
Abstract
Epidemiological studies show that type 2 diabetes (T2D) is inversely associated with intracranial aneurysms (IA) and abdominal aortic aneurysms (AAA). Although adiposity has not been considered a risk factor for IA, there have been inconsistent reports relating adiposity to AAA risk. We assessed whether these observations have a genetic, causal basis. To this end, we extracted genotypes of validated single-nucleotide polymorphisms associated with T2D (n=65), body mass index (BMI) (n=97) and waist–hip ratio adjusted for BMI (WHRadjBMI) (n=47) from genotype data collected in 717 IA cases and 1988 controls, and in 818 AAA cases and 3004 controls, all of Dutch descent. For each of these three traits, we computed genetic risk scores (GRS) for each individual in these case–control data sets by summing the number of risk alleles weighted by their published effect size, and tested whether these GRS were associated with risk of aneurysm. We divided the cohorts into GRS quartiles, and compared IA and AAA risk in the highest with the lowest GRS quartile using logistic regression. We found no evidence for association in IA or AAA risk between top and bottom quartiles for the genetic risk scores for T2D, BMI and WHRadjBMI. However, additional Mendelian randomization analyses suggested a trend to potentially causal associations between BMI and WHRadjBMI and risk of AAA. Overall, our results do not support epidemiological observations relating T2D to aneurysm risk, but may indicate a potential role of adiposity in AAA that requires further investigation.
Similar content being viewed by others

Introduction
Thanks to the success of genome-wide association studies (GWAS), we are rapidly gaining novel insights into the genetic basis of traits and diseases as well as the overlap between them. An important advantage compared with observational studies, genetic analysis can rigorously test whether one trait (eg, circulating levels of a biomarker) affects risk of disease in a causal manner. In a previous study, for example, we showed that genetic risk profiles for lipid factors and coronary artery disease susceptibility were associated with abdominal aortic aneurysms (AAA) but not intracranial aneurysms (IA), whereas the genetic risk profile for blood pressure was instead associated with IA but not AAA,1 consistent with epidemiological observations.2, 3, 4 This illustrated that a genetic approach can help assess the causal role of traditional risk factors in disease.
Here, we apply the same methodology to study the effect of other metabolic risk factors that do not yet have an established role in aneurysm pathology. Epidemiological studies have reported an inverse association between type 2 diabetes (T2D) and both IA2, 5, 6 and AAA,6, 7, 8 but it remains unclear whether these relations are truly causal or may have arisen due to confounding. Adiposity is not considered a bona fide risk factor for IA,2, 9 and for AAA the impact of adiposity is still unclear due to conflicting results from observational studies.10, 11 (eg, one study suggested that abdominal adiposity rather than total obesity may play a role in AAA).11 In this study, we aim to test whether there is a causal association between T2D or adiposity and risk of IA and AAA, by testing the combined effect of established genetic risk variants for these traits on the risk of IA and AAA. For adiposity, we tested genetic risk variants for both BMI as a measure of total adiposity, and for waist–hip ratio adjusted for BMI (WHRadjBMI) as a measure of central adiposity. In addition, we perform Mendelian randomization analyses to test whether any of these risk factors have causal effect on aneurysm risk.
Methods
Study populations
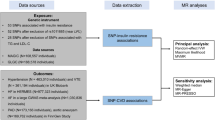
We used genotype data from Dutch IA and AAA patients and controls. These cohorts have been described in detail elsewhere.12, 13, 14, 15, 16 All studies have been approved by the relevant medical ethical committees and all participants provided written informed consent. Below is a brief description of each study population.
