
Abstract
Polymerase gamma (POLG) is the gene most commonly involved in mitochondrial disorders with mitochondrial DNA instability and causes a wide range of diseases with recessive or dominant transmission. More than 170 mutations have been reported. Most of them are missense mutations, although nonsense mutations, splice-site mutations, small deletions and insertions have also been identified. However, to date, only one large-scale rearrangement has been described in a child with Alpers syndrome. Below, we report a large cohort of 160 patients with clinical, molecular and/or biochemical presentation suggestive of POLG deficiency. Using sequencing, we identified POLG variants in 22 patients (18 kindreds) including five novel pathogenic mutations. Two patients with novel mutations had unusual clinical presentation: the first exhibited an isolated ataxic neuropathy and the second was a child who presented with endocrine signs. We completed the sequencing step by quantitative multiplex PCR of short fluorescent fragments (QMPSF) analysis in 37 patients with either only one POLG heterozygous variant or a family history suggesting a dominant transmission. We identified a large intragenic deletion encompassing part of intron 21 and exon 22 of POLG in a child with refractory epilepsia partialis continua. In conclusion, we describe the first large French cohort of patients with POLG mutations, expanding the wide clinical and molecular spectrum observed in POLG disease. We confirm that large deletions in the POLG gene are rare events and we highlight the importance of QMPSF in patients with a single heterozygous POLG mutation, particularly in severe infantile phenotypes.
Similar content being viewed by others

Introduction
Mitochondrial diseases are, a heterogeneous group, caused by genetic defects in mitochondrial DNA (mtDNA) or in nuclear genes that encode proteins essential to mitochondrial biogenesis. Polymerase gamma (POLG) is the gene most frequently involved in mitochondrial disorders with mtDNA instability. It encodes the catalytic subunit of the DNA polymerase gamma (POLG), the sole DNA polymerase found in mammalian mitochondria, which is essential for mtDNA replication and repair. POLG consists of a catalytic subunit and a homodimeric form of an accessory subunit encoded by POLG2. The catalytic subunit contains three different domains: a 3′→5′ exonuclease domain, a ‘linker’ domain and a highly conserved polymerase domain.1 Mutations in POLG have been identified in severe mtDNA depletion syndromes, such as Alpers syndrome, as well as in mtDNA multiple deletion disorders such as ataxia, chronic progressive external ophthalmoplegia (CPEO), mitochondrial recessive ataxia syndrome, sensory ataxic neuropathy with dysarthria and ophthalmoparesis (SANDO) and spinocerebellar ataxia with epilepsy (SCAE)2, 3, 4, 5 (http://tools.niehs.nih.gov/polg/). Most of these mutations were missense mutations. Nonsense mutations, splice-site mutations, small deletions and insertions have also been described.5 To date, only one large-scale rearrangement involving POLG has been identified by array-CGH, but in most laboratories, the detection of large-scale rearrangements is not performed as a routine analysis.6, 7
In this study, we performed POLG sequencing in 160 French patients presenting a phenotype compatible with POLG deficiency. We describe clinical and molecular data for 22 patients from 18 kindreds harboring POLG variants including two patients with unusual clinical presentations. Among the 22 patients, we identified 17 different variants among which five were novel pathogenic mutations. Twelve affected individuals had two mutations and 10 patients harbored only one mutation. We also designed a new quantitative multiplex PCR of short fluorescent fragments (QMPSF) approach to easily identify large-scale rearrangements, and this new technique was performed on DNA samples from 37 patients with either only one POLG variant or a family history suggesting a dominant transmission. This approach led us to identify a large deletion (846 bp in length) encompassing part of intron 21 and exon 22 in a 3-year-old patient with refractory epilepsia partialis continua (EPC).
Patients and methods
Patients
A total of 160 patients from 156 kindreds were analyzed for POLG mutations in this study. For each patient we obtained written informed consent. Patients were diagnosed in French referral centers and presented a phenotype compatible with POLG mutations.7
mtDNA molecular analysis
Total DNA was extracted using standard phenol–chloroform procedure. Long-range PCR was performed as described previously 8 and Southern blot was used when deletions were detected by PCR method.9 mtDNA quantification in the muscle was performed by real-time quantitative PCR as described by Rouzier et al.10 Primer sequences and PCR conditions are available on request.
