
Abstract
IgA nephropathy (IgAN) represents the most common primary glomerulonephritis worldwide with a prevalence of 25–50% among patients with primary glomerulopathies. In ~5–10% of the patients the disease segregates with an autosomal dominant (AD) pattern. Association studies identified loci on chromosomes 1q32, 6p21, 8p23, 17p13, 22q12, whereas classical linkage studies on AD families identified loci on chromosomes 2q36, 4q26-31, 6q22, 17q12-22. We have studied a large Sicilian family where IgAN segregates with an AD transmission. To identify the causal gene, the exomes of two affected and one unaffected individual have been sequenced. From the bioinformatics analysis a p.(Arg119Trp) variant in the SPRY2 gene was identified as the probable disease-causing mutation. Moreover, functional characterization of this variant showed that it is responsible for the inhibition of the MAPK/ERK1/2 pathway. The same effect was observed in two sporadic IgAN patients carriers of wild-type SPRY2, suggesting that downregulation of the MAPK/ERK1/2 pathway represents a common mechanism leading to IgAN.
Similar content being viewed by others

Introduction
IgA nephropathy (IgAN) represents the most common primary glomerulonephritis worldwide.1
The disease is characterized by the deposition of IgA immunocomplexes (IC) onto the renal glomeruli, activating mesangial proliferation and stimulating production of inflammatory cytokines, leading to progressive kidney failure. Although the pathogenesis of this condition is poorly understood, it is likely that kidneys, like in other IC related diseases, represent ‘innocent bystanders’.2, 3, 4, 5
Most IgAN patients are sporadic cases. When familial clustering is found, usually an autosomal dominant (AD) transmission with incomplete penetrance and variable age of onset is observed. Association studies identified distinct loci on chromosomes 6p21, 8p23, 17p13, 22q12, 1q32.6, 7 On the other hand, classical linkage studies performed on AD families identified additional loci on chromosomes 2q36, 4q26-31, 6q22, 17q12-22,8, 9, 10 lending further support to the complex etiology and broad genetic heterogeneity of the disease.
In this study an exome sequencing analysis was performed in a large Sicilian family where the disease segregates with an AD mode of inheritance. The analysis of the exome of one unaffected and two affected individuals identified several single nucleotide polymorphisms (SNP) and fewer deletions/insertions polymorphisms (DIP) segregating with the disease. When the segregation analysis of all these variants was extended to the remaining affected members of the family, only one novel missense variant in the SPRY2 gene perfectly segregated with the phenotype in all affected individuals. Functional analysis of the variant in B lymphoblastoid cell lines (LCL) from affected members of this family linked the SPRY2 mutation to inhibition of the MAPK/ERK pathway, ultimately leading to IgA nephropathy. When two LCL were analyzed from two different IgAN sporadic patients, wild type for SPRY2, the same reduced activity of the MAPK/ERK pathway was observed, suggesting that different variants in the same pathway, ultimately leading to the downregulation of the MAPK/ERK pathway, represent one of the causative lesions of IgA nephropathy.
Materials and methods
DNA extraction and exome sequencing
All IgAN patients were recruited from central Italy and from Sicily, after providing their informed consent. All sporadic patients had a diagnosis of IgAN based on an IgA-positive renal biopsy.
Blood samples were collected in lithium heparin and the DNA was extracted using a modified salting out method with a final purification step with phenol/chloroform. DNA was resuspended in TE buffer. The DNA of individuals II-7, III-23 and IV-44 from the family were sent to the Istituto di Genomica Applicata in Udine for exome sequencing (see Supplementary Appendix). II-7 and IV-44 were chosen because these two individuals were the two more distantly related individuals available when we identified this family.
Mutation analysis by Sanger sequencing
Variants identified by the bioinformatics analysis, after the exome sequencing, were confirmed by Sanger sequencing. In brief, a PCR product containing the identified mutation was performed under standard conditions. Primer sequences will be provided on request. Two and half microliters of each PCR product were purified and sequenced using the BigDye terminator v3.1 Cycle Sequencing Kit (Applied Biosystem, Life Technologies, Carlsbad, CA, USA) in a final volume of 10 μl. The electropherograms were analyzed by the Sequencing Analysis v5.2 software (Applied Biosystem). The c.355C>T SPRY2 variant was submitted to LOVD databases (http://www.lovd.nl/SPRY2 patient ID #25484). Additional methods are described in the Supplementary Methods.
