
Abstract
Over the past decades the notion of ‘inflammation’ has been extended beyond the original hallmarks of rubor (redness), calor (heat), tumor (swelling) and dolor (pain) described by Celsus. We have gained a more detailed understanding of the cellular players and molecular mediators of inflammation which is now being applied and extended to areas of biomedical research such as cancer, obesity, heart disease, metabolism, auto-inflammatory disorders, autoimmunity and infectious diseases. Innate cytokines are often central components of inflammatory responses. Here, we discuss how the type I interferon and interleukin-1 cytokine pathways represent distinct and specialized categories of inflammatory responses and how these key mediators of inflammation counter-regulate each other.
Similar content being viewed by others
Introduction
Inflammation is a highly complex response initiated by the host to a large variety of stimuli such as damaged and dying cells, chemical irritants, injuries and pathogens. Inflammatory responses are critical because they not only alert cells to mount effective immune responses during infections but also initiate wound repair and healing programs.1 In contrast, excessive un-resolved inflammation can lead to tissue damage and disease.2 Therefore, understanding the unique properties of a given inflammatory response is paramount to gain new perspectives on disease pathogenesis and novel treatment strategies.
Given the diversity of inflammation-inducing insults and variations in both exposure route and duration, qualitatively and quantitatively distinct inflammatory responses must be generated that are fine-tuned to achieve the optimal response to a given stimuli. Thus, each type of inflammatory response is comprised of unique sets of molecular events, lipid mediators, cytokines and specialized cell types that nucleate inflammation, followed by equally tuned steps to ensure resolution of inflammation. We suggest here that depending on the type of insult and the ensuing inflammatory response, distinct classes of inflammatory pathways can be delineated. Moreover, we propose that the innate cytokines interleukin-1 (IL-1) and type I interferons (IFNs) are the pillars of two major types of inflammatory responses. We discuss how the type I IFNs and IL-1 cytokine pathways represent distinct and specialized classes of innate inflammatory responses and how these mediators antagonize each other. Moreover, we highlight how studies on the innate inflammatory response to Mycobacterium tuberculosis (Mtb) infection, a major global health threat, uncovered key-aspects of this special antagonistic relationship between IL-1 and type I IFNs.
Interleukin-1 driven inflammatory responses
IL-1 is the prototypic pro-inflammatory cytokine and the classic fever-inducing endogenous pyrogen.3 IL-1 mediates highly inflammatory responses via two cytokine species, IL-1α and IL-1β, respectively, which can be expressed by most cell types and signals on cells of both hematopoietic and non-hematopoietic in origin. IL-1α and IL-1β generate a vast spectrum of biological responses spanning from effects on the central nervous, hematologic and metabolic systems, and are extensively reviewed elsewhere.4, 5 Although IL-1 signaling plays pivotal roles in immunity, sterile inflammation and metabolism1, 6, 7, 8 excessive overproduction of IL-1 is highly detrimental and contributes to auto-inflammatory diseases, autoimmune encephalomyelitis, rheumatoid arthritis and gout.4, 9, 10, 11, 12, 13 IL-1 production is therefore extensively regulated and the margin between clinical benefit and undesirable pathogenic effects for IL-1 is exceedingly narrow.
IL-1R1 cytokine system
IL-1α and IL-1β share little amino acid homology (26%) yet display similar secondary structures.14, 15, 16 In both human and mouse, the IL-1α and IL-1β genes are located next to each other on chromosome 2, and have conserved synteny in this region.17 IL-1α and IL-1β also appear to carry out similar biological functions by binding to a common receptor comprised of the IL-1 receptor type I (IL-1RI) and IL-1 receptor accessory protein (IL-1RAcP) chains.18 The third ligand for IL-1R1, IL-1 receptor antagonist (IL-1Ra), is a naturally occurring specific IL-1R1 antagonist and prevents IL-1α and IL-1β mediated signaling. Such endogenous antagonism in form of a dedicated soluble secreted protein appears to be a unique feature of IL-1 cytokine family members and highlights the extraordinary tight regulation of the biological activity of IL-1. In addition, a second IL-1R chain, the IL-1RII, is both a surface and soluble receptor that lacks a signaling-competent cytosolic domain and therefore functions as an additional decoy receptor in limiting IL-1 driven responses.18, 19, 20, 21, 22 Finally, both IL-1α and IL-1β are regulated at the post-transcriptional and translational level as outlined below. Thus, expression, generation and signaling of IL-1 are among the most highly and complex regulated checkpoints of any cytokine system.