Sequencing analysis of POLG
The coding regions of POLG (NM_002693.2) were sequenced as described previously.11 A mutation was considered as new if it was neither present in the Human DNA Polymerase Gamma Mutation Database (http://tools.niehs.nih.gov/polg/), in the NCBI (http://www.ncbi.nlm.nih.gov/sites/), UCSC (http://genome.ucsc.edu/cgi-bin/hgGateway) and EVS databases (http://evs.gs.washington.edu/EVS/) nor published. To determine the pathogenicity of new variants, we used the following criteria: (1) the evolutionary conservation of the amino-acid residue, (2) the location of the amino-acid residue in an important functional domain, (3) the cosegregation of the variant with the disease within the family, (4) in silico predictions by PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/) and (5) the absence of the variant in 100 normal controls from the Caucasian population.
Crystal structure and structural insight of mutations
The 2.2 Å coordinate sets for the holoenzyme (pdb code: 3ikm) and Swiss Pdb Viewer 4.1 (http://spdbv.vital-it.ch) were used to visualize the structure and analyze the structural insight into PolγA mutations.
QMPSF
QMPSF is a semiquantitative method based on a simultaneous amplification by PCR (multiplex PCR) of short exonic fragments in a single tube using dye-labeled primers.12, 13 The PCR is performed with a limited number of cycles so that DNA yield generated in the exponential step may be comparable between different samples. Universal extensions are added at 5′ of sense and antisense primers, so that it homogenizes melting temperatures (Tm) between the various hybridization segments used in multiplex. An additional fragment from the SNRPN gene (NM_022807.2), which is not involved in the critical regions, is coamplified as a control. After PCR, amplicons are size differentiated by capillary electrophoresis using an ABI PRISM 3130 DNA analyser sequencer (Life Technologies, Saint Aubin, France). Data are analyzed using genemapper software (Life Technologies). The areas of the peaks corresponding to the regions to be explored are compared between the patient sample and the DNA of control individuals. Quantitative changes are detected by an increase or decrease in the areas of the corresponding fluorescent peaks.14
Short fragments of the 22 POLG coding exons (NM_002693.2) were simultaneously amplified in three multiplex PCR using dye-labeled primers. For exon 2, we used two primer sets to reduce amplicon size. When two small exons were separated by small introns, they were amplified in the same amplicon (exons 5–6, 8–9, 15–16 and 19–20). We designed primers using Primer 3 software (http://frodo.wi.mit.edu/) (Supplementary Table 1). As a positive control, we used DNA from a patient with a large deletion encompassing exons 15–21, kindly provided by Dr Alison Compton (Murdoch Childrens Research Institute, The Royal Children's Hospital, Parkville, Victoria, Australia).6
Molecular characterization of exon 22 deletion (patient 12)
First, we performed a PCR on genomic DNA (forward primer, 5′-CTTTGGGTGAGCGTTCCTTG-3′; reverse primer, 5′-AAGCACAGTTTGTTTTTCTGTCA-3′) and the amplified product was sequenced using BigDye Terminator v.3.1 Cycle Sequencing Kit (Life Technologies) according to the manufacturer’s instructions. We supplemented genomic DNA analysis with RNA analysis. Total RNAs were extracted from primary fibroblast culture using Trizol reagent (Life Technologies) according to the manufacturer’s instructions. Reverse transcription was performed with the First Strand cDNA Synthesis kit for RT-PCR (Roche Applied Sciences, Meylan, France) and was followed by PCR amplification (forward flanking primer, 5′-ATGACCAGGTTCGCTCCTG-3′; reverse, 5′-GGAGCCTTTGGTGAGTTCAA-3′).
RT-PCR analysis of the full-length POLG cDNA (patients 13, 14, 15)
RNA extraction from muscle or fibroblast cultures and cDNA synthesis were performed as described above. PCR amplification of the POLG coding region was performed in one step with a forward primer in exon 2 (5′-GAGACAGGACGTGTCTCTCT-3′) and a reverse primer in exon 23 (5′-GCTACTGAAAAATGGCTGGCC-3′). PCR conditions are available on request.
Results
Among the 160 patients analyzed, POLG sequencing identified variants in a total of 22 individuals, corresponding to 18 families. Clinical, biochemical, histopathological and molecular data from these patients are summarized in Table 1. Twelve patients, from 11 families, carried two deleterious mutations, among which four were novel. Ten patients, from seven families, carried only one heterozygous variant. Data from the 138 patients (corresponding to 138 kindreds) in whom we suspected but did not find a POLG mutation are described in Supplementary Table 2.
Identification of 12 patients harboring two POLG mutations
Among the 12 patients with two POLG mutations (Table 1), 10 had age-of-onset and clinical features similar to the previous reported series.7 Children (age of onset 15 months–10 years) presented seizures, developmental delay and hepatopathy, whereas peripheral neuropathy, ataxia, myopathy and CPEO/ptosis were found in adult individuals (age of onset 15–63 years). Five patients presented with a refractory EPC or recurrent status epilepticus, among which only one had hepatic involvement.