IA cases in the Dutch cohort (n=1080) were admitted to the University Medical Center Utrecht, the Netherlands between 1997 and 2011. The cohort both included cases with ruptured and unruptured IA. Ruptured IA cases were defined by symptoms suggestive of subarachnoid hemorrhage (SAH) combined with subarachnoid blood on a computed tomography (CT) scan and a proven IA at angiography (conventional angiogram, CT- or magnetic resonance (MR)-angiogram). Unruptured IA cases were identified by CT or MR angiography or conventional angiography in the absence of clinical or radiological signs of SAH. Patients with fusiform IA, possible traumatic SAH and polycystic kidney disease were excluded. All cases were genotyped on Illumina CNV370 Duo BeadChips (Illumina, San Diego, CA, USA).12 Controls (n=2089) were ascertained via the Rotterdam Study, a population-based cohort of subjects aged 45 years and older recruited from a district in Rotterdam (The Netherlands). These controls were genotyped on Illumina HumanHap550 BeadChips (Illumina).17
AAA patients (n=859) were recruited from eight medical centers in the Netherlands, mainly when individuals visited their vascular surgeon in the outpatient clinic or, in some cases, during hospital admission for elective or emergency AAA surgery. An AAA was defined as an infrarenal aorta diameter of ≥30 mm. The mean AAA diameter was 58.4 mm. Of these patients 530 had received surgery, of which 43 after rupture. Genotyping was performed on Illumina HumanHap610 chips (Illumina).15 As controls, we included 3110 Dutch subjects who were recruited as part of the Nijmegen Biomedical Study (n=1832) and the Nijmegen Bladder Cancer Study (n=1278).13, 14 These controls were genotyped on Illumina CNV370 Duo BeadChips (Illumina).
Quality control, imputation and association analysis
We performed quality control of samples and single-nucleotide polymorphisms (SNPs) as described previously.1 For each case–control data set, genotype imputation was performed in Ricopili (Rapid Imputation Consortium Pipeline, https://sites.google.com/a/broadinstitute.org/ricopili/). For imputation, the prephasing/imputation stepwise approach implemented in IMPUTE2 and SHAPEIT was used (chunk size of 3 Mb and default parameters).18, 19 The imputation reference set consisted of 2184 phased haplotypes from the full 1000 Genomes Project data set (February 2012; 40 318 253 variants).20 All genomic locations are given in NCBI Build 37/UCSC hg19 coordinates. After imputation, SNPs with an imputation accuracy score <0.6 or minor allele frequency <0.5% were excluded. Next, we performed an association analysis in both the IA and AAA cohorts using PLINK.21 We used logistic regression models where we included the allelic dosage (0–2) of each SNP as an independent variable, and case–control status as the dependent variable (outcome). The first four principal components (PCs) in each study cohort were included as covariates to correct for population stratification.
Genetic risk scores
From the literature, we found 65 established risk SNPs for T2D,22 97 risk SNPs for BMI23 and 49 SNPs for WHRadjBMI.24 One BMI risk SNP (rs12016871:C>T) was not present in our association study, and was replaced by a proxy SNP (rs9581854:C>T; r2=1). One WHRadjBMI risk SNP (rs7759742:A>T) was excluded from further analyses, because it was not present in our association study, and no proxy SNPs (r2>0.8) were available. For each of these SNPs, we looked up the corresponding risk allele and its published effect size. We extracted the results of these SNPs from our IA and AAA association analysis, and computed genetic risk scores for T2D, BMI and WHRadjBMI in each individual of the IA and AAA cohorts as follows:

where xi is the estimated allele dosage (between 0 and 2) in a given individual, and βi is the published effect size (BMI and WHRadjBMI) or the natural log of the reported odds ratio (T2D) for the ith SNP. We tested the resulting genetic risk scores for association with IA and AAA using logistic regression, adjusting for sex and the first four PCs in the study cohorts. To evaluate the magnitude of effect of the genetic risk scores on IA or AAA disease risk, we divided the IA and AAA cohorts into quartiles on the basis of the genetic risk score distributions in these two cohorts, and calculated odds ratios and corresponding 95% confidence intervals (CI) for IA or AAA disease risk in the highest quartile compared with that in the lowest quartile, using logistic regression, adjusting for sex and the first four PCs in the study cohorts.
Mendelian randomization analyses
To assess any causal association between T2D and adiposity (indexed by BMI and WHRadjBMI) on risk of aneurysms, we performed a series of complementary Mendelian randomization (MR) analyses.25 Conventional MR findings (based on an inverse variance weighted (IVW) approach) were contrasted with results from sensitivity analyses, including MR-Egger, MR-Egger adjusted for simulation extrapolation and weighted median MR.26, 27, 28 MR-Egger tests for the presence of unbalanced pleiotropic effects of the SNPs under analysis that may distort the causal effect estimate. For all MR analyses, SNPs were oriented towards an increase in the exposure. The standard error was obtained by bootstrap resampling 10 000 times. IVW was performed using linear regression of the SNPs-exposure (T2D, BMI or WHRadjBMI) estimates against SNPs-outcome (IA or AAA) estimates, weighted by the minor allele frequencies of each SNP and forced to pass through the origin.26 MR-Egger is similar to IVW but the constant was not constrained to pass through the origin.26 A significant departure from the origin provides evidence for the presence of unbalanced pleiotropy (tested using the Egger test; ie, a P-value significant at the 5% level indicates presence of pleiotropic effects). For MR-Egger adjusted for simulation extrapolation using I2-statistics, we first quantified any violation of the NOME (NO Measurement Error (NOME)) assumption (ie, testing the fact that the SNP–exposure association is true).28 We then applied simulation extrapolation (SIMEX; implemented in R using the simex package) to adjust the MR-Egger causal estimates to account for a potential NOME violation (results based on 1000 simulations).28 Weighted median MR yields robust and precise results even when up to 50% of the weight in the analysis comes from invalid genetic variants.27 We specifically performed a penalized weighted median MR analysis (implemented in Stata using the mrrobust package; available at: https://github.com/remlapmot/mrrobust), which favors SNPs with causal estimates close to the median causal estimate.