Post-transcriptional and -translational regulation of IL-1α and IL-1β
A key feature in the regulation of IL-1α and IL-1β is that they are both translated as pro-proteins without leader sequences that require further proteolytic cleavage to gain optimal biological activity.5, 23 Processing of the IL-1α precursor is accomplished by calpain II, a membrane-associated, calcium-dependent cysteine protease,24 and calcium influx induces IL-1α secretion of the processed form.25, 26 Pro-IL-1β is typically cleaved following activation of intracellular cysteine protease caspase-1 or caspase-11 via aggregation of intracellular multiprotein complexes called inflammasomes.27 IL-1 release by inflammasomes is a two-step process. A Signal 1 event typically represents pro-IL-1β protein transcription and translation often as a result of nuclear-factor-kb (NF-κb) activation by toll-like receptor (TLR) ligands or IL-1 itself.28, 29 Signal 2, in contrast, is an activation step that differs for the respective inflammasome sensors such as NOD-like receptors (NLRs) and AIM2-like receptors (ALRs), and ultimately leads to the assembly of inflammasome complexes which are comprised of the NLR/ALR and adapter molecules such as apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) or NLRC4.30, 31 The inflammasome platform recruits and activates caspase-1, the enzyme that in turn coverts the 31 kD immature pro-IL-1β polypeptide to a 17-kD mature IL-1β.32, 33 The cleavage of IL-1β has been suggested to be required for its active secretion via unconventional, endoplasmatic reticulum–Golgi independent, ill-defined processes thought to involve secretory autophagosomes, cytolysis, multi-vesicular body formation and micro-vesicle shedding.34, 35, 36, 37, 38, 39, 40
Increasing evidence suggests that inflammasomes and caspase-1/11 are not the only mechanism for processing IL-1 cytokines. Several studies have identified neutrophil- and macrophage-derived serine proteases such as proteinase 3, elastase and cathepsin-G, as enzymes that can process pro-IL-1β into the 17-kDa bioactive fragment.18, 41, 42 In addition, two other serine proteinases, chymase and chymotrypsin, can also cleave pro-IL-1β into bioactive IL-1β. Apart from serine proteases, metalloproteinases such as Meprin can process pro-IL-1β as well as proteases released by invading pathogens.43, 44, 45, 46 Alternatively, it has also been suggested that inflammasome-mediated IL-1β release can be a strictly cytolysis-driven event, through necrosis or pyroptosis.47 Pyroptosis is a form of caspase-1-dependent programmed cell death that is initiated downstream of inflammasome activation and can contribute to IL-1β release. Moreover, immature pro-IL-1 is often released by cells undergoing cytolysis and present in vast excess of mature IL-1 from cells undergoing inflammasome activation. Although the biological activity of mature IL-1β is 600 times that of pro-IL-1β, pro-IL-1β can still bind to its receptor and it remains unclear what the relative contribution of pro-IL-1β is to IL-1R1 mediated signals in vivo.19, 48, 49
IL-1 signaling pathway
Once IL-1α or IL-1β binds to the IL-1R1 chain, a ligand-induced conformational change facilitates recruitment of IL-1RAcP, the receptor chain required to form a functioning signaling IL-1 R complex.50, 51, 52 Subsequently, the trimeric IL-1 R complex recruits the myeloid differentiation primary response gene 88 (Myd88) via its C-terminal Toll-and IL-1 R-like (TIR) domains.53, 54 MyD88 oligomerizes via its death domain (DD) and TIR domain, and interacts with the interleukin-1 receptor-associated kinase 4 (IRAK4) to form the Myddosome complex that serves as a platform to phosphorylate IRAK4, as well as IRAK2 and IRAK1.55, 56, 57 IRAK phosphorylation is then followed by the recruitment and oligomerization with tumor-necrosis factor-associated factor (TRAF) TRAF6.58 IRAK1 and IRAK2 serve as both adapters and protein kinases to propagate downstream signals with TRAF6 resulting in NF-κB activation.56, 58, 59
Role of IL-1 in host resistance to infection
IL-1 is most widely studied and implicated in host resistance to acute bacterial infections, such as Staphylococcus aureus, where rapid inflammatory responses and IL-1-induced chemokines are required for optimal neutrophil-dependent control60, 61 (also see Table 1). Indeed, this is the classic scenario for IL-1-mediated host control of acute bacterial infections and mice deficient in caspase-1 or IL-1 display increased susceptibility and mortality to infections with Francisella tularensis, Legionella pneumonia, Shigella, Salmonella typhimurium, Bacillus anthracis or Pseudomonas aeruginosa.62, 63, 64, 65, 66, 67, 68, 69 For instance, Gram-negative bacteria, such as Legionella pneumophila and Salmonella typhimurium, trigger IL-1 responses required for host resistance by injecting virulence factors into the host cell cytosol with a specialized type III secretion system.70, 71, 72
In the context of chronic bacterial infections, such as with the intracellular pathogen Mtb, IL-1α and IL-1β are each critically required for host resistance.73, 74, 75, 76, 77, 78, 79, 80, 81 Il1a,b−/− doubly deficient mice and Il1r1−/− mice develop significantly larger granulomatous lesions with neutrophil infiltration in their lungs compared with wild-type (WT) mice. Mice deficient in IL-1 signaling are more susceptible to pulmonary tuberculosis, as reflected by an increased mortality and an enhanced mycobacterial growth in lungs and spleens.73, 74, 75, 80 However, consistent with the detrimental effects of uncontrolled IL-1β production, mice deficient in inducible nitric oxide synthase (iNOS) have dysregulated inflammasome activation and IL-1 production leading to increased pathology and mortality during Mtb infection.82 On the opposing role, the exact mechanisms by which IL-1 mediates protection against bacterial pathogens have only recently been studied. In the case of Mtb, the major protective role of IL-1 during Mtb infection was shown to be linked to its ability to trigger arachidonic acid derived lipid mediator prostaglandin E2 (PGE2) synthesis and COX-2 activation.78 Thus, mice deficient in IL-1 or IL-1 signaling display major defects in PGE2 production in the lungs and increased extracellular bacteria and necrosis. Add-back of PGE2 reduces pulmonary Mtb loads and extends survival78 indicating that IL-1-induced PGE2 is required for bacterial containment and control inside Mtb-infected macrophages.