Eight patients had muscle biopsies and a histological pattern of mitochondrial myopathy with ragged-red fibers (RRFs) and cytochrome c oxidase (COX)-negative fibers, and/or lipid accumulation was found in six out of the eight cases. mtDNA instability, with either multiple deletions or depletion, was present in all but one patients in muscle or liver (9/10). Biochemical respiratory chain (RC) analysis was performed on nine samples (muscle, liver or fibroblasts) and a deficiency was observed on only four samples. Interestingly, two patients with novel mutations had uncommon clinical features.
Patient 6 had developed a slowly progressive isolated neuropathy. At 5 years of age, she presented generalized areflexia, suggesting childhood onset. She became symptomatic at 20 years of age with sensory ataxia followed by distal muscle weakness and hypoesthesia of the lower limbs. At 53 years of age, the neuropathy was still isolated with no other neurological or muscular clinical signs. Electromyography showed axonal sensorimotor polyneuropathy and a muscle biopsy revealed numerous RRF, COX-negative fibers (5%) and mtDNA multiple deletions. This patient was compound heterozygous for two novel missense mutations: p.Met464Thr and p.Lys768Glu. The patient’s mother was heterozygous for the p.Lys768Glu variant, suggesting that both mutations were in trans.
The crystal structure of human DNA Polγ holoenzyme is a heterotrimer containing one PolγA subunit and a dimeric PolγB subunit. The PolγA active site domain adopts a canonical ‘right-hand’ polymerase configuration with subdomains of ‘fingers’, ‘palm’ and ‘thumb’ that bind template DNA and substrate nucleotide triphosphate, as well as catalyze phosphodiester bond formation.15 Between the polymerase (pol) and exonuclease (exo) domains lies a spacer domain containing a single fold that interacts primarily with only one PolγB monomer. The spacer domain is located far from the exo and pol domains and connects to them only through the long helices of the ‘thumb’ subdomain. The spacer has two obvious subdomains, a globular intrinsic processivity (IP) subdomain (residues 475–510 and 571–785) and an extended accessory-interacting determinant (AID) subdomain (residues 511–570). IP subdomain explains the IP of PolγA, and the AID subdomain forms an important interface with PolγB that is essential for increased processivity of the holoenzyme. Methionine at position 464 is located in the catalytic pol domain at the interface of interaction with the accessory subunit monomer, and may alter the oligomerization. Lysine at position 768 is located in the globular IP subdomain in the spacer region. The replacement of a basic with an acidic residue will destabilize the spacer region spatially connecting the exo and pol domains, and thus functionally altering the POLG enzymatic activity (Supplementary Figure 1).
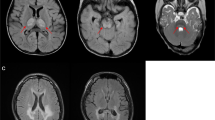
A second individual (patient 10a) also had uncommon clinical features. This young girl had suffered from migraines and lack of coordination since early childhood. At 7 years of age, she developed a multiendocrine disease with adrenocortical insufficiency and hypothyroidy. At 8 years of age, she developed a refractory status epilepticus followed by coma. Brain magnetic resonance imaging (MRI) showed white matter involvement. She died a few months later. Her younger sister, patient 10b, had suffered from lack of coordination since childhood. At 8 years of age, she developed left partial seizures leading to refractory EPC during an episode of asthenia and headache. No endocrine dysfunction was noticed. A few weeks later, clinical examination revealed cerebellar ataxia, slight ptosis, left hemiparesis and lateral homonymous hemianopsia. After initial improvement, she developed contralateral EPC with cortical blindness. MRI showed initially lesions of right thalamus and right parieto-occipital cortex, and then bilateral and symmetrical lesions. Two novel missense mutations, p.Leu79Phe and p.Ser1080Ile, were identified in the two sisters. The healthy parents were both heterozygous carriers. Replacement of leucine at position 79 adjacent to DNA-binding channel with a bulkier phenylalanine residue is predicted to result in reduced DNA binding (Supplementary Figure 1). The serine at position 1080 is located in a small subdomain (residues 1050–1095) that partially blocks the DNA-binding channel. Thus, the p.Ser1080Ile mutation located at the DNA–POLG interaction channel will probably alter the proper positioning of the DNA template.
Identification of seven patients harboring one POLG mutation
Among the seven patients with only one heterozygous POLG mutation, six carried a mutation previously reported in recessive cases (http://tools.niehs.nih.gov/polg/), and one patient (patient 16) had a novel nonsense mutation, p.Gln975* (Table 1).