Data availability
The study data (summary-level data of primary study results) have been made publicly available. They can be obtained from GWAS Central: http://www.gwascentral.org/study/HGVST1837.
Results
IA and AAA cohorts
After quality control and imputation, the IA cohort consisted of 717 cases and 1988 controls, and the AAA cohort comprised 818 cases and 3004 controls. Table 1 shows the baseline characteristics of the IA and AAA cohorts.
Genetic risk scores
The association results of the individual T2D, BMI and WHRadjBMI-associated SNPs in the IA and AAA cohorts are listed in Supplementary Table S1. None of the T2D, BMI and WHRadjBMI SNPs were significantly associated with IA or AAA (P>0.002).
The association results of the T2D, BMI and WHRadjBMI genetic risk scores with IA risk are shown in Table 2 and with AAA in Table 3. We did not detect any evidence for associations between the weighted GRS and IA/AAA case status (P≥0.06). There was no evidence for association in IA risk between top and bottom quartiles for the T2D score (odds ratio (OR)=1.03, 95% confidence interval (CI)=0.95–1.11), for the BMI score (OR=1.04, 95% CI=0.96–1.13) or for the WHRadjBMI score (OR=0.96, 95% CI=0.88–1.04). Also for AAA risk no evidence for association was found between top and bottom quartiles for the T2D score (OR=1.03, 95% CI=0.96–1.11), for the BMI score (OR=1.05, 95% CI=0.97–1.13) or for the WHRadjBMI score (OR=1.04, 95% CI=0.96–1.13).
Mendelian randomization analyses
MR analyses showed consistent directions of effect of BMI on AAA and WHRadjBMI on AAA, but the 95% CI of all effect sizes included the value of 1 (Table 4). The MR analysis of BMI was suggestive of a causal relationship with risk of AAA on conventional (IVW) MR analysis (OR for 1−SD increase in BMI on risk of AAA=1.63, 95% CI 0.99–2.61). The causal effect estimates of the associations between T2D and aneurysms were close to the null. In the Egger tests we did not find evidence against the null hypothesis of no pleiotropy of the SNPs under analysis, except for WHRadjBMI on IA analysis. Supplementary Figure S1 presents the effect estimates from conventional and Egger Mendelian randomization as regression slopes, for each combination of a risk trait and IA or AAA. These slopes are all parallel, except for the analysis of WHRadjBMI on IA, indicating the presence of pleiotropic effects in this analysis.
Discussion
In this study, we found no evidence for a genetic overlap between genetic variants associated with T2D, BMI and WHRadjBMI and risk of IA and AAA. Nonetheless, Mendelian randomization analyses suggest a potentially causal effect of BMI and WHRadjBMI on AAA risk. The lack of a genetic association between T2D and IA or AAA, further supported by Mendelian randomization analyses, suggests that the observed inverse relation between T2D and aneurysms is likely confounded and not real. Several explanations for this inverse relation have been proposed. For example, hyperglycemia and hyperinsulinemia could lead to biological changes protecting the aortic wall from AAA development.8, 29 Confounding factors have also been suggested, like competing morbidity from other (cardiovascular) diseases induced by T2D.2, 30 Furthermore, effects of medication used in the management of T2D could protect against aneurysm development.8, 29
The absence of a genetic association between adiposity and IA in this study is consistent with epidemiological studies, which have ruled BMI out as a risk factor for IA.2, 9 The association between adiposity and AAA is not clear, however, and our data suggest that there may well be a causal relationship.