Despite the importance of inflammasome activation in certain experimental models of inflammation in vitro, certain bacterial infection models in mice deficient in inflammasome components show intriguing results that question the importance of inflammasome-mediated processing of IL-1β in vivo.43, 83 For example, Mtb-infected Il1r1−/− or Il1b−/− deficient mice both display significantly increased mortality with highly increased pulmonary bacterial burden, suggesting a major role for IL-1β signaling in determining the MyD88-dependent phenotype.77, 81 However, Mtb-infected mice deficient in caspase-1/11, ASC or NLRP3, which have critical functions in inflammasome-mediated IL-1β maturation in vitro, showed unimpaired IL-1β production and importantly, were considerably less susceptible to infection than IL-1β-deficient mice.77, 84, 85, 86 The exact mechanisms of IL-1β activation in vivo during Mtb infection remain to be elucidated. Similarly, caspase-1 appears to be dispensable in host resistance against Chlamydia trachomatis, although IL-1β is critical for host defense against this pathogen.87, 88 Together these findings suggest that the production of mature host protective IL-1β during infections in vivo can occur independently of caspase-1/11 activation and ASC-containing inflammasomes. Possible mechanisms could involve inflammasome and caspase-1/11-independent processing of pro-IL-1β by innate immune cells derived serine proteases as mentioned above.18, 41, 42, 43, 44
Although IL-1 mediates host resistance most commonly in bacterial infections, IL-1 signaling can also protect against certain viral infections, including Influenza. Il1r1−/− deficient mice show significantly increased mortality but markedly reduced inflammatory pathology in the lung after Influenza virus infection.89 IL-1α/β appears not to influence the killing of virus infected cells per se but to enhance antibody responses and recruitment of CD4+ T cells and neutrophils to the site of infection.89 Interestingly, data from genome-wide association studies show that genetic variants in IL-1α and IL-1β contributed to the susceptibility to 2009 pandemic H1N1 influenza A virus.90 A more recent study using human pulmonary microvascular endothelial cells showed that IL-1β secreted by the endothelial cells contributes to influenza-induced inflammation, and blockade of IL-1β signaling is a potential treatment or therapeutic target for influenza-induced inflammation and pathology.91 Furthermore, human immunodeficiency virus (HIV) infection IL-1 induces viral gene expression in chronically infected U1 cells and viral replication is inhibited by addition of IL-1RA.92, 93 IL-1RA gene polymorphisms have also been reported to be linked with circulating levels of HIV viral titers in Brazilian women,94 while caspase-1-dependent pyroptosis has been suggested to play a detrimental role during HIV infection.95, 96
During fungal infections, both IL-1α and IL-1β have been shown to play critical roles in host resistance.23, 97 Both IL-1 species are necessary for host resistance against Candida albicans and in the absence of IL-1α or IL-1β growth of C. albicans in the kidneys as well as mortality is significantly increased.98 During pulmonary Aspergillus fumigatus infection, it is IL-1α rather than IL-1β that is crucial for optimal leukocyte recruitment after challenge with the fungal pathogen.99
Type I IFN driven inflammatory responses
Although IL-1 protects most commonly against bacterial infections, type I IFNs belong to a family of cytokines that are specialized to be highly protective against viral infections. In 1957 Isaacs and Lindenmann100 observed that heat-inactivated influenza virus interfered with subsequent viral replication and identified a secreted factor responsible for this phenomenon that they called interferon. Interferons can be separated into three sub-families designated as types 1−3 IFNs. In humans and mice, the type I IFN family is composed of 13 IFNα subtypes, IFNβ, IFNɛ, IFNκ and IFNω.101 The type II IFN group is comprised of one cytokine, IFNγ. The third type of IFNs are members of the IFNλ family, which includes IFNλ1 (also known as IL-29), IFNλ2 (also known as IL-28 A) and IFNλ3 (also known as IL-28B).102
Type I IFN induction
Most cell types express type I IFNs after detection of pathogen-associated molecular patterns by membrane bound or cytosolic pattern recognition receptors.103 Cytosolic receptors are mainly responsible for triggering type I IFN secretion, through recognition of viral RNA or DNA. Such receptors include the retinoic acid–inducible gene I (RIG-I), melanoma differentiation-associated gene 5 (MDA5), NOD1, NOD2 receptors, interferon gamma-inducible protein 16, DEAD-box helicase 41 and RNA polymerase III amongst others.104, 105, 106, 107, 108 Cytosolic nucleotidyltransferase GAMP synthase (cGAS) detects cytosolic DNA and stimulates cyclic GAMP (cGAMP) synthesis.109 cGAMP engages stimulator of interferon genes (STING) as a secondary receptor and further stimulates STING-dependent inflammatory cytokine production including type 1 IFNs.109 In addition to cytosolic signaling events, type I IFNs can also be produced in response to TLR signaling in macrophages and dendritic cells (DCs). TLR3 and TLR4 sense double-stranded (ds) RNA and lipopolysaccharide (LPS), respectively, and via TIR-domain-containing adapter-inducing interferon-β and TANK-binding kinase 1 (TBK1) activate interferon regulatory transcription factor 3 to trigger type I IFN inductions.103 Although most cell types can produce type I IFN, plasmacytoid DCs (pDCs) represent a major source of type I IFNs in mice110 and men.111, 112 pDCs abundantly express the TLR9 subfamily members TLR7, TLR8 and TLR9 which recognize viral single-stranded RNA (TLR7,8) or ds CpG-rich DNA (TLR9), respectively.113
Type I IFN signaling pathway
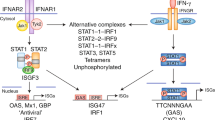
Type 1 IFNs bind to a heterodimeric transmembrane receptor interferon (alpha and beta) receptor 1 (IFNAR), comprised of two chains, IFNAR1 and IFNAR2. Once bound IFNAR activates Tyk2 and Jak1, which results in signal transducer and activator of transcription (STAT) STAT1-STAT2 hetero-dimer formation and subsequent translocation to the nucleus. In the nucleus, dimeric STATs recruit an additional transcriptional factor, IFN regulatory factor 9, forming a trimeric complex called IFN-stimulated gene factor 3 (ISGF3).114 ISGF3 then binds to interferon-stimulated response elements, inducing hundreds of IFN-stimulated genes (ISGs).115, 116 Depending on the cell type, IFNAR-mediated activation of Tyk2 and Jak1 can promote homo-dimerization of other STATs including the formation of STAT1 and STAT3 dimers that bind to IFNγ -activated site (GAS) enhancer elements and STAT3-biding elements, respectively. It can also result in STAT4 activation, leading to IFNγ production during viral infection.117 In addition, type I IFN can activate mitogen-activated protein kinases and phosphatidylinositol-3 kinase (MAPK) signaling pathways that contribute to antiviral effects.102 Type I IFNs also signal through other STATs, including STAT3, STAT4, STAT5A and STAT5B.118 The phosphoinositide 3-kinase (PI3K)–mammalian target of rapamycin (mTOR) and MAPK pathways can also be activated by IFNAR1 signaling. This large diversity of signaling pathways may contribute to the pleiotropic effects of type I IFN-driven responses, as it allows transcription of a broad range of genes besides those intended for viral restriction, such as cytokines, chemokines, pro-apoptotic and anti-apoptotic molecules, and molecules involved in lipid metabolism.118, 119