Patient 12 was the first child of unrelated healthy parents. Pregnancy and birth were unremarkable. She had had a moderately delayed psychomotor development with sitting posture at 9 months of age and walking at 20 months of age. At 3 years old, she presented with acute partial status epilepticus. After this first episode, she developed refractory EPC with neurological regression leading to a severe encephalopathy. At 4 years, she was unable to hold her head up or to sit and had poor contact. Muscle biopsy showed lipid accumulation and spectrophotometric measurements of the individual RC complexes revealed CIII deficiency, associated with decreased activitiy of complexes II and V. In the liver, specific activity of complexes I, III and V was also affected. mtDNA depletion was identified by qPCR (25% of the mean normal control amount of mtDNA relative to nuclear DNA in the muscle and 30% in the liver). Sequencing identified only one heterozygous mutation (p.Trp748Ser) in POLG, inherited from the mother.
At 56 years old, patient 13 developed a motor neuron disease with severe tetraparesis, dysphonia and respiratory insufficiency leading to death a few years later. Muscular biopsy was strongly evocative of mitochondrial disease with a generalized RC deficiency, multiple mtDNA deletions and decreased mtDNA copy number in the muscle (39%). This patient was heterozygous for p.Ala467Thr,5 the most frequent recessive mutation found in POLG.
Patient 14 was a 5-year-old girl presenting with muscle weakness, amyotrophy, myoclonic epilepsy and renal tubulopathy. Cerebrospinal fluid and blood lactate concentrations were elevated. Muscle biopsy showed lipid myopathy with no biochemical RC deficiency and neither depletion nor deletions of mtDNA. She was heterozygous for the p.Pro1073Leu mutation, previously described in recessive hepatic encephalopathy.7, 16
Patient 15 was a 14-year-old boy born of healthy consanguineous parents. He presented delayed psychomotor development and muscular weakness associated with hepatopathy. He had recurrent episodes of cholestasis and cytolysis and liver biopsy showed fibrosis and severe steatosis. He was heterozygous for the p.Arg1187Trp mutation, previously identified in a patient with mitochondrial depletion syndrome and T-cell immunodeficiency.17 However, the involvement of this mutation in the patient’s disease is not obvious given that the pathogenesis of this mutation is still unclear and the parents are consanguineous.
Patient 16 was a 6-year-old girl with psychomotor delay and generalized seizures. Her development was marked by the appearance of abnormal movements. Brain MRI showed lesions of thalami. A novel variant, p.Gln975*, was identified in this patient and was inherited from the father.
Patients 17a, b and c were three siblings with axonal sensorimotor neuropathy, ataxic or not, one of whom presented with parkinsonism for one of them. Patient 17a underwent a muscle biopsy, which showed lipid accumulation and multiple mtDNA deletions. At first, the absence of family background suggested recessive transmission but the identification of a very mild axonal neuropathy in patient 17c’s daughter could argue in favor of dominant transmission. The three patients were heterozygous for the p.Arg232His and shared the same POLG polymorphisms. The p.Arg232His mutation has previously been described in trans with three other mutations in patients with Leigh, Alpers or Charcot–Marie–Tooth disease.18, 19, 20
Patient 18a exhibited late-onset CPEO with a sensory neuronopathy and a Parkinsonian syndrome. Her sister, patient 18b, had a similar phenotype without Parkinsonian syndrome. Both were found to be heterozygous for the p.Asp1184Asn mutation previously found in trans with p.Asn468Asp in a patient with PEO and tetraparesis and in a severe childhood multisystem disorder.21, 22 As in the previous family, all affected individuals shared the same POLG polymorphisms (6 in family 17 and 10 in family 18) and recessive transmission seemed likely. However, both parents died before the age at which the first clinical symptoms developed in patients 18a and b.
Three other individuals were found to be heterozygous for the p.Gly517Val, p.Gly268Ala and p.Tyr831Cys polymorphisms found at a high percentage in control populations (2.2%, 1.7% and 1.1%, respectively) (http://evs.gs.washington.edu/EVS/).