Results
Family study
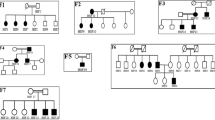
This IgAN family was identified ~10 years ago at the Columbus Hospital of the Catholic University in Rome. The diagnosis was made through a renal biopsy positive for IgA in individuals II-10, III-24 and III-21 (Figure 1a). Individuals II-7 and II-10 are currently in end-stage renal disease. All the remaining individuals are still routinely monitored with urine analysis yearly. A clinical summary of this family is described in Supplementary Table S1.

Identification of the SPRY2 mutation. (a) Pedigree of the family. Black symbols indicate affected individuals. B letter indicates a renal biopsy positive for IgA deposits. The * indicates individuals with the exome analyzed. M or W under each symbol indicates a SPRY2 mutation carrier or wild type. (b) The heterozygous mutation c.355C>T of one of the affected individuals (II-10) was sub-cloned to isolate the two alleles. Electropherograms of the SPRY2 gene segment containing the missense mutation changing an arginine (R) into a tryptophan (W) are shown. (c) Multiple sequence alignment generated by PolyPhen2. The R (indicated by the arrow) is a highly conserved amino-acid residue throughout evolution.
All individuals indicated with a number provided their DNA and their consent to participate to this study. This study was approved by the bioethical committee of the Catholic University.
In this family IgAN shows an AD transmission without any apparent penetrance defect. We considered definitely affected, even without of a renal biopsy, all individuals with constant proteinuria (>1 g/die) or microhaematuria. Microhaematuria was considered positive only when red blood cells had an altered morphology as they were derived from the glomeruli.
Exome sequencing and bioinformatics analysis
To identify the gene responsible for IgAN in this family, the exomes of two distantly related affected individuals (II-7 and IV-44) were sequenced along with the exome of an ‘apparently’ unaffected individual (III-23, at age 43 has a normal urine analysis without proteinuria or hematuria), to rule out potential benign variants. The first bioinformatics analysis was performed to identify all the mutations shared by the two affected individuals and absent in the unaffected individual, assuming AD transmission (Supplementary Tables S2 and S3).
This analysis identified 59 novel variants (55 SNPs and 4 DIPs within coding regions, including flanking introns) shared by the two affected individuals, but absent in the healthy relative. To identify a variant exclusively associated with the disease phenotype, all 59 mutations were clustered to perform a low-coverage total genome linkage analysis. Forty-six of those variants identified 10 loci on 9 different chromosomes (Supplementary Table S4). A second bioinformatics analysis was performed assuming that III-23 was ‘affected’ and 24 new non synonymous variants and 14 DIPs were identified (Supplementary Tables S5 and S6). Then, to identify the locus and the gene responsible for the disease, all 35 non synonymous variants and 18 DIPs, obtained from the two different bioinformatics analyses, were analyzed in all remaining affected individuals. A single novel missense mutation c.355C>T in the SPRY2 gene (NM_005842.2), causing a p.(Arg119Trp) change (Figure 1b) was identified on chromosome 13, perfectly segregating with the disease. This variant is also present in some of the healthy younger individuals in generation IV (IV-42, IV-43 and IV-45), but it is absent in all the healthy individuals older than 20 years, including III-23 (Figure 1a). This variant has never been reported in any publicly available database such as the 1000genomes and the ESP6500.
To define its pathogenic role, the p.(Arg119Trp) variant was run on different online software (SIFT, PolyPhen2, pMut, Panther, Mutation Taster). All the results clearly point to a pathogenic role for this variant (Table 1), indicating a non conservative nucleotide change.
Arginine 119 is a conserved amino-acid residue throughout evolution (Figure 1c) and is located in the N-terminal region of the protein close to serine 121 that has been shown to be directly phosphorylated by MNK1 kinase.11 Phosphorylation of serines 112/121 is crucial for the inhibitory interaction with BRAF kinase12 and to modulate protein degradation by NEDD4.13 The p.(Arg119Trp) variant was run on NetPhos2.0 software to evaluate the phosphorylation score of different serines around the Arg119Trp variant and the results showed that the presence of tryptophan lowered the phosphorylation score of serine 121 from 0.996 to 0.602 (Figure 2a), suggesting that this variant, in addition to changing a highly conserved amino-acid residue has also an impact on the consensus sequence of the kinase phosphorylating SPRY2 at position 121.

