
Abstract
Chromosome 1p32-p31 deletion syndrome involving the Nuclear factor I/A (NFIA) gene is characterized by corpus callosum hypoplasia or defects and urinary tract defects. Herein we report on a case resembling the 1p32-p31 deletion syndrome carrying a de novo truncating mutation (c.1094delC; p.Pro365Hisfs*32) in the NFIA gene, confirming that haploinsufficiency of the NFIA gene is a major determinant of this syndrome.
Similar content being viewed by others

Chromosome 1p32-p31 deletion syndrome (OMIM #613735) involving the Nuclear factor I/A (NFIA) gene is characterized by corpus callosum hypoplasia or defects, hydrocephalus or ventricular enlargement and urinary tract defects.1 Only six cases of this contiguous gene-deletion syndrome have been reported in the literature.1–5 Additionally, Lu et al.1 reported two patients showing a similar phenotype, but with balanced translocations breakpoints in the NFIA gene.6 These authors also demonstrated ventricular enlargement, callosal agenesis and urinary tract defects in homozygous Nfia−/− mice and heterozygous Nfia+/− mice.1 Recently, Rao et al.7 reported a case exhibiting a similar phenotype with an intragenic deletion in the NFIA gene, but no structural chromosomal abnormalities detected by CGH microarray. Although haploinsufficiency of the NFIA gene is considered the main contributor to the phenotype of this chromosome 1p32-p31 deletion syndrome, the evidence is not conclusive because no single nucleotide variant (SNV) in the NFIA gene has been identified and chromosomal rearrangements including deletion or translocation could have a position effect disturbing the proper expression of the neighboring genes.
We herein report on a case of an individual showing interhemispheric cysts, ventricular enlargement, callosal agenesis and urinary tract defects, and carrying a heterozygous de novo frameshift mutation in the NFIA gene. These findings provide further strong evidence that haploinsufficiency of the NFIA gene is a main contributor of 1p32-p31 deletion syndrome, and the NFIA gene has a fundamental role in development of brain as well as urinary tract.
This study was approved by the institutional review board of Nagoya City University Graduate School of Medical Sciences.
The proband is a 5-year-old boy with no family history of the relevant diseases. Callosal agenesis was suspected from the 28th gestational week of the fetal period. The boy was born by cesarean section on the 41st gestational week due to enlargement of the head circumference and post-term pregnancy. His Apgar score was 9 at 5 min post partum, his weight was 3180 g (+0.4 s.d.) and his head circumference was 38.2 cm (+3.3 s.d.). No apparent external malformations were observed. Head magnetic resonance imaging (MRI) on the third day of life revealed interhemispheric cysts, ventricular enlargement and callosal agenesis; however, he was in good general condition. Regarding his developmental milestones, he was able to hold his head up by 4 months and started walking without support at 1 year and 3 months. He was observed speaking meaningful words at 2 years and 1 month, showing a slight delay in language, and his intelligence quotient at 4 years measured by the Tanaka–Binet Intelligence Scale was 75. No epileptic seizures have been observed to date, although electroencephalogram detected sharp waves in the frontal head area at 11 months. A follow-up MRI at 4 years revealed polymicrogyria in the right frontal lobe, while the size of the interhemispheric cysts, longitudinal cerebral fissure and ventricular system remained unchanged (Figures 1a,b). Although no abnormal signals were observed in the spinal cord in a spine MRI carried out at 5 years, cystectasia and left hydronephrosis were observed. A voiding cysturethrogram performed at 5 years showed bilateral grade IV vesicoureteral reflux (Figure 1c). His current head circumference is 56.1 cm (+3.8 s.d.) showing non-progressive enlargement of head circumference. He showed only a little dysmorphic facial features including mild macrocephaly, high forehead, and thin upper lip (Figure 1d).