QMPSF analysis allowed identification of a large deletion in patient 12
QMPSF analysis was performed in the seven unrelated patients carrying only one POLG mutation and in the 30 patients with no identified mutation but with a family history suggesting dominant transmission. Among the 37 samples analyzed, we identified a novel large POLG deletion. In patient 12, QMPSF analysis showed a 50% decrease in exon 22 peak area, which suggested a heterozygous deletion of exon 22 region. Exon 21 and 23 area peaks were not decreased and heterozygous polymorphisms were present in these exons, ruling out deletion of exons 21 and 23 (Figure 1 and Supplementary Table 3). We designed a PCR assay to characterize the breakpoints of the deleted allele. Using a forward primer in the 5′ part of intron 21 and a reverse primer in intron 22, we identified a large 846-bp deletion encompassing the 3′ part of intron 21 and the 49 first nucleotides of exon 22 (c.3482+208 to c.3531) (Figure 2). The deletion was inherited from the father, thus confirming the localization in trans of the two mutations. Sequencing of the patient’s cDNA showed that both wild-type and deleted alleles were expressed and that nonsense-mediated mRNA decay was not occurring. The deletion resulted in the use of a downstream cryptic acceptor splice site, leading to an in-frame skipping of the first 20 amino acids encoded by exon 22, corresponding to part of the highly conserved C-terminal polymerase domain (p.Arg1161_Phe1180del20) (Figure 3).

QMPSF profiles corresponding to the three multiplex PCR. N: Normal control DNA; D: Deleted control with deletion of introns 14–21 (Exon 14 also appears deleted because the reverse primer used to amplify amplicon 14 spans the 5' breakpoint of the deletion.); P12: Patient 12 with deletion of exon 22 (Two peaks are observed for exon 23 corresponding to a wildtype allele and an allele with a G insertion corresponding to the rs3087377 polymorphism.); M1: multiplex 1; M2: multiplex 2; M3: multiplex 3. Arrows mark the deleted exons. The heterozygous deletions are detected on the basis of a twofold reduction of the peak area.

Genomic DNA deletion analysis in patient 12. (a) PCR analysis using primers located in introns 21 and 22. M: molecular weight marker. In control (C) a band of 1433 bp corresponding to the full-length product is detected. In patient 12 (P12), a smaller band of 587 bp is observed. The non-deleted allele is not amplified in these PCR conditions. (b) Sequencing of the PCR deletion product (P12). The 846-bp deletion is located between the breakpoints c.3482+208 and c.3531.

cDNA analysis in patient 12. Upper panel: wild-type (wt) sequence of a control sample. The red dashed line corresponds to the junction between exons 21 and 22. Lower panel: sequencing of patient 12 cDNA showed the wt and the deleted alleles. The deletion resulted in the use of a downstream cryptic acceptor splice site (black rectangle) leading to an in-frame skipping of the first 20 amino acids encoded by exon 22 (p.Arg1161_Phe1180del20). E21: exon 21; E22: exon 22.
RT-PCR analysis of the full-length POLG cDNA in patients 13, 14 and 15 revealed no abnormalities
To supplement the QMPSF approach, we analyzed the POLG full-length transcript in patients carrying one mutation when samples from muscle or fibroblast culture were available. cDNA analysis revealed no abnormalities and demonstrated a biallelic expression in all three patients.
Discussion
In this study, we performed POLG analysis in 160 French patients, corresponding to 156 families, who presented a phenotype evocative of a POLG deficiency. Sequencing identified 12 patients (11 families out of 156, 7%) who carried two pathogenic recessive mutations and 10 (7 families out of 156, 4.5%) who harbored a single deleterious mutation. These results are consistent with previous reports.5, 22 Using QMPSF, we identified a second mutation in only one additional patient. Our results confirm the previously described mutation frequencies, the four most common mutations p.Ala467Thr, p.Trp748Ser, p.Gly848Ser and p.Thr914Pro accounting for 60% of disease alleles in cases with confirmed autosomal recessive phenotypes due to POLG mutations.5 However, the p.Trp748Ser mutation is the most common mutation in our cohort (5/20 alleles, 25% of all mutant alleles) contrary to previous articles, which reported the p.Ala467Thr to be the most common POLG mutation in patients with autosomal recessive inheritance.5, 23, 24 The p.Ala467Thr mutation was the second most common mutation in our cohort (4/20 alleles, 20% of all mutant alleles).
We identified five novel variants including one nonsense mutation and four missense mutations, which fitted the required pathogenicity criteria and corresponded to damaging changes predicted by PolyPhen-2 (Table 2; Supplementary Figure 1). The p.Met464Thr and p.Lys768Glu novel mutations were identified in patient 6 who presented an isolated sensory ataxia. To our knowledge, POLG mutations have never previously been described in isolated sensory ataxic neuropathy. Following these results, we performed POLG sequencing in 30 patients with isolated axonal sensory neuropathy and a negative causal analysis. We identified no POLG mutation (data not shown), thus confirming that POLG is not classically responsible for isolated neuropathy. A special feature of this case is the early onset at 5 years of age and the mitochondrial pattern observed in the muscle biopsy with mtDNA multiple deletions. RC deficiency was also observed on fibroblasts. To conclude, POLG sequencing should not be used as a systematic analysis in isolated neuropathies but must be performed in early-onset forms with negative causal analysis and if additional arguments, in favor of mitochondrial disease, are found in tissue biopsies. However, it is important to keep in mind that children with recessive POLG disease who show no clinical signs of peripheral neuropathy can have subclinical peripheral nerve involvement, which can be detected by electrophysiological testing.