Functional analysis of the SPRY2 mutation. (a) The p.(Arg119Trp) mutation lowers the phosphorylation score of serine 121, as shown by the NetPhos2.0 analysis. The arrow indicates the serine in position 121, in bold R and W, the wild-type and the mutated amino-acid residues. (b) qRT-PCR showing the SPRY2 mRNA levels in LCLII7, LCL156 and LCL157 compared with five different wild-type controls. Per each experiment three different RNA preparations, each one run in triplicates, were used. For the graphical representation were used the data of just one wild-type control. (c) Representative image of a western blot on the whole cell lysates extracted from the lymphoblasts and probed with an anti-SPRY2 antibody. The upper band represents the phosphorylated form of the protein. (d) Western blot analysis on the whole cell lysates of different cell lines. The loading was not equal because of the large amount of IgA present in the LCLII7. The membrane was also exposed for a short period of time, for this reason in the wild-type cell lines there are no visible bands.
To identify the biological role of SPRY2 in other cases of IgAN, a mutation analysis was performed on the SPRY2 single coding exon of 70 apparently sporadic cases with IgAN. No additional mutations in this cohort of patients were found, ruling out that SPRY2 mutations represent a common cause of IgAN among sporadic patients. As the c.355C>T variant abrogates a BstUI restriction site, we rapidly screened a cohort of 52 Sicilian controls without finding the T355 allele. This observation confirms that this variant is extremely rare/private.
Functional characterization of the T355 variant
B LCL were established from individuals II-7, III-26, IV-44, IV-46 and IV-43 (henceforth LCLII7, III-26, IV-44, IV-46, IV-43), from five different controls (LCLWT1 to LCLWT5) and from two sporadic IgAN patients, negative for SPRY2 mutations (LCL156 and LCL157).
A direct analysis of the SPRY2 cDNA extracted from the LCLs confirmed that this gene is expressed in B cells14 and the T355 allele is present in transcripts from LCLII7 (data not shown). The same finding was obtained on RNA freshly extracted from white blood cells from individuals II-10, II-7, III-24 and III-26 (data not shown). To investigate the levels of SPRY2 mRNA in LCLs, quantitative real-time PCR was performed on three different RNA preparations. A clear reduction in the amount of SPRY2 transcript was observed in LCLII7 compared with the other cell lines (Figure 2b). LCLII7 showed the lowest SPRY2 levels among all LCLs investigated, with a statistically significant difference, whereas LCL156 and LCL157 showed a slight reduction. The same reduction of the SPRY2 transcript was also observed in III-26 (a SPRY2 variant carrier) compared with IV-46 (wild type for SPRY2) (Supplementary Figure S1). In spite of the drastic reduction in mRNA expression, the amount of SPRY2 protein, evaluated by western blot, at the same time points was similar among all cell lines (Figure 2c). The antibody against SPRY2 recognizes two bands, the upper one corresponding to the phosphorylated protein.15 No difference in the ratio between the phosphorylated and non-phosphorylated SPRY2 forms was observed in LCLII7 or in other affected individuals from the family.
It could be hypothesized that this variant could have an effect on the stability of the mutated protein. To evaluate the stability of SPRY2 protein the mutated and wild-type SPRY2 cDNA were tagged with MYC and then transfected in HEK293 cells. Forty-eight hours upon transfection, cells were treated with cycloheximide and then harvested at three different time points. The cell lysates were analyzed by western blot and probed for the MYC tag, showing an increased stability of the mutated protein compared with the wild-type protein (Supplementary Figure S2).