In our study we chose to index adiposity by BMI and WHRadjBMI. In particular, we favored WHRadjBMI over WHR alone as the former represents a better surrogate of body fat distribution (ie, central obesity)31 and previous studies suggest that the risk of AAA is influenced by central obesity rather than total obesity.11 Nevertheless, it is important to realize that the adjustment for the heritable BMI trait in WHRadjBMI may have created spurious associations, a phenomenon referred to as collider bias.32 To investigate this further, future studies should investigate WHR and BMI separately using multivariable MR approaches.33
MR analysis has the advantage of reducing bias from confounding and reverse causation. Inherited alleles from parents remain stable throughout life and are not influenced by environmental factors. Whereas standard genetic association studies (using GRS) help identify common genetic determinants between traits and disease outcomes, MR more formally investigates this and synthesizes a causal estimate for a given difference in genetically instrumented exposure. Thus, a causal effect discovered by MR analysis is likely to reflect a true association.34 Here, we did not observe strong evidence for causal relationships between T2D, BMI or WHRadjBMI and risk of aneurysms, although our MR analyses consistently suggest a potential association between adiposity (particularly BMI) and risk of AAA. We emphasize that these results are hampered by limited statistical power and studies involving larger sample sizes would be required to provide a definitive answer. Even so, we were previously able to show significant associations between genetic risk scores for well-established risk factors (hypertension and serum lipid levels) and IA or AAA risk, respectively, using the same data.1 This suggests that the power of the present study should have been sufficient under the assumption that the effect is real and has a comparable magnitude of effect.
In summary, we found no evidence for a causal role of T2D in aneurysms, and only suggestive evidence for a potentially causal role of adiposity traits in AAA. Future studies with larger data sets would be needed to further investigate the nature of this relationship.
References
van't Hof FN, Ruigrok YM, Baas AF et al: Impact of inherited genetic variants associated with lipid profile, hypertension, and coronary artery disease on the risk of intracranial and abdominal aortic aneurysms. Circ Cardiovasc Genet 2013; 6: 264–270.
Feigin VL, Rinkel GJ, Lawes CM et al: Risk factors for subarachnoid hemorrhage: an updated systematic review of epidemiological studies. Stroke 2005; 36: 2773–2780.
Forsdahl SH, Singh K, Solberg S, Jacobsen BK : Risk factors for abdominal aortic aneurysms: a 7-year prospective study: the Tromso Study, 1994–2001. Circulation 2009; 119: 2202–2208.
Cornuz J, Sidoti Pinto C, Tevaearai H, Egger M : Risk factors for asymptomatic abdominal aortic aneurysm: systematic review and meta-analysis of population-based screening studies. Eur J Public Health 2004; 14: 343–349.
Inagawa T : Risk factors for aneurysmal subarachnoid hemorrhage in patients in Izumo City, Japan. J Neurosurg 2005; 102: 60–67.
Shah AD, Langenberg C, Rapsomaniki E et al: Type 2 diabetes and incidence of cardiovascular diseases: a cohort study in 1.9 million people. Lancet Diabetes Endocrinol 2015; 3: 105–113.
Ohrlander T, Merlo J, Ohlsson H, Sonesson B, Acosta S : Socioeconomic position, comorbidity, and mortality in aortic aneurysms: a 13-year prospective cohort study. Ann Vasc Surg 2012; 26: 312–321.
Shantikumar S, Ajjan R, Porter KE, Scott DJ : Diabetes and the abdominal aortic aneurysm. Eur J Vasc Endovasc Surg 2010; 39: 200–207.
Vlak MH, Rinkel GJ, Greebe P, Algra A : Independent risk factors for intracranial aneurysms and their joint effect: a case-control study. Stroke 2013; 44: 984–987.
Kent KC, Zwolak RM, Egorova NN et al: Analysis of risk factors for abdominal aortic aneurysm in a cohort of more than 3 million individuals. J Vasc Surg 2010; 52: 539–548.
Stackelberg O, Bjorck M, Sadr-Azodi O, Larsson SC, Orsini N, Wolk A : Obesity and abdominal aortic aneurysm. Br J Surg 2013; 100: 360–366.
Bilguvar K, Yasuno K, Niemela M et al: Susceptibility loci for intracranial aneurysm in European and Japanese populations. Nat Genet 2008; 40: 1472–1477.
Kiemeney LA, Thorlacius S, Sulem P et al: Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nat Genet 2008; 40: 1307–1312.