Two other novel mutations, p.Leu79Phe and p.Ser1080Ile, were identified in patients 10a and b, who both exhibited refractory status epilepticus and EPC. Interestingly, before the first symptoms evocative of POLG deficiency, patient 10a developed an unusual multiendocrine disease, which delayed diagnosis. To date, only one report has described multiendocrine disease in a family with a dominant POLG mutation.25 However, the role of POLG in this study is not confirmed given that the identified mutation was recently reclassified as an ‘unclassified variant’ (p.Gly517Val).26 Moreover, contrary to a previous report which suggested that POLG was not a common cause of isolated epilepsy,27 in our cohort we found a high frequency of epilepsy, EPC or refractory recurrent status, without hepatic involvement (five out of 13 cases with two confirmed mutations). Histological or biological signs of mitochondrial disease were not always observed (patient 8). Our data suggest that such types of epilepsy are strongly evocative of POLG mutations and, when faced with EPC or refractory recurrent status, POLG analysis should be performed in children even without hepatic involvement and before tissue biopsies. The analysis of key symptoms regarding the presence of POLG mutations found in Supplementary Table 4 is in favor of this conclusion; CPEO being significantly associated with POLG mutations in adult patients.
A novel p.Gln975* mutation was identified in patient 16. This variant is located in the polymerase domain leading to a truncated protein without the last 264 amino acids. This mutation is likely recessive, as demonstrated for the p.Lys965* mutation,28 but we failed to identify a second one. As haploinsufficiency of POLG has not been associated with a dominant disease phenotype, we cannot exclude that this heterozygous nonsense mutation is a coincidental finding.
Sequencing identified a single heterozygous variant in half of patients with identified POLG variants as reported previously.5, 7 This method only detects point mutations or small deletions/duplications in the coding sequence. However, mutations located in introns or promoter regions or large rearrangements could be missed. On the hypothesis that a second mutation could be a large heterozygous deletion on the other allele, Tang et al7 performed oligonucleotide array CGH analyses on 81 samples and identified the first heterozygous large intragenic deletion of POLG encompassing exons 15–21 in a patient with Alpers syndrome because of mtDNA depletion.6 This patient was compound heterozygous for the common p.Ala467Thr mutation and a large 4.7-kb length deletion leading to an in-frame skipping of all three of the C-terminal polymerase motifs in a truncated POLG protein. These results suggest that large POLG rearrangements could be rare events.
QMPSF was initially developed for the detection of deletions and duplications in genes involved in nonpolyposis colorectal syndrome.29 Since this first report, this simple, rapid and reproducible method has been used in diagnostic laboratories to detect rearrangements in numerous genes such as CFTR30 or SMN1.31 Herein, we designed a QMPSF strategy to identify POLG large-scale rearrangements. Among the seven patients with only one identified mutant POLG allele, we identified a large intragenic deletion in a young child carrying a single heterozygous p.Trp748Ser mutation. In this case, as well in the case reported by Compton et al,6 deletions result in a truncated POLG protein lacking part of the C-terminal polymerase domain. Although no clear genotype–phenotype correlation could be established in patients with POLG mutations, both patients had a severe phenotype with epilepsy and mtDNA depletion in liver (8% and 30%, respectively). Large deletions associated with other mutations resulting in very low wild-type DNA polymerase activity, such as p.Ala467Thr or p.Trp748Ser,32, 33 could lead to severe cases of childhood disease.
We also analyzed POLG full-length cDNA when material allowing RNA extraction was available (patients 13–15) without finding any abnormality. In terms of diagnostic strategy, transcript analysis is an excellent first-line approach. However, QMPSF should be available in a diagnostic laboratory when material allowing RNA study is lacking.
The possibility of a locus heterogeneity involving other functionally related genes, such as POLG2, TWINKLE, ANT1, DGUOK, MPV17, TK2 and RRM2B, has also been suspected,5 and a digenic inheritance involving POLG and TWINKLE has been reported previously.34 We sequenced allother genes involved in mtDNA instability in all patients from our cohort and identified no mutations, suggesting that digenism is also a rare event.