To characterize the LCLs and to show that they could represent a valuable in vitro model to investigate the molecular mechanisms of the disease, a western blot was performed on the whole cell lysate using an anti human IgA antibody. LCLII7 as well as the LCL156 and LCL157 showed increased levels of IgA compared with two wild-type cell lines (Figure 2d). This finding suggests that increased IgA expression can be considered a marker for the IgAN molecular phenotype in IgAN cell lines. To analyze whether the increased production was caused by expansion of the IgA+ B cells, flow cytometry analysis was performed on LCLII7, LCL156, LCL157 and two LCLWTs. In the control cell lines, surface IgA antibody staining identified two populations (Supplementary Figure S3A), one IgA+, representing about 1–2% of the total cells, whereas 98–99% were IgA. LCL156 and LCL157 had approximately the same fractions of IgA-positive cells as the two LCLWT, but their mean fluorescence intensity was higher, suggesting that in the two sporadic IgAN patients, the increased amount of IgA was probably caused by an increased IgA expression in the same fraction of the B cells. On the other hand, LCLII7 contained only a single IgA+ population (Supplementary Figure S3B). Fresh blood samples from III-26, IV-44, IV-45 and IV-46 were stained with an anti-CD19 and anti-IgA antibodies and the fraction of CD19+IgA+ B cells was within the normal range (Supplementary Figure S4). The apparent increase in IgA+ lymphoblasts in vitro is probably related to the aspecific binding to the B cells of the IgA secreted in the culture media.
An immuno-cytochemical analysis using the anti-SPRY2 antibody did not show significant differences in the localization and intensity of the signal between LCLII7 and the other cell lines (data not shown).
Functional characterization of the SPRY2 pathway
SPRY2 is part of the MAPK/ERK pathway, where it has a prevalent role as inhibitor of the ERK1/2 activation through its interaction with the BRAF kinase.16 To test the biological function of SPRY2 W119 variant, we investigated the MAPK/ERK pathway by western blot in the available cell lines. Because in EBV-transformed B lymphoblasts the ERK1/2 pathway is constitutively active,17 the level of phosphoERK1/2 (pERK1/2) was directly analyzed. pERK1/2 was hypophosphorylated in all mutation carriers as well as in the LCL156 and LCL157, compared with the control cell lines and to IV-46, negative for SPRY2 mutations (Figures 3a and b). The finding of reduced pERK1/2 activation in the two other LCLIgAs was intriguing.

Characterization of the MAPK/ERK1/2 pathway. (a) Western blot on the whole cell lysate showing reduction of phosphoERK1/2 in the three IgAN cell lines compared with the constitutive activation in the control cell line. Total ERK1/2 and ACTIN were used as loading control. In the graph is presented the ratio between pERK1/2 and actin in the LCL analyzed. (b) Western blot on the whole cell lysate showing reduction of phosphoERK1/2 in all SPRY2 mutation carriers, II-7, III-26, IV-44 and IV-43 cell lines, compared with the constitutive activation in the control cell line and in IV-46, negative for SPRY2 mutations. GAPDH was used as loading control. In the graph is presented the ratio between pERK1/2 and GAPDH in the LCL analyzed. (c) After 7 days of sorafenib treatment LCL157 showed complete block of pERK1/2, increased production of IgA and inhibition of SPRY2 transcription. (d) Cartoon showing the MAPK/ERK pathway with SPRY2 and sorafenib and the effect of the hypo-phosporylated SPRY2 protein on the pathway. In affected individuals mutant SPRY2 is constantly inhibitory, owing to hypophosphorylation.
The most plausible explanation for this finding would be a mutation in one of the genes of the same pathway. This pathway is mutated in cancer18 and in a group of genetic conditions collectively known as RASopathies,19 where all the mutations lead to an inappropriate activation of the pathway itself. For this reason a candidate gene analysis was performed in the two LCLIgAs for some of the genes involved in the RASopathies with the goal of identifying a mutation leading to silencing of ERK1/2, but no mutations were found in RAF1 and BRAF genes.
On the basis of what was found in the IgAN family and in two sporadic IgAN patients, a suggestive hypothesis leads to the possibility that ERK1/2 inhibition could be the common lesion in all IgAN cases. Support to this hypothesis is derived from the observation of sudden worsening of kidney function because of the IgA deposits in a patient treated with Sorafenib,20 an inhibitor of the ERK pathway, as well as other kinases. Therefore, the LCL157 was treated in vitro for 7 days with sorafenib, followed by the assessment of SPRY2 transcript levels, IgA and ERK1/2 phosphorylation. After 3 days lymphoblasts started to die as a probable result of a multiple kinase inhibition. Treatment was maintained until the 7th day. In spite of the reduced cell number and block of protein synthesis21 IgA production increased, and, as a result of ERK1/2 inhibition, expression of SPRY2 transcript was reduced (Figure 3c). This finding supports the hypothesis that a block in the MAPK/ERK leads to an increased IgA production.