Head MRI, voiding cysturethrogram (VCUG) and craniofacial appearance of our patient. (a) Axial T2-weighted image showing interhemispheric cysts, ventricular enlargement and polymicrogyria (arrow). (b) Mid-sagittal T1-weighted image showing callosal agenesis (asterisk). (c) The VCUG showed bilateral grade IV vesicoureteral reflux. (d) Representative photograph of the patient showing a little dysmorphic facial features including mild macrocephaly, high forehead, and thin upper lip. His parents gave informed consent for publication of this image.
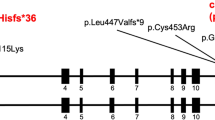
We performed a whole-exome sequencing on the proband and his parents (Figure 2a). To do this, genomic DNA was extracted from peripheral blood using the QIAamp DNA Blood Midi Kit according to the manufacturer’s instructions (Qiagen, Tokyo, Japan). Three micrograms of DNA was sheared into 150-200-bp fragments using the Covaris DNA Shearing service (Covaris, Woburn, MA, USA). To capture the exonic DNA, we used the SureSelect XT Human All Exon V5 capture library (Agilent Technologies, Santa Clara, CA, USA). We then constructed a sequence library using the SureSelect XT Target Enrichment System for Illumina Paired-End Sequencing Library kit (Agilent Technologies), and performed DNA sequencing of 100-bp paired-end reads by using the Illumina HiSeq 2000 sequencer (Illumina, San Diego, CA, USA). On average, we obtained 5.85 Gb of sequence reads. The sequencing data was mapped to a reference genome (GRCh37/hg19) using Burrows-Wheeler Alignment tool (ver.0.6.1; http://www.bio-bwa.sourceforge.net/), and the average read depth of targeted regions was 67.5. Variant calling was performed using SAMtools (ver.0.1.16; http://www.samtools.sourceforge.net/) and GATK (ver.1.6; http://www.broadinstitute.org/gatk/) software. To identify the disease causative mutations, we excluded known variants found in public databases (dbSNP138, 1000 Genomes Project, and NHLBI ESP6500) and a control in-house database (154 Japanese individuals of normal and other diseases control), except for those also identified as pathogenic mutations in the NCBI ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) and HGMD databases (http://www.hgmd.org/). We focused on non-synonymous SNVs, insertions and deletions (indels), and splice-site variants (Figure 2b). This analysis revealed a heterozygous frameshift mutation (c.1094delC; p.Pro365Hisfs*32) in the NFIA gene (NM_001134673.3), which is absent in his parents, indicating that the mutation arose de novo (Figure 2c). The mutation was confirmed by Sanger sequencing (Figure 2d). This deletion led to an open reading frameshift that introduced an early stop codon which truncated 114 aminoacids. Consequently, a truncated NFIA protein is generated by this mutation.

Genetic analysis of the pedigree. (a) Family tree of the pedigree. (b) Filtering the candidate mutations. Numbers show the patient result. The top numbers indicate number of called variants by whole-exome sequencing. The second numbers indicate number of variants after filter out known variants in databases, except for those which were also known pathogenic mutations. The third number indicates number of variants after excluded synonymous change variants. The bottom numbers indicate number of variants consistent with the phenotype in the pedigree (that is, total of the de novo, autosomal recessive, X-linked and compound heterozygous variants). Finally, only one deletion variant was remained. (c) Identified frameshift mutation in the NFIA gene. (d) Sanger sequencing of the NFIA mutation. Patient had a heterozygous c.1094delC mutation (arrow) not found in his parents.
The NFIA gene has four transcriptional variants in human. Functional significance of each isoform has not been clarified. Only isoform 2 lacks an alternative exon downstream of the identified deletion site (Supplementary Figure a). The c.1094delC mutation would create protein truncation in each isoform. To examine the expression level of each transcript PCR with reverse transcription reaction was performed by using RNA isolated from normal human brain tissues (adult cortex, cerebellum, spinal cord and fetal brain). NFIA isoform 2 transcript was less predominant compared with isoforms 1, 3 and 4 (Supplementary Figure b). Therefore, the c.1094delC mutation would induce more effects on longer isoforms 1, 3 and 4 than on isoform 2, and the exon of deletion site and/or next exon which are used in all wild-type isoforms as translation regions would have an important role in NFIA protein.