Finally, given the size of the cohort studied and the carrier frequency of the recessive POLG mutations, estimated around 1:50–1:100 (http://www.ncbi.nlm.nih.gov/books/NBK26471/), we cannot exclude that identification of only one recessive POLG mutation in patients 13, 14 and 15 could represent an incidental finding. Concerning patients 17 and 18, we do not have sufficient evidence to establish whether these mutations have a role in determining the clinical phenotype either directly, because of an unidentified mutation in the other allele or by acting in a semidominant manner, or indirectly, acting as a modifier of a defect in another unidentified gene.
In the 30 patients in whom dominant transmission was suspected, we identified no large rearrangement, suggesting that other genes are likely involved, although we did not screen these patients for intronic and promoter mutations. This result is in phase with the fact that, to date, POLG haploinsufficiency has never been reported with dominant transmission of the disease.
In conclusion, in this study, we report the first large French cohort of patients with POLG mutations. We described patients with uncommon features, thus expanding the wide spectrum of clinical phenotypes observed in patients with POLG mutations. We also added six novel pathogenic mutations, including the second large intragenic deletion, to the growing list of POLG gene mutations. Finally, we showed that QMPSF is of great interest in patients with a single heterozygous POLG mutation, mainly in severe infantile phenotypes.
References
Graziewicz MA, Longley MJ, Copeland WC : DNA polymerase gamma in mitochondrial DNA replication and repair. Chem Rev 2006; 106: 383–405.
Van Goethem G, Dermaut B, Löfgren A : Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet 2001; 28: 211–212.
Lamantea E, Tiranti V, Bordoni A et al: Mutations of mitochondrial DNA polymerase gammaA are a frequent cause of autosomal dominant or recessive progressive external ophthalmoplegia. Ann Neurol 2002; 52: 211–219.
Winterthun S, Ferrari G, He L et al: Autosomal recessive mitochondrial ataxic syndrome due to mitochondrial polymerase gamma mutations. Neurology 2005; 64: 1204–1208.
Wong L-JC, Naviaux RK, Brunetti-Pierri N et al: Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum Mutat 2008; 29: E150–E172.
Compton AG, Troedson C, Wilson M et al: Application of oligonucleotide array CGH in the detection of a large intragenic deletion in POLG associated with Alpers syndrome. Mitochondrion 2011; 11: 104–107.
Tang S, Wang J, Lee N-C et al: Mitochondrial DNA polymerase gamma mutations: an everexpanding molecular and clinical spectrum. J Med Genet 2011; 48: 669–681.
Paul R, Santucci S, Saunières A et al: Rapid mapping of mitochondrial dna deletions by large-fragment PCR. Trends Genet 1996; 12: 131–132.
Moraes CT, DiMauro S, Zeviani M et al: Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns–Sayre syndrome. N Engl J Med 1989; 320: 1293–1299.
Rouzier C, Le Guédard-Méreuze S, Fragaki K et al: The severity of phenotype linked to SUCLG1 mutations could be correlated with residual amount of SUCLG1 protein. J Med Genet 2010; 47: 670–676.
Naïmi M, Bannwarth S, Procaccio V et al: Molecular analysis of ANT1, TWINKLE and POLG in patients with multiple deletions or depletion of mitochondrial DNA by a dHPLC-based assay. Eur J Hum Genet 2006; 14: 917–922.
Gouas L, Goumy C, Véronèse L et al: Gene dosage methods as diagnostic tools for the identification of chromosome abnormalities. Pathol Biol 2008; 56: 345–353.
Ceulemans S, van der Ven K, Del-Favero J : Targeted screening and validation of copy number variations. Methods Mol Biol 2012; 838: 311–328.
Saugier-Veber P, Goldenberg A, Drouin-Garraud V et al: Simple detection of genomic microdeletions and microduplications using QMPSF in patients with idiopathic mental retardation. Eur J Hum Genet 2006; 14: 1009–1017.
Lee YS, Kennedy WD, Yin YW : Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell 2009; 139: 312–324.
Kurt B, Jaeken J, Van Hove J et al: A novel POLG gene mutation in 4 children with Alpers-like hepatocerebral syndromes. Arch Neurol 2010; 67: 239–244.
Reichenbach J, Schubert R, Horvàth R et al: Fatal neonatal-onset mitochondrial respiratory chain disease with T cell immunodeficiency. Pediatr Res 2006; 60: 321–326.
Kollberg G, Moslemi A-R, Darin N et al: POLG mutations associated with progressive encephalopathy in childhood. J Neuropathol Exp Neurol 2006; 65: 758–768.
Harrower T, Stewart JD, Hudson G et al: POLG mutations manifesting as autosomal recessive axonal Charcot–Marie–Tooth disease. Arch Neurol 2008; 65: 133–136.