Role of type I IFNs in host resistance
Type I IFNs are the prototypical cytokines associated with control of viral infections as they successfully restrict viral replication by an acute induction of specific sets of several hundreds of ISGs inside infected cells that can directly interrupt viral gene transcription and translation.120 These genes are induced by type I IFNs in response to innate viral recognition and also promote an antiviral state in bystander cells that limits viral spread. In fact, most viruses devote a significant part of their limited genome to mechanisms that modulate type I IFNs pathways so ISG induction is limited, highlighting the importance of IFNα/β in host cell protection from viral infection.121 In most pathogenic virus infections, early and rapid production of type I IFN is required to limit initial viral replication before effective humoral or cellular adaptive immune mechanisms become operational. This is exemplified by the fact that mice deficient in IFNAR1 are highly susceptible to viral infections as extensively reviewed elsewhere.122, 123
In addition to the protective role for type I IFNs during viral infections, they are also involved in immunity against fungal pathogens, these cytokines can have both detrimental and beneficial roles. In one study with C. albicans, type I IFNs were required to induce reactive oxygen species important for killing of yeast cells by infected phagocytes,124 whereas in another study the absence of type I IFN signaling did not alter fungal burden but instead lead to lethal immunopathology.125 During infections with Cryptococcus neoformans mice lacking either Ifnar1−/− or Ifnb−/− have been shown to die from unrestrained pneumonia and encephalitis when compared with control animals.126 Consistent with this, C. neoformans or C. gattii-infected mice showed increased resistance to infection when they were intranasally administered polyinosinic-polycytidylic acid stabilized with poly-l-lysine (polyinosinic -polycytidylic acid-polylysine-carboxymethylcellulose (poly-ICLC)), a double-stranded RNA homolog which is a potent inducer of type I IFNs.127 Although Type I IFN signaling was also reported to be required for optimal host resistance in mice infected with Aspergillus fumigatus,128, 129 there are also reports of a detrimental role of type I IFNs in defense against Candida glabrata and Histoplasma capsulatum.130, 131
Albeit most critically required for host resistance against viral infections, Type I IFNs can contribute to protection against some bacterial infections. For example, Legionella pneumophila remains an important opportunistic pathogen and type I IFNs have been shown to limit its replication. IFNα significantly suppressed Legionella pneumophila growth in WT but not in Ifnar1−/− deficient macrophages and Legionella was able to trigger type I IFN production in an autocrine manner.132 Treatment of mice with exogenous IFN or poly-ICLC protected mice from Bacillus anthracis and Chlamydia trachomatis infection, and inhibited intracellular replication in both human and mouse cells.133, 134, 135 Type I IFNs may also contribute to host resistance against Salmonella typhimurium infection by promoting strong IFN-γ production in an IL-12-independent manner.136
Although in some instances, type I IFNs contribute to host resistance against bacterial pathogens, there is mounting evidence that the detrimental and pro-bacterial effects of type I IFN are far reaching.122 The pro-bacterial effects of an inappropriate or excessive type I IFN responses accelerate the pathogenesis of disease by both intracellular and extracellular bacteria. For example, the hyper-virulence of certain Mtb strains correlates with enhanced type I IFN synthesis and Ifnar1−/− mice infected with Mtb display lower bacterial loads when compared with WT animals.137, 138, 139, 140 Mtb-infected mice intranasally treated with poly-ICLC exhibit exacerbated lung pathology and increased bacterial burden.141 The relevance of these observations to human tuberculosis is supported by whole-blood transcriptional profiles of TB patients, which were found to be dominated by a type I IFNs gene signature that closely correlated with disease severity.142 In line with these observations, viral co-infection with influenza A virus increases susceptibility to Mtb infection in a type I IFN-dependent manner.143
Listeria monocytogenes is another intracellular bacteria that infects primarily macrophages and is most commonly associated with food-borne illness in immune compromised individuals. Ifnar1−/− mice are resistant to Listeria monocytogenes infection and display increased survival and lower spleen, and liver bacterial loads compared with WT animals.144, 145, 146 The major mechanism attributed to this was reduced apoptotic cell death, particularly of lymphocytes, with IFNAR1 signaling sensitizing cells to the bacterial virulence factor listeriolysin O and resultant cell death in WT mice.144, 145 Moreover, Ifnar1−/− deficient mice were also resistant to infection with S. aureus, where lethal pneumonia was observed in only 10% of Ifnar1−/− mice as compared with 80% in the WT animals.147 Increased resistance against Pseudomonas aeruginosa has also been reported in Ifnar1−/− mice.148
IL-1 and Type I IFN Cross talk
Both the IL-1 and type I IFNs pathways can cause great harm to the human body when dysregulated or activated untimely in an inappropriate context. Studies in infants and adolescents with inborn errors of innate immunity have revealed key roles for both IL-1 and type I IFN in auto-inflammatory and immunodeficiency disorders. Mutations in NLRP3 and IL1RN lead to increased IL-1 activity such as Cryopyrin-associated periodic syndromes and deficiency in IL-1RA.149 These diseases are characterized by episodes of strong inflammation, including high fevers, urticaria-like rashes, joint pain and malaise, and present in patients early in life leading to stunted growth and high mortality.149 In recent years it has also become evident that inborn errors in type I IFN regulation can cause inflammatory syndromes termed interferonopathies.150 Specifically, conditions such as Aicardi–Goutieres syndrome, STING-associated vasculopathy with onset in infancy as well as certain types of systemic lupus erythematosus are characterized by an increased and dysregulated type I IFN response that underlies these diverse pathological syndromes. The severity of these inborn errors of IL-1 and type I IFN inflammation highlights the critical importance of these inflammatory pathways in the physiological state in addition to their role in response to bacterial or viral infections. Moreover, these syndromes strongly underscore the notion of distinct types of inflammatory classes and increasing evidence suggests that cross-regulation by IL-1 and type I IFN plays an important role in balancing the innate inflammatory equilibrium in both physiological homeostasis as well as infection.