Wetzels JF, Kiemeney LA, Swinkels DW, Willems HL, den Heijer M : Age- and gender-specific reference values of estimated GFR in Caucasians: the Nijmegen Biomedical Study. Kidney Int 2007; 72: 632–637.
Gretarsdottir S, Baas AF, Thorleifsson G et al: Genome-wide association study identifies a sequence variant within the DAB2IP gene conferring susceptibility to abdominal aortic aneurysm. Nat Genet 2010; 42: 692–697.
Helgadottir A, Thorleifsson G, Magnusson KP et al: The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet 2008; 40: 217–224.
Hofman A, van Duijn CM, Franco OH et al: The Rotterdam Study: 2012 objectives and design update. Eur J Epidemiol 2011; 26: 657–686.
Howie B, Marchini J, Stephens M : Genotype imputation with thousands of genomes. G3 (Bethesda) 2011; 1: 457–470.
Delaneau O, Marchini J, Zagury JF : A linear complexity phasing method for thousands of genomes. Nat Methods 2012; 9: 179–181.
Abecasis GR, Altshuler D, Auton A et al: A map of human genome variation from population-scale sequencing. Nature 2010; 467: 1061–1073.
Purcell S, Neale B, Todd-Brown K et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575.
Morris AP, Voight BF, Teslovich TM et al: Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 2012; 44: 981–990.
Locke AE, Kahali B, Berndt SI et al: Genetic studies of body mass index yield new insights for obesity biology. Nature 2015; 518: 197–206.
Shungin D, Winkler TW, Croteau-Chonka DC et al: New genetic loci link adipose and insulin biology to body fat distribution. Nature 2015; 518: 187–196.
Smith GD, Ebrahim S : 'Mendelian randomization': can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 2003; 32: 1–22.
Bowden J, Davey Smith G, Burgess S : Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015; 44: 512–525.
Bowden J, Davey Smith G, Haycock PC, Burgess S : Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 2016; 40: 304–314.
Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR : Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int J Epidemiol 2016, pii dyw220.
Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM : Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol 2011; 8: 92–102.
Rodin MB, Daviglus ML, Wong GC et al: Middle age cardiovascular risk factors and abdominal aortic aneurysm in older age. Hypertension 2003; 42: 61–68.
Heid IM, Jackson AU, Randall JC et al: Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet 2010; 42: 949–960.
Aschard H, Vilhjálmsson BJ, Joshi AD, Price AL, Kraft P : Adjusting for heritable covariates can bias effect estimates in genome-wide association studies. Am J Hum Genet 2015; 96: 329–339.
Burgess S, Thompson SG : Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol 2015; 181: 251–260.
Smith GD, Lawlor DA, Harbord R, Timpson N, Day I, Ebrahim S : Clustered environments and randomized genes: a fundamental distinction between conventional and genetic epidemiology. PLoS Med 2007; 4: e352.
Acknowledgements
FNGVH is supported by a grant of the Dutch Heart Foundation (NHS) (project no. 2008B004). JV works for the CoLaus|PsyCoLaus study which was and is supported by research grants from GlaxoSmithKline, the Faculty of Biology and Medicine of Lausanne, and the Swiss National Science Foundation (grants 3200B0-105993, 3200B0-118308, 33CSCO-122661, 33CS30-139468 and 33CS30-148401). YMR is supported by a clinical fellowship grant of the Netherlands Organization for Scientific Research (NWO) (project no. 40-00703-98-13533). AFB is supported by a grant from the Dr E Dekker Program of the NHS (2009T001). The generation of the GWAS data sets is supported by the Netherlands Organisation for Scientific Research NWO Investments (no. 175.010.2005.011, 911-03-012), the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the Research Institute for Diseases in the Elderly (014-93-015; RIDE2), the Netherlands Genomics Initiative (NGI)/Netherlands Organisation for Scientific Research (NWO) Netherlands Consortium for Healthy Aging (NCHA), project no. 050-060-810. The Rotterdam Study is funded by the Erasmus Medical Center and Erasmus University, Rotterdam, Netherlands Organization for Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry of Health, Welfare and Sport, the European Commission (DG XII) and the Municipality of Rotterdam.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
van 't Hof, F., Vaucher, J., Holmes, M. et al. Genetic variants associated with type 2 diabetes and adiposity and risk of intracranial and abdominal aortic aneurysms. Eur J Hum Genet 25, 758–762 (2017). https://doi.org/10.1038/ejhg.2017.48
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.48