To see whether the ERK1/2 block has an effect on cell proliferation, the proliferative kinetics was evaluated in all cell lines after a BrdU pulse-chase, but no significant differences were found among the different cell lines (Supplementary Figure S5).
Discussion
A heterozygous missense variant, p.(Arg119Trp) in the SPRY2 gene was identified in a family with an AD IgAN phenotype. The SPRY2 mutation is the only novel variant found in the chromosome 13 region and is predicted to be pathogenic by bioinformatics analysis. The latter indicated that it falls in a highly conserved amino-acid residue. The frequency of the W119 allele is estimated to be lower than 1/15 000 as it was not found in dbSNP132, in 52 controls from the same geographic area, and in 1000genomes and ESP6500. All these data combined strongly suggest that the SPRY2 is the gene responsible for the IgAN phenotype in this family.
To confirm the role of SPRY2 in IgAN, the 1000genomes and ESP6500 databases were interrogated looking for SPRY2 missense and null variants and their relative frequency. This search showed that germline null mutations have never been reported in the general population. Moreover, except for two common missense polymorphisms, also found in our cohorts of patients and controls, only 12 missense rare mutations were found in SPRY2 in the ESP6500 database, 3 of which had a frequency of ~1/3 000 and 9 of 1/13 000. These observations suggest that this gene is mutation intolerant, and any dosage/functional imbalance could lead to an observable phenotype or early lethality.
To support our conclusions the coding sequence of SPYR2 was also investigated in a cohort of sporadic IgAN patients, from Sicily and central Italy, but no additional mutations were identified. This is not surprising because several linkage and association studies identified several loci for IgAN across the genome, suggesting a strong genetic heterogeneity for this condition. Moreover, these same studies suggest that familial cases and sporadic cases do not share mutations in the same genes, as indicated by the lack of overlap among loci identified in AD families and loci identified in association studies in sporadic patients.
IgA-producing B lymphocytes probably represent the affected tissue lineage in IgAN. SPRY2 is expressed in B cells and in B lymphoblasts, and its relevant role in B lymphocytes is well documented.14 SPRY2 has a prevalent role as an inhibitor of the MAPK/ERK pathway (Figure 3d), mostly through its interaction with BRAF, a kinase activated by several receptors with tyrosine kinase activity. SPRY2 transcription is part of a feedback loop, mediated by SPRY2 itself as well as by pERK1/2, which finely modulates/represses the MAPK/ERK pathway activation.22 SPRY2 is also post-transcriptionally regulated by serine and tyrosine phosphorylation. Several studies have shown that serine at positions 112 and 121 are important for its inhibitory interaction, when de-phosphorylated, with BRAF.12 Instead, serine 112/121 phosphorylation releases the inhibitory block of BRAF and consequently leads to SPRY2 ubiquitination and degradation.12 The W119 variant not only affects a highly conserved amino-acid residue, but it is located in the recognition sequence, RXS, of MNK1, one of the SPRY2 kinases, suggesting that its presence is associated with S121 hypophosphorylation. As a consequence, mutated SPRY2 could be more stable, as we showed in Supplementary Figure S2, and lock BRAF in an inactive form. This inhibition in turn could block MAPK/ERK activation.22 This hypothesis was verified showing that in EBV-transformed B LCL ERK1/2 is hypo-phosphorylated in SPRY2 variant carriers and normal in the only family member, who tested negative for the variant. Moreover, as mentioned above, pERK1/2 is a strong transcriptional activator of SPRY2,20, 23, 24 whereas its block causes SPRY2 transcriptional silencing. This tight transcriptional feedback explains the strong reduction observed in SPRY2 transcript, without apparent reduction in the protein levels.