Type I IFN regulation of IL-1
Inhibition of IL-1 by type I IFNs was first described in the 1990s when multiple groups observed that IL-1 levels were reduced and IL-1Ra simultaneously upregulated in in vitro studies of IFN-treated cells, and later, in patients who received a single dose of type I IFN.151, 152, 153, 154 These early observations were the foundation for subsequent studies on the anti-inflammatory properties of type I IFNs,155 and exemplified the multi-faceted antagonism between IL-1 and type I IFNs. Importantly, both IFNα and IFNβ can suppress IL-1α and IL-1β transcription and translation in various cell types.76, 77, 78, 79, 156 The absence of type I IFN-mediated IL-1 inhibition in Ifnar1 or Ifnb deficient animals, after infection with a variety of pathogens, results in increased IL-1 concentrations in tissues and circulation and elevated IL-1-dependent IL-17 responses.78, 157, 158, 159 IL-1 and type I IFNs play divergent roles in host resistance to infections and while type I IFNs can enhance or impede host resistance, type IFN-mediated IL-1 inhibition has been observed in both scenarios. Therefore IL-1 inhibition by type I IFNs can both impair host resistance, as in the case of Mtb infection78, 79 as well as limit IL-1 driven immunopathology as shown recently during S. pyogenes infection.157 Besides direct regulation of IL-1 protein expression, type I IFNs also potently induce anti-inflammatory IL-1Ra and IL-10, which in turn can inhibit IL-1 signaling effects.79, 160, 161, 162, 163, 164, 165 In addition, 25-hydroxycholesterol has been implicated as a downstream effector mechanism of type I IFN-mediated inhibition of IL-1β expression and inflammasome activation.166 More recently, with increased understanding of the complex regulation of IL-1β processing, we now know that type IFNs and IL-10 also potently regulate NLRP1 and NLRP3 inflammasome activation thereby further modulating IL-1 activity.164, 165
Interestingly, antagonism of IL-1 does not seem to be limited to type I IFNs. Indeed, type II IFN, IFN-γ and recently type III IFNs, IFN-λ as well as IFN-ταυ have been reported to be able to inhibit IL-1 expression.79, 167, 168, 169 Of note, there is also evidence that type I IFN can positively regulate IL-1 expression via the AIM2 inflammasome, which is primarily activated by cytosolic DNA and operational during some select bacterial and viral infections.170, 171, 172, 173
IL-1 regulation of type I IFNs
Much less is known about how IL-1 could in turn regulate type I IFN production and/or effector functions. Until recently there was limited evidence for IL-1 mediated inhibition of type I IFNs with one study showing that IL-1β was able to attenuate IFNα/β-induced STAT1 phosphorylation in hepatocytes via a proteasome-dependent mechanism.174 Another study observed IL-1-dependent and TNF-independent inhibition of IFN-β production in a human fibroblast line, suggesting that IL-1 driven inhibition of type I IFNs is an effect unique to IL-1 rather than a general effect due to NF-κb activation via TNFα.175 PGE2 has been shown to suppress type I IFN production in the context of LPS-induced responses in mice and more recently during Influenza infection.176, 177 However, a link to IL-1 has not been generated in these studies and remains to be elucidated further. In fact, recent work in Mtb-infected cells and animals revealed that IL-1 potently antagonizes type I IFN responses by directly regulating both transcription and translation of IFN-β via induction of PGE2 and PGE2 by itself is able to inhibit and antagonize type I IFN.78 Moreover, limiting excessive and detrimental type I IFN expression via PGE2 has proven to be a promising host-directed therapeutic approach in Mtb infection.78 Although IL-1 and type I IFN cross-regulation is an emerging research area, it has become clear that the contextual differences in pathogenesis will likely determine whether the cross-regulation results in beneficial outcomes with a given pathogen or contributes to inflammatory pathology and susceptibility.
Mtb: a case study for IL-1 and type I IFN cross talk
The relevance of IL-1 and type I IFN cross talk is perhaps best studied and exemplified in the context of mycobacterial infections, where IL-1 plays a major protective and type I IFN a primarily detrimental role in host resistance to Mtb. Here it has been shown that, in human monocyte-derived DC and macrophages as well as murine bone marrow derived dendritic cells and bone marrow derived macrophages, IFNβ or poly-ICLC were able to potently inhibit both IL-1α and IL-1β in response to Mtb infection in vitro.76, 79 This inhibition was at least in part due to type I IFN-dependent induction of IL-10, an important anti-inflammatory cytokine,79, 178 previously reported to inhibit IL-1 production downstream of type I IFNs.164 Besides inhibiting IL-1α and IL-1β cytokine expression directly, type I IFNs also potently upregulated expression of IL-1Ra during Mtb infection in vitro and in vivo, further amplifying the negative effects on IL-1 activity,78, 79 a central feature in type I IFN opposition of IL-1.151 In addition to modulating IL-1RA and IL-1α, and IL-1β expression, type I IFNs also regulated expression of the decoy receptor IL-1R2 (ref. 78; Figure 1). Moreover, the type I IFN-mediated inhibition of IL-1 cytokine production observed in vitro was confirmed in vivo in Mtb-infected lungs using single-cell analysis of IL-1 producing myeloid subsets.79

Cross-regulation between interleukin-1 (IL-1) and type-1 interferon (IFN) inflammatory pathways as exemplified during Mtb infection. Mtb infection triggers both cytosolic nucleotidyltransferase GAMP synthase-STING-TBK1-interferon regulatory transcription factor 3-interferon-β (cGAS-STING-TBK1-IRF3-IFN-β) axis and AIM2/NLRP3-IL-1β pathway in myelophagocytic cells. These two pathways exert opposite biological outcomes for host defense against Mtb: IL-1 is recognized as beneficial with anti-bacterial effects while type I IFNs are considered largely detrimental with replication promoting properties. IL-1α and IL-1β limit type I IFN production through direct transcriptional downregulation and PTGS2-mediated PGE2 production, which in turn inhibits type I IFN. Type I IFNs attenuate IL-1α/β signaling through induction of IL-10, IL-1R2, IL-1RA and CH25H.
A potential molecular mechanism was recently uncovered that could shed some light on the reciprocal relationship between IL-1 and type I IFNs and extends previous findings that implicated the ESAT6-secretion system (ESX)-1 in type I IFN expression and inflammasome activation.140, 179 Multiple independent groups have shown that the nucleotidyltransferase cGAS is a central component in the cytosolic surveillance pathway and recognizes Mtb, leading to type 1 interferon induction and autophagy.180, 181, 182, 183 The most recent studies identify cGAS as the sensor for Mtb DNA in the cytosol leading to STING activation and autophagy induction.180, 181, 182 Wasserman et al. in addition showed that AIM2 recognizes cytosolic Mtb DNA and triggered AIM2-dependent inflammasome activation and IL-1 production. They found that virulent and attenuated Mtb can engage distinct cytosolic pattern recognition receptor systems, namely the cGAS-IFN-axis vs the AIM2/NLRP3-IL-1β-pathway, and that the decision as to which pathway is triggered is determined by the relative abundance of EsxA and/or by additional ESX-1/EsxA-dependent effectors. Thus, the tug of war between IFN and IL-1 exists even at the single-cell level inside infected macrophages and centers around innate sensing of cytosolic DNA of Mtb.