Taanman J-W, Rahman S, Pagnamenta AT et al: Analysis of mutant DNA polymerase gamma in patients with mitochondrial DNA depletion. Hum Mutat 2009; 30: 248–254.
González-Vioque E, Blázquez A, Fernández-Moreira D et al: Association of novel POLG mutations and multiple mitochondrial DNA deletions with variable clinical phenotypes in a Spanish population. Arch Neurol 2006; 63: 107–111.
Blok MJ, van den Bosch BJ, Jongen E et al: The unfolding clinical spectrum of POLG mutations. J Med Genet 2009; 46: 776–785.
Nguyen KV, Østergaard E, Ravn SH et al: POLG mutations in Alpers syndrome. Neurology 2005; 65: 1493–1495.
Ferrari G, Lamantea E, Donati A et al: Infantile hepatocerebral syndromes associated with mutations in the mitochondrial DNA polymerase-gamma A. Brain 2005; 128: 723–731.
Hopkins SE, Somoza A, Gilbert DL : Rare autosomal dominant POLG mutation in a family with metabolic strokes, posterior column spinal degeneration, and multi-endocrine disease. J Child Neurol 2010; 25: 752–756.
Kasiviswanathan R, Copeland WC : Biochemical analysis of the Gly517Val POLG variant reveals wild-type like activity. Mitochondrion 2011; 11: 929–934.
Isohanni P, Hakonen AH, Euro L et al: POLG manifestations in childhood. Neurology 2011; 76: 811–815.
Horvath R, Hudson G, Ferrari G et al: Phenotypic spectrum associated with mutations of the mitochondrial polymerase gamma gene. Brain 2006; 129 (Part 7): 1674–1684.
Charbonnier F, Raux G, Wang Q et al: Detection of exon deletions and duplications of the mismatch repair genes in hereditary nonpolyposis colorectal cancer families using multiplex polymerase chain reaction of short fluorescent fragments. Cancer Res 2000; 60: 2760–2763.
Férec C, Casals T, Chuzhanova N et al: Gross genomic rearrangements involving deletions in the CFTR gene: characterization of six new events from a large cohort of hitherto unidentified cystic fibrosis chromosomes and meta-analysis of the underlying mechanisms. Eur J Hum Genet 2006; 14: 567–576.
Amara A, Adala L, Ben Charfeddine I et al: Correlation of SMN2, NAIP, p44, H4F5 and Occludin genes copy number with spinal muscular atrophy phenotype in Tunisian patients. Eur J Paediatr Neurol 2012; 16: 167–174.
Chan SSL, Longley MJ, Copeland WC : The common Ala467Thr mutation in the human mitochondrial DNA polymerase (POLG) compromises catalytic efficiency and interaction with the accessory subunit. J Biol Chem 2005; 280: 31341–31346.
Chan SSL, Longley MJ, Copeland WC : Modulation of the Trp748Ser mutation in DNA polymerase gamma by the E1143G polymorphism in mitochondrial disorders. Hum Mol Genet 2006; 15: 3473–3483.
Van Goethem G, Löfgren A, Dermaut B, Ceuterick C, Martin J-J, Van Broeckhoven C : Digenic progressive external ophthalmoplegia in a sporadic patient: recessive mutations in POLG and C10orf2/Twinkle. Hum Mutat 2003; 22: 175–176.
Acknowledgements
We thank Christelle Camuso and Bernadette Chafino for technical help. This work was made possible by grants to VP-F from the Association Française contre les Myopathies (AFM) and to CR from the CHU of Nice.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Rights and permissions
About this article
Cite this article
Rouzier, C., Chaussenot, A., Serre, V. et al. Quantitative multiplex PCR of short fluorescent fragments for the detection of large intragenic POLG rearrangements in a large French cohort. Eur J Hum Genet 22, 542–550 (2014). https://doi.org/10.1038/ejhg.2013.171
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.171
Keywords
This article is cited by
-
Clinical and Molecular Features of POLG-Related Sensory Ataxic Neuropathy with Dysarthria and Ophthalmoparesis
Journal of Molecular Neuroscience (2021)
-
Clinico-pathological and Molecular Spectrum of Mitochondrial Polymerase γ Mutations in a Cohort from India
Journal of Molecular Neuroscience (2021)
-
Novel biallelic mutations in POLG gene: large deletion and missense variant associated with PEO
Neurological Sciences (2021)
-
Contribution of nuclear and mitochondrial gene mutations in mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome
Journal of Neurology (2021)
-
Rod bipolar cell dysfunction in POLG retinopathy
Documenta Ophthalmologica (2021)

{kind=link}