Usually the MAPK/ERK pathway is found activated in cancer and in errors of development. For this reason it was surprising to find that it is downregulated in EBV-transformed immortal cell lines from IgAN patients. This finding implies that lack of activation or permanent inhibition of the MAPK/ERK pathway could be the final pathway where different mutations converge in different patients with IgAN.
Three clinical reports25 provide support to this hypothesis: these concerned two patients treated with EGFR inhibitors (cetuximab and gefitinib) and one patient treated with sorafenib, who showed a marked worsening of their kidney function because of IgA deposits. This very rare complication for cetuximab, gefitinib and sorafenib has never been reported during clinical trials and it is probably related to the fact that the drug triggered or exacerbated a latent IgAN, already present in those patients, by blocking the MAPK/ERK pathway. The same phenotype was reproduced in vitro treating LCL157 with sorafenib for 7 days showing an increase in the amount of IgA produced.
How the inhibition of the MAPK/ERK causes the IgAN phenotype is still unknown. Except for hypo-galactosylated IgA, also present in the normal population, there are no clear pathogenic mechanisms for this disease. The LCLs carrying a SPRY2 mutation and LCL156/157 cell lines shared two common characteristics: ERK1/2 reduced phosphorylation and an increased IgA production. These two phenotypes were inversely correlated, as the lower was the phosphorylation level of ERK1/2 the higher was IgA production. For this reason it will be pivotal to investigate the role of the MAPK/ERK pathway in a more physiological setting in B lymphocytes and to verify whether increased local IgA production consequent to MAPK/ERK blockage leads to IgA nephropathy in a larger cohort of IgAN patients.
References
Wyatt RJ, Julian BA : IgA nephropathy. N Engl J Med 2013; 368: 2402–2414.
Cuevas X, Lloveras J, Mir M, Aubia J, Masramon J : Disappearance of mesangial IgA deposits from the kidneys of two donors after transplantation. Transplant Proc 1987; 19: 2208–2209.
Iwata Y, Wada T, Uchiyama A et al: Remission of IgA nephropathy after allogeneic peripheral blood stem cell transplantation followed by immunosuppression for acute lymphocytic leukemia. Intern Med 2006; 45: 1291–1295.
McDonald SP, Russ GR : Recurrence of IgA nephropathy among renal allograft recipients from living donors is greater among those with zero HLA mismatches. Transplantation 2006; 82: 759–762.
Meng H, Ohtake H, Ishida A, Ohta N, Kakehata S, Yamakawa M : IgA production and tonsillar focal infection in IgA nephropathy. J Clin Exp Hematopathol 2012; 52: 161–170.
Gharavi AG, Kiryluk K, Choi M et al: Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet 2011; 43: 321–327.
Yu XQ, Li M, Zhang H et al: A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat Genet 2011; 44: 178–182.
Bisceglia L, Cerullo G, Forabosco P et al: Genetic heterogeneity in Italian families with IgA nephropathy: suggestive linkage for two novel IgA nephropathy loci. Am J Hum Genet 2006; 79: 1130–1134.
Gharavi AG, Yan Y, Scolari F et al: IgA nephropathy, the most common cause of glomerulonephritis, is linked to 6q22-23. Nat Genet 2000; 26: 354–357.
Paterson AD, Liu X-Q, Wang K et al: Genome-Wide Linkage Scan of a Large Family with IgA Nephropathy Localizes a Novel Susceptibility Locus to Chromosome 2q36. J Am Soc Nephrol 2007; 18: 2408–2415.
DaSilva J, Xu L, Kim HJ, Miller WT, Bar-Sagi D : Regulation of sprouty stability by Mnk1-dependent phosphorylation. Mol Cell Biol 2006; 26: 1898–1907.
Brady SC, Coleman ML, Munro J, Feller SM, Morrice NA, Olson MF : Sprouty2 association with B-Raf is regulated by phosphorylation and kinase conformation. Cancer Res 2009; 69: 6773–6781.
Edwin F, Anderson K, Patel TB : HECT domain-containing E3 ubiquitin ligase Nedd4 interacts with and ubiquitinates Sprouty2. J Biol Chem 2010; 285: 255–264.
Frank MJ, Dawson DW, Bensinger SJ et al: Expression of sprouty2 inhibits B-cell proliferation and is epigenetically silenced in mouse and human B-cell lymphomas. Blood 2009; 113: 2478–2487.
