newly described genera Conoideocrella and Orbiocrella, while spider-pathogenic species (largest group) and hopper pathogens (minor group) were retained as Torrubiella, but they were assigned to family Cordycipitaceae. Two scale insect pathogenic species, Conoideocrella luteorostrata (formerly T. luteorostrata) and C. tenuis (formerly T. tenuis) have recently been investigated as sources of novel bioactive compounds, such as paecilodepsipeptides2 and conoideocrellides3 (hexadepsipeptides), luteorides4 (prenylated tryptophan analogs), isocoumarin glucosides,3, 5 bioxanthracenes,3 and hopane-type triterpenes.3 In contrast, there has been only one report on bioactive compounds from the spider-pathogenic Torrubiella, torrubiellones A–D (pyridone and tetramic acid derivatives),6 and no report on leafhopper pathogens. As part of our research program on novel bioactive compounds from arthropod-pathogenic fungi, we selected a leafhopper-pathogenic Torrubiella sp., strain BCC 28517, for large-scale fermentation and chemical studies. A mycelial extract of this strain exhibited cytotoxicity to oral cavity cancer cells (KB) and small-cell lung cancer cells (NCI-H187) with respective IC50 values of 1.66 and 0.79 μg ml−1. We report here the isolation, structure elucidation and biological activities of two new anthraquinone dimers, torrubiellins A (1) and B (2), together with the known anthraquinones, chrysophanol (3),7, 8 aloe-emodin (4)7 and emodin (5) (Figure 1).7, 9

Structures of compounds 1–5.
Results and discussion
Torrubiellin A (1) was obtained as a dark-brown solid, and the molecular formula was established as C30H20O8, from the protonated quasi-molecular ion peak in the HRESIMS. The 13C NMR, DEPT135 and HMQC spectroscopic data indicated the presence of 30 carbons categorized as three conjugated ketones (δC 192.5, 190.1 and 183.1), fourteen quaternary sp2 carbons (δC 172.3–102.6), eight sp2 methines (δC 142.4–119.0), an oxygenated methine (δC 71.9), an aliphatic quaternary carbon, a methine, a methylene and a methyl group (Table 1). In addition, the 1H NMR spectrum showed the presence of four chelated phenolic hydroxy groups. The structure of the A-ring was deduced from the COSY correlations of three aromatic protons H-5/H-6/H-7, and the HMBC correlations from these protons and a chelated phenolic OH (δC 12.12, 8-OH) to carbons of the trisubstituted benzene (A-ring) (Figure 2). Intense HMBC correlation from H-5 to a conjugated ketone carbon at δC 183.1 (C-10) required the connection of this ketone to C-10a. The presence of a chelated OH (8-OH) suggested that C-8a was also connected to a ketone (C-9, δC 192.5 or 190.1). The structure of the CDEFG-ring system was established on the basis of the HMBC correlations. Thus, HMBC correlations from 1′-OH, H-2′, H-4′ and H3-11′ to carbons of the tetrasubstituted benzene revealed the G-ring. HMBC correlations from an oxymethine H-10′ to C-4′, C-9a′, C-5′, C-8a′ and C-10a′, and the correlations from H-4′ and H-5′ to the methine carbon (C-10′) indicated that this methine linked G-ring (C-4a′) and E-ring (C-10a′). The chelated hydroxy groups, 1′-OH and 8′-OH, requested the presence of a ketone (δC 190.1 or 192.5, C-9′) functionality to link C-9a′ and C-8a′. The remaining pentasubstituted benzene (C-ring) was suggested based on the HMBC correlations from H-2 to C-1, C-2 and C-4, and the correlations from 1-OH to C-1, C-2 and C-9a, although no correlation to the quaternary sp2 carbon at δC 129.5 (C-4a) was observed. The connection of C-4 (C-ring) and C-5′ (E-ring) was revealed by the HMBC correlations from H-5′ and H-6′ to C-4. HMBC correlations from nonequivalent methylene protons H2-11 to C-2, C-3, C-4, C-5′, C-8a′, C-10′ and C-10a′ demonstrated that this methylene group linked C-3 and C-10a′ to constitute the D-ring. On the basis of these data, two fragments, A-ring (attached with two ketone carbons) and CDEFG-ring, covering all atoms required from the molecular formula. Two possible linkage patterns to constitute the ABC-ring, 1,8-dihydroxyanthraquinone and 1,5-dihydroxyanthraquinone, were proposed. The latter structure should be ruled out due to the large chemical shift differences of the two ketone carbons; δC 183.1 (C-9) and δC 192.5 or 190.1 (C-10). These carbon chemical shifts were consistent with those of monomeric 1,8-dihydroxyanthraquinone derivatives, such as 4 (C-9 δC 191.5, C-10 δC 181.4, in DMSO-d6).8 The relative configuration of 1 was assigned on the basis of the NOESY correlations (Figure 3). The cross-peak for H-2/Hα-11 (δH 2.89) was much more intense than that for H-2/Hβ-11 (δH 3.07). Intense NOESY correlations H-10′/Hα-11 and H-10′/H-5′ demonstrated the relative configuration of the three contiguous chiral carbon centers C-10′, C-10a′ and C-5′ (Figure 3).