Three molecular mechanisms are known to cause the complex central nervous system malformation syndrome associated with the NFIA gene: (1) interstitial deletion of chromosome 1p32-p31 involving the NFIA gene; (2) translocations of chromosome 1p32-p31 involving the NFIA gene; and (3) intragenic deletion in the NFIA gene. Nine cases (6 deletions, 2 translocations and 1 intragenic deletion) of this syndrome have been reported to date,1–7 with corpus callosum hypoplasia or defects and hydrocephalus, ventricular enlargement and developmental delays observed in all cases. Urinary tract defects, tethered spinal cord and type 1 Chiari malformation were also observed in six, four and three cases, respectively. Additionally, abnormal facies and marble skin have been reported. The present case showed ventricular enlargement, callosal agenesis, urinary tract defects, mildly dysmorphic facial features and an intelligence quotient in the borderline range. Thus, our case showed virtually the same phenotype as 1p32-p31 deletion syndrome. Since the single nucleotide deletion detected in our case is not likely to cause a position effect affecting surrounding genes, it indicates that haploinsufficiency of the NFIA gene is a major determinant of this syndrome.
A truncating mutation in the NFIA gene was previously reported in one patient with autistic spectrum disorder (c.112C>T; p.R38*),8 but detailed clinical information was not available. Since urinary tract involvement is easily missed, it is conceivable that this previous patient may have the same phenotype, and comprehensive evaluation of the patient might uncover the underlying defects.
The NFIA gene encodes a member of the nuclear factor I (NFI) family of transcription factors.9 NFI proteins control a range of key processes in central nervous system development including axon guidance and outgrowth, glial and neuronal cell differentiation, and neuronal migration.10 Additionally, these molecules regulate midline glia formation in the cortex11 and gliogenesis within the spinal cord.12 Thus, NFI functional defects could result in abnormal brain formation, especially in midline structures, and spinal cord defects leading to neurogenic urinary tract dysfunction and defects. Interestingly, haploinsufficiency of the NFIB and NFIX genes, which belong to the same NFI family, also cause callosal agenesis,13–15 and missense mutations in the NFIX gene cause Sotos-like syndrome.14,16 Our case also demonstrated prominent macrocephaly, which is a major feature of Sotos syndrome. Therefore, mutations in NFI family genes may give rise to similar clinical presentations that encompass callosal agenesis and macrocephaly.
In our case, identification of the mutation by whole-exome sequencing led us to identify urinary tract defects in the presymptomatic period, and untreated higher grades of vesicoureteral reflux may have resulted in renal scar formation.17 Therefore, whole-exome sequencing can be a powerful tool in clinical practice for early diagnosis of congenital disorders.
References
References
Lu W, Quintero-Rivera F, Fan Y, Alkuraya FS, Donovan DJ, Xi Q et al. NFIA haploinsufficiency is associated with a CNS malformation syndrome and urinary tract defects. PLoS Genet 2007; 3: e80.
Campbell CG, Wang H, Hunter GW . Interstitial microdeletion of chromosome 1p in two siblings. Am J Med Genet 2002; 111: 289–294.
Koehler U, Holinski-Feder E, Ertl-Wagner B, Kunz J, von Moers A, von Voss H et al. A novel 1p31.3p32.2 deletion involving the NFIA gene detected by array CGH in a patient with macrocephaly and hypoplasia of the corpus callosum. Eur J Pediatr 2010; 169: 463–468.
Chen CP, Su YN, Chen YY, Chern SR, Liu YP, Wu PC et al. Chromosome 1p32-p31 deletion syndrome: prenatal diagnosis by array comparative genomic hybridization using uncultured amniocytes and association with NFIA haploinsufficiency, ventriculomegaly, corpus callosum hypogenesis, abnormal external genitalia, and intrauterine growth restriction. Taiwan J Obstet Gynecol 2011; 50: 345–352.