Lao DH, Yusoff P, Chandramouli S et al: Direct binding of PP2A to Sprouty2 and phosphorylation changes are a prerequisite for ERK inhibition downstream of fibroblast growth factor receptor stimulation. J Biol Chem 2007; 282: 9117–9126.
Tsavachidou D, Coleman ML, Athanasiadis G et al: SPRY2 is an inhibitor of the ras/extracellular signal-regulated kinase pathway in melanocytes and melanoma cells with wild-type BRAF but not with the V599E mutant. Cancer Res 2004; 64: 5556–5559.
Clybouw C, McHichi B, Mouhamad S et al: EBV infection of human B lymphocytes leads to down-regulation of Bim expression: relationship to resistance to apoptosis. J Immunol 2005; 175: 2968–2973.
Dhillon AS, Hagan S, Rath O, Kolch W : MAP kinase signalling pathways in cancer. Oncogene 2007; 26: 3279–3290.
Tidyman WE, Rauen KA : The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev 2009; 19: 230–236.
Jonkers IJ, van Buren M : Nephrotic-range proteinuria in a patient with a renal allograft treated with sorafenib for metastatic renal-cell carcinoma. Clin Exp Nephrol 2009; 13: 397–401.
Rahmani M, Davis EM, Crabtree TR et al: The kinase inhibitor sorafenib induces cell death through a process involving induction of endoplasmic reticulum stress. Mol Cell Biol 2007; 27: 5499–5513.
Ozaki K, Kadomoto R, Asato K, Tanimura S, Itoh N, Kohno M : ERK pathway positively regulates the expression of Sprouty genes. Biochem Biophys Res Commun 2001; 285: 1084–1088.
Sasaki K, Anderson E, Shankland SJ, Nicosia RF : Diffuse proliferative glomerulonephritis associated with cetuximab, an epidermal growth factor receptor inhibitor. Am J Kidney Dis 2013; 61: 988–991.
Masutani K, Fujisaki K, Maeda H et al: Tubulointerstitial nephritis and IgA nephropathy in a patient with advanced lung cancer treated with long-term gefitinib. Clin Exp Nephrol 2008; 12: 398–402.
Suzuki H, Moldoveanu Z, Hall S et al: IgA1-secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J Clin Invest 2008; 118: 629–639.
Acknowledgements
We thank Elisabetta Tabolacci, Noemi Michetti and Maurizio Genuardi for critical reading of the manuscript and the rest of the Institute of Medical Genetics for sharing discussions and ideas. We would like to acknowledge with this paper the scientific life and work of Dr Giovanni Neri. He retired as head of our department in October 2013, but his guidance and scientific leadership, during the last 30 years transformed many undergraduate medical students in doctors and researchers and his legacy will last forever through them. ES is supported by the AIRC grant MFAG 9141 and received an award from the Fondazione ‘Guido Berlucchi’. AM received an award from the association A.Ma.R.T.I. to support this project.
Web resources: www.1000genomes.org/; evs.gs.washington.edu/; sift.jcvi.org/; genetics.bwh.harvard.edu/pph2/; mmb2.pcb.ub.es:8080/PMut; www.mutationtaster.org/; www.pantherdb.org/pathway/; www.cbs.dtu.dk/services/NetPhos/.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Milillo, A., La Carpia, F., Costanzi, S. et al. A SPRY2 mutation leading to MAPK/ERK pathway inhibition is associated with an autosomal dominant form of IgA nephropathy. Eur J Hum Genet 23, 1673–1678 (2015). https://doi.org/10.1038/ejhg.2015.52
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2015.52
This article is cited by
-
Retinoblastoma gene expression profiling based on bioinformatics analysis
BMC Medical Genomics (2023)
-
Increased retinal drusen in IgA glomerulonephritis are further evidence for complement activation in disease pathogenesis
Scientific Reports (2022)
-
Expert consensus guidelines for the genetic diagnosis of Alport syndrome
Pediatric Nephrology (2019)
-
Defective activation of the MAPK/ERK pathway, leading to PARP1 and DNMT1 dysregulation, is a common defect in IgA nephropathy and Henoch-Schönlein purpura
Journal of Nephrology (2018)