HMBC correlations for 1.

Key NOESY correlations for 1.
The molecular formula of torrubiellin B (2) was determined by HRESIMS as C30H20O10, which has two more oxygen atoms than 1. The 1H and 13C NMR spectra suggested close structural resemblance to 1, although the 1H NMR spectrum of 2 in DMSO-d6 (used due to the solubility reason) did not exhibit any peaks for phenolic OH. Interpretation of the 2D NMR (COSY, HMQC and HMBC) data revealed that the BCDEFG-ring structure was identical to 1. The significant differences were the 1H and 13C NMR spectroscopic data for the A-ring, consisting of a methine (δH 7.27, s; δC 110.0) and five quaternary carbons. The methine proton was assigned to H-5 on the basis of its HMBC correlations to four quaternary carbons (C-10a, C-6, C-7 and C-8a) and C-10 (δC 182.6). In addition, a weak4 J correlation from H-5 to C-9 (δC 190.9) further supported the anthraquinone structure. Although HMBC correlation to C-8 was absent, the chemical shifts of C-6 (δC 153.6), C-7 (δC 139.9) and C-8 (δC 151.8) were consistent with the 6,7,8-trihydroxy functionalities. The NOESY spectrum of 2 exhibited similar key correlations as 1, H-2/Hα-11, H-10′/Hα-11 and H-10′/H-5′, which indicated the identical relative configuration. Torrubiellin B (2) was therefore identified as the 6,7-dihydroxy derivative of 1.
The co-occurrence of anthraquinone monomers 3–5 strongly suggested that 1 and 2 were biogenetically derived from two anthraquinones (ABC-ring and EFG-ring). Although a variety of dimeric anthraquinones have been reported as fungal secondary metabolites, the linkage pattern as shown for torrubiellins, condensation at C-4–C-5′ and C-11–C-10a′, is quite rare. The only but closely related compounds are uredinorubellins I and II, which were isolated from the plant pathogenic fungus Ramuralia uredinicola.10 Uredinorubellin II is the 8′,8′a-dihydro-8′a-hydroxy derivative of 1, while uredinorubellin I possesses additionally 6-OH functionality. Rubellins A–D, isolated from Ramuralia collo-cygni and reported before uredinorubellins, are F-ring modified (degraded) analogs probably derived from uredinorubellins.10, 11, 12
As torrubiellins A and B possess a rare chemical skeleton, they were subjected to several bioassay protocols in our research center (Table 2). Torrubiellin B (2) exhibited higher biological activity than 1 in all the tests. Although torrubiellin B (2) exhibited significant antimalarial (Plasmodium falciparum), antifungal (Candida albicans) and antibacterial (Bacillus cereus) activities, it also showed comparable magnitude of cytotixicity to cancer cell lines. Related compounds, uredinorubellins I and II, are reported to exhibit photodynamic activity influencing cell viability in three mammalian cell lines, HIG82, HT29 and J774A.1, as well as antibacterial activity against Staphylococcus aureus.10
Methods
General experimental procedures
Melting points were measured with an IA9100 digital melting point apparatus (Electrothermal, Essex, UK). Optical rotations were measured with a P-1030 digital polarimeter (JASCO, Tokyo, Japan). UV spectra were recorded on a Cintra 404 spectrophotometer (GBC Scientific Equipment, Braeside, VIC, Australia). FTIR spectra were taken on an ALPHA spectrometer (Bruker, Bremen, Germany). NMR spectra were recorded on DRX400 and AV500D spectrometers (Bruker). ESITOF mass spectra were measured with a micrOTOF mass spectrometer (Bruker).