Ji J, Salamon N, Quintero-Rivera F . Microdeletion of 1p32-p31 involving NFIA in a patient with hypoplastic corpus callosum, ventriculomegaly, seizures and urinary tract defects. Eur J Med Genet 2014; 57: 267–268.
Shanske AL, Edelmann L, Kardon NB, Gosset P, Levy B . Detection of an interstitial deletion of 2q21-22 by high resolution comparative genomic hybridization in a child with multiple congenital anomalies and an apparent balanced translocation. Am J Med Genet A 2004; 131: 29–35.
Rao A, O'Donnell S, Bain N, Meldrum C, Shorter D, Goel H et al. An intragenic deletion of the NFIA gene in a patient with a hypoplastic corpus callosum, craniofacial abnormalities and urinary tract defects. Eur J Med Genet 2014; 57: 65–70.
Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012; 74: 285–299.
Qian F, Kruse U, Lichter P, Sippel AE . Chromosomal localization of the four genes (NFIA, B, C, and X) for the human transcription factor nuclear factor I by FISH. Genomics 1995; 28: 66–73.
Mason S, Piper M, Gronostajski RM, Richards LJ . Nuclear factor one transcription factors in CNS development. Mol Neurobiol 2009; 39: 10–23.
Shu T, Butz KG, Plachez C, Gronostajski RM, Richards LJ . Abnormal development of forebrain midline glia and commissural projections in Nfia knock-out mice. J Neurosci 2003; 23: 203–212.
Deneen B, Ho R, Lukaszewicz A, Hochstim CJ, Gronostajski RM, Anderson DJ et al. The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron 2006; 52: 953–968.
Sajan SA, Fernandez L, Nieh SE, Rider E, Bukshpun P, Wakahiro M et al. Both rare and de novo copy number variants are prevalent in agenesis of the corpus callosum but not in cerebellar hypoplasia or polymicrogyria. PLoS Genet 2013; 9: e1003823.
Malan V, Rajan D, Thomas S, Shaw AC, Louis Dit Picard H, Layet V et al. Distinct effects of allelic NFIX mutations on nonsense-mediated mRNA decay engender either a Sotos-like or a Marshall-Smith syndrome. Am J Hum Genet 2010; 87: 189–198.
Steele-Perkins G, Plachez C, Butz KG, Yang G, Bachurski CJ, Kinsman SL et al. The transcription factor gene Nfib is essential for both lung maturation and brain development. Mol Cell Biol 2005; 25: 685–698.
Yoneda Y, Saitsu H, Touyama M, Makita Y, Miyamoto A, Hamada K et al. Missense mutations in the DNA-binding/dimerization domain of NFIX cause Sotos-like features. J Hum Genet 2012; 57: 207–211.
Peters CA, Skoog SJ, Arant BS Jr, Copp HL, Elder JS, Hudson RG et al. Summary of the AUA Guideline on Management of Primary Vesicoureteral Reflux in Children. J Urol 2010; 184: 1134–1144.
Data Citations
Saitoh, Shinji HGV Database http://dx.doi.org/10.6084/ m9.figshare.hgv.574 (2015)
Acknowledgements
This study was supported in part by a grant for Research on Applying Health Technology from the Ministry of Health, Labour and Welfare of Japan to FM, NO, MK, MY, YK, KK and SS. We thank KA Boroevich for English proofreading.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if thematerial is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Negishi, Y., Miya, F., Hattori, A. et al. Truncating mutation in NFIA causes brain malformation and urinary tract defects. Hum Genome Var 2, 15007 (2015). https://doi.org/10.1038/hgv.2015.7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.7
This article is cited by
-
A novel homozygous missense mutation in the SH3-binding motif of STAMBP causing microcephaly-capillary malformation syndrome
Journal of Human Genetics (2018)
-
A combination of genetic and biochemical analyses for the diagnosis of PI3K-AKT-mTOR pathway-associated megalencephaly
BMC Medical Genetics (2017)