Fungal material
The fungus used in this study was isolated from a leafhopper (Hemiptera) collected in Khlong Lan National Park, Kamphaeng Phet Province, Thailand, by one of the authors (KT). This fungus was deposited in the BIOTEC Culture Collection (BCC) on 27 November 2007 as BCC 28517. On the basis of the sequence data of the ITS rDNA, the strain was assigned to the genus Torrubiella within the family Cordycipitaceae.1
Fermentation and isolation
The fungus BCC 28517 was maintained on potato dextrose agar at 25 °C. The agar plugs (1 × 1 cm) were cut into small pieces and inoculated into 4 × 250 ml Erlenmeyer flasks containing 25 ml of potato dextrose broth (potato starch 4.0 g l−1, dextrose 20.0 g l−1) and incubated at 25 °C for 4 days on a rotary shaker (200 r.p.m.). Each primary seed culture was transferred into a 1 l Erlenmeyer flask containing 250 ml of potato dextrose broth, and incubated at 25 °C for 4 days on a rotary shaker (200 r.p.m.). Each 25 ml portion of the seed culture was added into 40 × 1 l Erlenmeyer flasks containing 250 ml of M102 medium (sucrose 30 g l−1, malt extract 20 g l−1, Bacto-peptone 2.0 g l−1, yeast extract 1.0 g l−1, KCl 0.5 g l−1, MgSO4·7H2O 0.5 g l−1 and KH2PO4 0.5 g l−1), and the final fermentation was carried out at 25 °C for 15 days on rotary shakers (200 r.p.m.). The culture was filtered to separate broth (filtrate) and mycelia (residue). The filtrate was extracted with EtOAc (2 × 6 l), and the combined organic layer was concentrated under reduced pressure to obtain a dark-brown gum (2.36 g, broth extract). The wet mycelia were macerated in MeOH (1 l, RT, 2 days) and filtered. Hexanes (800 ml) and H2O (40 ml) were added to the filtrate, and the layers were separated. The aqueous MeOH layer was concentrated by evaporation, and H2O (150 ml) was added to the residue, which was then extracted with EtOAc (2 × 1.2 l). The EtOAc solution was concentrated under reduced pressure to obtain a brown gum (3.66 g, mycelial extract). The mycelial extract was triturated in MeOH (2 ml)/CH2Cl2 (5 ml) and filtered. The filtrate was concentrated to leave a brown gum (3.50 g), which was subjected to fractionation by column chromatography on Sephadex LH-20 (3.5 × 50 cm, MeOH) to obtain four pooled fractions, wherein fractions 3 (20.9 mg) and 4 (34.2 mg) were identified as 2. Fraction 2 (240 mg) was subjected to preparative high performance liquid chromatography (Grom-Sil 120 ODS-4 HE, 20 × 150 mm, 5 μm; isocratic elution with MeCN/H2O=65:35, 0–13 min, then MeCN/H2O=90:10; flow rate 8 ml min−1) to furnish 4 (3.4 mg, tR 7 min), 5 (2.6 mg, tR 10 min), 2 (27.2 mg, tR 12 min), 3 (13.9 mg, tR 15 min) and 1 (6.1 mg, tR 20 min). The broth extract was also fractionated using similar procedures to obtain 2 (12.5 mg), 3 (2.3 mg) and 1 (2.0 mg).
Torrubiellin A ( 1): dark-brown solid; m.p. 215–217 °C; [α]25D +148 (c 0.075, CHCl3); UV/vis (MeOH) λmax (log ɛ) 227 (4.45), 253 (4.21), 277 sh (4.01), 289 sh (3.93), 396 (3.96), 416 (3.97), 438 sh (3.89), 463 sh (3.78) nm; IR (ATR) νmax 1626, 1572, 1281 cm−1; 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3) data, see Table 1; HRMS (ESI-TOF, positive) m/z 509.1228 [M+H]+ (calculated for C30H21O8, 509.1231).
Torrubiellin B ( 2): dark-brown solid; m.p. 251–253 °C (dec.); [α]27D +189 (c 0.025, CHCl3); UV/vis (MeOH) λmax (log ɛ) 218 (4.40), 287 (4.31), 395 (4.20), 417 (4.18), 460 sh (3.98) nm; IR (ATR) νmax 3424, 1623, 1588, 1572, 1275 cm−1; 1H NMR (400 MHz, DMSO-d6) and 13C NMR (100 MHz, DMSO-d6) data, see Table 1; HRMS (ESI-TOF, negative) m/z 539.0971 [M − H]− (calculated for C30H19O10, 539.0984).
Biological assays
Cytotoxic activities against human cancer cell lines (KB, NCI-H187 and MCF-7), antifungal (Candida albicans) and antibacterial (Bacillus cereus) activities were evaluated using the resazurin microplate assay.13 Antiplasmodial activity (Plasmodium falciparum K1, multi-drug resistant strain) was performed using the microculture radioisotope technique.14 Antimycobacterial activity (Mycobacterium tuberculosis H37Ra) was performed using the green fluorescent protein microplate assay.15
References
Johnson, D. et al. Systematics and evolution of the genus Torrubiella (Hypocreales, Ascomycota). Mycol. Res. 113, 279–289 (2009).
Isaka, M., Palasarn, S., Kocharin, K. & Hywel-Jones, N. L. Comparison of the bioactive secondary metabolites from the scale insect pathogens, anamorph Paecilomyces cinnamomeus, and teleomorph Torrubiella luteorostrata. J. Antibiot. 60, 577–581 (2007).
Isaka, M., Palasarn, S., Supothina, S., Komwijit, S. & Luangsa-ard, J. J. Bioactive compounds from the scale insect pathogenic fungus Conoideocrella tenuis BCC 18627. J. Nat. Prod. 74, 782–789 (2011).
Asai, T., Yamamoto, T. & Oshima, Y. Histone deacetylase inhibitor induced the production of three novel prenylated tryptophan analogs in the entomopathogenic fungus, Torrubiella luteorostrata. Tetrahedron Lett 52, 7042–7045 (2011).
Kornsakulkarn, J., Thongpanchang, C., Lapanun, S. & Srichomthong, K. Isocoumarin glucosides from the scale insect fungus Torrubiella tenuis BCC 12732. J. Nat. Prod. 72, 1341–1343 (2009).
Isaka, M., Chinthanom, P., Supothina, S., Tobwol, P. & Hywel-Jones, N. L. Pyridone and tetramic acid alkaloids from the spider pathogenic fungus Torrubiella sp. BCC 2165. J. Nat. Prod. 73, 2057–2060 (2010).
Danielsen, K., Aksnes, D. W. & Francis, G. W. NMR study of some anthraquinones from rhubarb. Magn. Reson. Chem. 30, 359–360 (1992).
Kuhnert, N. & Molod, H. Y. An efficient total synthesis of chrysophanol and the sennoside C aglycon. Tetrahedron Lett. 46, 7571–7573 (2005).
Toma, F. et al. Carbon-13 NMR spectroscopy of the biological pigments luteoskyrin and rugulosin and some polyhydroxyanthraquinone analogues. Org. Magn. Reson. 7, 496–503 (1975).
Miethbauer, S. et al. Uredinorubellins and caeruleoramularin, photodynamically active anthraquinone derivatives produced by two species of the genus. Ramularia. J. Nat. Prod. 71, 1371–1375 (2008).
Miethbauer, S. et al. Biosynthesis of photodynamically active rubellins and structure elucidation of new anthraquinone derivatives produced by Ramularia collo-cygni. Phytochemistry 67, 1206–1213 (2006).
Heiser, I. et al. Fatty acid peroxidation by rubellin B, C, and D, phytotoxins produced by Ramularia collo-cygni (Sutton et Waller). Physiol. Mol. Plant Pathol. 64, 135–143 (2004).
O’Brien, J., Wilson, I., Orton, T. & Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 267, 5421–5426 (2000).
Desjardins, R. E., Canfield, C. J., Haynes, J. D. & Chulay, J. D. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 16, 710–718 (1979).
Changsen, C., Franzblau, S. G. & Palittapongarnpim, P. Improved green fluorescent protein reporter gene-based microplate screening for antituberculosis compounds by utilizing an acetaminase promoter. Antimicrob. Agents Chemother. 47, 3682–3687 (2003).
Acknowledgements
Financial support from the Bioresources Research Network, National Center for Genetic Engineering and Biotechnology (BIOTEC), is gratefully acknowledged. We thank Dr J Jennifer Luangsa-ard for identification of the fungus used in this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article
Cite this article
Isaka, M., Palasarn, S., Tobwor, P. et al. Bioactive anthraquinone dimers from the leafhopper pathogenic fungus Torrubiella sp. BCC 28517. J Antibiot 65, 571–574 (2012). https://doi.org/10.1038/ja.2012.76
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2012.76