Recent work uncovered that IL-1 can in turn counter-regulate type I IFN driven detrimental responses during Mtb infection.78 In murine and human macrophages IL-1α and IL-1β potently inhibit type I IFN induction at both the mRNA and protein level, and similarly IFNβ mRNA and protein levels are upregulated in the lungs of Mtb-infected Il1r1−/− deficient mice.78 This inhibition is of functional importance because mice doubly deficient in Il1r1,Ifnar1-/- are partially protected while Il1r1−/− singly deficient animals succumb rapidly to Mtb aerosol challenge. Moreover, when IL-1 is present in type I IFN-treated cultures, it even suppresses the pro-bacterial effects downstream of IFN that lead to increased bacterial replication. Interestingly, IL-1-induced PGE2 is also able to potently inhibit type I IFNs in a dose-dependent manner. Targeting PGE2 during Mtb infection, either via direct administration or its enhancement by 5 lipoxygenase blockade with Zileuton, reversed poly-ICLC-mediated type I IFN driven mortality.78 These data highlighted and provided proof-of-concept that the cross talk of IL-1 and type I IFN provides a valuable target for host-directed therapies of Mtb and plays a major role during infection in mice.78 Thus, the above findings generated during the study of Mtb infection played into a broader context for previous studies that showed that IL-1 and PGE2 can inhibit type I IFN production.176, 177
Concluding remarks
Most of our insights into IL-1 driven inflammatory processes are based on studies during acute inflammation (or infection), where a trigger appears suddenly and leads to a rapid onset of innate and adaptive immune responses. Perhaps a key determinant in whether a particular inflammatory pathway such as IL-1 or type I IFN dominates, or the nature of the inflammatory cross talk, is whether the inflammatory stimuli is temporally limited or persistently present. For example, it has become increasingly appreciated that in particular IFNα/β can also be harmful during chronic viral infections, either by immunosuppressive effects that impair viral control or by triggering inflammation and tissue damage that exacerbates disease.184, 185, 186, 187 Perhaps a contributing factor for the pronounced interplay between IL-1 and type I IFNs during pulmonary Mtb infection in contrast to other acute bacterial infections is the chronic nature of the infection and the slow replication time of the bacterium. The inability of a host to clear chronic pathogens promotes immunosuppressive programs that lead to sustained expression of both pro- and anti-inflammatory cytokines and vastly change the rules of engagement between inflammatory pathways. Understanding the rules of engagement, how they are influenced by both magnitude and quality of a given inflammatory trigger and how acute vs persistent stimuli influence the inflammatory equilibrium, will be key to develop novel anti-inflammatory agents and host-directed therapies for a variety of diseases.
References
Chen GY, Nunez G . Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 2010; 10: 826–837.
Wallach D, Kang TB, Kovalenko A . Concepts of tissue injury and cell death in inflammation: a historical perspective. Nat Rev Immunol 2014; 14: 51–59.
Dinarello CA . Interleukin-1 and the pathogenesis of the acute-phase response. N Engl J Med 1984; 311: 1413–1418.
Dinarello CA . A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur J Immunol 2011; 41: 1203–1217.
Dinarello CA . Biology of interleukin 1. FASEB J 1988; 2: 108–115.
Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL . Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med 2007; 13: 851–856.
Yan B, Han P, Pan L, Lu W, Xiong J, Zhang M et al. Il-1β and reactive oxygen species differentially regulate neutrophil directional migration and basal random motility in a Zebrafish injury–induced inflammation model. J Immunol 2014; 192: 5998–6008.
Garlanda C, Dinarello CA, Mantovani A . The interleukin-1 family: back to the future. Immunity 2013; 39: 1003–1018.
Hoffman HM, Wanderer AA . Inflammasome and IL-1beta-mediated disorders. Curr Allergy Asthma Rep 2010; 10: 229–235.
Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G et al. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest 2006; 116: 2262–2271.
Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH . Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 2009; 31: 331–341.
Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC . A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med 2006; 203: 1685–1691.
Voronov E, Dayan M, Zinger H, Gayvoronsky L, Lin JP, Iwakura Y et al. IL-1 beta-deficient mice are resistant to induction of experimental SLE. Eur Cytokine Netw 2006; 17: 109–116.
March CJ, Mosley B, Larsen A, Cerretti DP, Braedt G, Price V et al. Cloning, sequence and expression of two distinct human interleukin-1 complementary DNAs. Nature 1985; 315: 641–647.
Lomedico PT, Gubler U, Hellmann CP, Dukovich M, Giri JG, Pan YC et al. Cloning and expression of murine interleukin-1 cDNA in Escherichia coli. Nature 1984; 312: 458–462.
Auron PE, Webb AC, Rosenwasser LJ, Mucci SF, Rich A, Wolff SM et al. Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc Natl Acad Sci USA 1984; 81: 7907–7911.
Taylor SL, Renshaw BR, Garka KE, Smith DE, Sims JE . Genomic organization of the interleukin-1 locus. Genomics 2002; 79: 726–733.
Dinarello CA . Biologic basis for interleukin-1 in disease. Blood 1996; 87: 2095–2147.
Jobling SA, Auron PE, Gurka G, Webb AC, McDonald B, Rosenwasser LJ et al. Biological activity and receptor binding of human prointerleukin-1 beta and subpeptides. J Biol Chem 1988; 263: 16372–16378.
Symons JA, Eastgate JA, Duff GW . Purification and characterization of a novel soluble receptor for interleukin 1. J Exp Med 1991; 174: 1251–1254.
Colotta F, Re F, Muzio M, Bertini R, Polentarutti N, Sironi M et al. Interleukin-1 type II receptor: a decoy target for IL-1 that is regulated by IL-4. Science 1993; 261: 472–475.
Watkins LR, Hansen MK, Nguyen KT, Lee JE, Maier SF . Dynamic regulation of the proinflammatory cytokine, interleukin-1beta: molecular biology for non-molecular biologists. Life Sci 1999; 65: 449–481.
Dinarello CA, Wolff SM . The role of interleukin-1 in disease. N Engl J Med 1993; 328: 106–113.
Miller AC, Schattenberg DG, Malkinson AM, Ross D . Decreased content of the IL1 alpha processing enzyme calpain in murine bone marrow-derived macrophages after treatment with the benzene metabolite hydroquinone. Toxicol Lett 1994; 74: 177–184.
Kavita U, Mizel SB . Differential sensitivity of interleukin-1 alpha and -beta precursor proteins to cleavage by calpain, a calcium-dependent protease. J Biol Chem 1995; 270: 27758–27765.
Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G et al. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 2012; 36: 388–400.
Martinon F, Mayor A, Tschopp J . The inflammasomes: guardians of the body. Annu Rev Immunol 2009; 27: 229–265.
Latz E, Xiao TS, Stutz A . Activation and regulation of the inflammasomes. Nat Rev Immunol 2013; 13: 397–411.
Latz E . The inflammasomes: mechanisms of activation and function. Curr Opin Immunol 2010; 22: 28–33.
Guo H, Callaway JB, Ting JP . Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 2015; 21: 677–687.
Ratsimandresy RA, Dorfleutner A, Stehlik C . An update on PYRIN domain-containing pattern recognition receptors: from immunity to pathology. Front Immunol 2013; 4: 440.
Sollberger G, Strittmatter GE, Garstkiewicz M, Sand J, Beer HD . Caspase-1: the inflammasome and beyond. Innate immunity 2014; 20: 115–125.
Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G . The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 2009; 10: 241–247.
Lopez-Castejon G, Brough D . Understanding the mechanism of IL-1beta secretion. Cytokine Growth Factor Rev 2011; 22: 189–195.
Shirasaki Y, Yamagishi M, Suzuki N, Izawa K, Nakahara A, Mizuno J et al. Real-time single-cell imaging of protein secretion. Sci Rep 2014; 4: 4736.
Liu T, Yamaguchi Y, Shirasaki Y, Shikada K, Yamagishi M, Hoshino K et al. Single-cell imaging of caspase-1 dynamics reveals an all-or-none inflammasome signaling response. Cell Rep 2014; 8: 974–982.
Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem 2011; 286: 9587–9597.
Zhang M, Kenny S, Ge L, Xu K, Schekman R . Translocation of interleukin-1beta into a vesicle intermediate in autophagy-mediated secretion. Elife 2015; 4: e11205.
Jiang S, Dupont N, Castillo EF, Deretic V . Secretory versus degradative autophagy: unconventional secretion of inflammatory mediators. J Innate Immun 2013; 5: 471–479.
Piccioli P, Rubartelli A . The secretion of IL-1beta and options for release. Semin Immunol 2013; 25: 425–429.
Coeshott C, Ohnemus C, Pilyavskaya A, Ross S, Wieczorek M, Kroona H et al. Converting enzyme-independent release of tumor necrosis factor alpha and IL-1beta from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc Natl Acad Sci USA 1999; 96: 6261–6266.
Sugawara S, Uehara A, Nochi T, Yamaguchi T, Ueda H, Sugiyama A et al. Neutrophil proteinase 3-mediated induction of bioactive IL-18 secretion by human oral epithelial cells. J Immunol 2001; 167: 6568–6575.
Netea MG, Simon A, van de Veerdonk F, Kullberg BJ, Van der Meer JW, Joosten LA . IL-1beta processing in host defense: beyond the inflammasomes. PLoS Pathog 2010; 6: e1000661.
Dinarello CA, Bendtzen K, Wolff SM . Studies on the active site of human leukocytic pyrogen. Inflammation 1982; 6: 63–78.
Schonbeck U, Mach F, Libby P . Generation of biologically active IL-1 beta by matrix metalloproteinases: a novel caspase-1-independent pathway of IL-1 beta processing. J Immunol 1998; 161: 3340–3346.
Herzog C, Haun RS, Kaushal V, Mayeux PR, Shah SV, Kaushal GP et al. and meprin alpha generate biologically functional IL-1beta from pro-IL-1beta. Biochem Biophys Res Commun 2009; 379: 904–908.
Cullen SP, Kearney CJ, Clancy DM, Martin SJ . Diverse activators of the NLRP3 inflammasome promote IL-1beta secretion by triggering necrosis. Cell Rep 2015; 11: 1535–1548.
Mosley B, Urdal DL, Prickett KS, Larsen A, Cosman D, Conlon PJ et al. The interleukin-1 receptor binds the human interleukin-1 alpha precursor but not the interleukin-1 beta precursor. J Biol Chem 1987; 262: 2941–2944.
Hazuda D, Webb RL, Simon P, Young P . Purification and characterization of human recombinant precursor interleukin 1 beta. J Biol Chem 1989; 264: 1689–1693.
Wang D, Zhang S, Li L, Liu X, Mei K, Wang X . Structural insights into the assembly and activation of IL-1beta with its receptors. Nat Immunol 2010; 11: 905–911.
Huang J, Gao X, Li S, Cao Z . Recruitment of IRAK to the interleukin 1 receptor complex requires interleukin 1 receptor accessory protein. Proc Natl Acad Sci USA 1997; 94: 12829–12832.
Greenfeder SA, Nunes P, Kwee L, Labow M, Chizzonite RA, Ju G . Molecular cloning and characterization of a second subunit of the interleukin 1 receptor complex. J Biol Chem 1995; 270: 13757–13765.
Loiarro M, Volpe E, Ruggiero V, Gallo G, Furlan R, Maiorino C et al. Mutational analysis identifies residues crucial for homodimerization of myeloid differentiation factor 88 (MyD88) and for its function in immune cells. J Biol Chem 2013; 288: 30210–30222.
Radons J, Dove S, Neumann D, Altmann R, Botzki A, Martin MU et al. The interleukin 1 (IL-1) receptor accessory protein Toll/IL-1 receptor domain: analysis of putative interaction sites in vitro mutagenesis and molecular modeling. J Biol Chem 2003; 278: 49145–49153.
Yamin TT, Miller DK . The interleukin-1 receptor-associated kinase is degraded by proteasomes following its phosphorylation. J Biol Chem 1997; 272: 21540–21547.
Cao Z, Henzel WJ, Gao X . IRAK: a kinase associated with the interleukin-1 receptor. Science 1996; 271: 1128–1131.
Kawagoe T, Sato S, Matsushita K, Kato H, Matsui K, Kumagai Y et al. Sequential control of toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat Immunol 2008; 9: 684–691.
Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV . TRAF6 is a signal transducer for interleukin-1. Nature 1996; 383: 443–446.
Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, Irie K et al. TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol Cell 2000; 5: 649–658.
Miller LS, O'Connell RM, Gutierrez MA, Pietras EM, Shahangian A, Gross CE et al. MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against Staphylococcus aureus. Immunity 2006; 24: 79–91.
Miller LS, Cho JS . Immunity against Staphylococcus aureus cutaneous infections. Nat Rev Immunol 2011; 11: 505–518.
Schultz MJ, Rijneveld AW, Florquin S, Edwards CK, Dinarello CA, van der Poll T . Role of interleukin-1 in the pulmonary immune response during Pseudomonas aeruginosa pneumonia. Am J Physiol Lung Cell Mol Physiol 2002; 282: L285–L290.
Joosten LA, Van De Veerdonk FL, Vonk AG, Boerman OC, Keuter M, Fantuzzi G et al. Differential susceptibility to lethal endotoxaemia in mice deficient in IL-1alpha, IL-1beta or IL-1 receptor type I. APMIS 2010; 118: 1000–1007.
Hultgren OH, Svensson L, Tarkowski A . Critical role of signaling through IL-1 receptor for development of arthritis and sepsis during Staphylococcus aureus infection. J Immunol 2002; 168: 5207–5212.
Sansonetti PJ, Phalipon A, Arondel J, Thirumalai K, Banerjee S, Akira S et al. Caspase-1 activation of IL-1β and IL-18 are essential for Shigella flexneri–induced inflammation. Immunity 2000; 12: 581–590.
Barry KC, Fontana MF, Portman JL, Dugan AS, Vance RE . IL-1alpha signaling initiates the inflammatory response to virulent Legionella pneumophila in vivo. J Immunol 2013; 190: 6329–6339.
Mariathasan S, Weiss DS, Dixit VM, Monack DM . Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J Exp Med 2005; 202: 1043–1049.
Verdrengh M, Thomas JA, Hultgren OH . IL-1 receptor-associated kinase 1 mediates protection against Staphylococcus aureus infection. Microbes infect 2004; 6: 1268–1272.
Moayeri M, Crown D, Newman ZL, Okugawa S, Eckhaus M, Cataisson C et al. Inflammasome sensor Nlrp1b-dependent resistance to anthrax is mediated by caspase-1, IL-1 signaling and neutrophil recruitment. PLoS Pathog 2010; 6: e1001222.
Zhao Y, Shao F . Diverse mechanisms for inflammasome sensing of cytosolic bacteria and bacterial virulence. Curr Opin Microbiol 2015; 29: 37–42.
van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LA . Inflammasome activation and IL-1beta and IL-18 processing during infection. Trends Immunol 2011; 32: 110–116.
Vladimer GI, Marty-Roix R, Ghosh S, Weng D, Lien E . Inflammasomes and host defenses against bacterial infections. Curr Opin Microbiol 2013; 16: 23–31.
Yamada H, Mizumo S, Horai R, Iwakura Y, Sugawara I . Protective role of interleukin-1 in mycobacterial infection in IL-1 alpha/beta double-knockout mice. Lab Invest 2000; 80: 759–767.
Sugawara I, Yamada H, Hua S, Mizuno S . Role of interleukin (IL)-1 type 1 receptor in mycobacterial infection. Microbiol Immunol 2001; 45: 743–750.
Juffermans NP, Florquin S, Camoglio L, Verbon A, Kolk AH, Speelman P et al. Interleukin-1 signaling is essential for host defense during murine pulmonary tuberculosis. J Infect Dis 2000; 182: 902–908.
Novikov A, Cardone M, Thompson R, Shenderov K, Kirschman KD, Mayer-Barber KD et al. Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1beta production in human macrophages. J Immunol 2011; 187: 2540–2547.
Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A et al. Caspase-1 independent IL-1beta production is critical for host resistance to mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol 2010; 184: 3326–3330.
Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 2014; 511: 99–103.
Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P et al. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity 2011; 35: 1023–1034.
Di Paolo NC, Shafiani S, Day T, Papayannoupoulou T, Russell DW, Iwakura Y et al. Interdependence between interleukin-1 and tumor necrosis factor regulates TNF-dependent control of Mycobacterium tuberculosis Infection. Immunity 2015; 43: 1125–1136.
Fremond CM, Togbe D, Doz E, Rose S, Vasseur V, Maillet I et al. IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J Immunol 2007; 179: 1178–1189.
Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta. Nat Immunol 2013; 14: 52–60.
Netea MG, van de Veerdonk FL, van der Meer JW, Dinarello CA, Joosten LA . Inflammasome-independent regulation of IL-1-family cytokines. Annu Rev Immunol 2015; 33: 49–77.
McElvania Tekippe E, Allen IC, Hulseberg PD, Sullivan JT, McCann JR, Sandor M et al. Granuloma formation and host defense in chronic Mycobacterium tuberculosis infection requires PYCARD/ASC but not NLRP3 or caspase-1. PLoS One 2010; 5: e12320.
Dorhoi A, Nouailles G, Jorg S, Hagens K, Heinemann E, Pradl L et al. Activation of the NLRP3 inflammasome by Mycobacterium tuberculosis is uncoupled from susceptibility to active tuberculosis. Eur J Immunol 2012; 42: 374–384.
Walter K, Holscher C, Tschopp J, Ehlers S . NALP3 is not necessary for early protection against experimental tuberculosis. Immunobiology 2010; 215: 804–811.
Prantner D, Darville T, Sikes JD, Andrews CW Jr, Brade H, Rank RG et al. Critical role for interleukin-1beta (IL-1beta) during Chlamydia muridarum genital infection and bacterial replication-independent secretion of IL-1beta in mouse macrophages. Infect Immun 2009; 77: 5334–5346.
Cheng W, Shivshankar P, Zhong Y, Chen D, Li Z, Zhong G . Intracellular interleukin-1alpha mediates interleukin-8 production induced by Chlamydia trachomatis infection via a mechanism independent of type I interleukin-1 receptor. Infect immun 2008; 76: 942–951.
Schmitz N, Kurrer M, Bachmann MF, Kopf M . Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol 2005; 79: 6441–6448.
Liu Y, Li S, Zhang G, Nie G, Meng Z, Mao D et al. Genetic variants in IL1A and IL1B contribute to the susceptibility to 2009 pandemic H1N1 influenza A virus. BMC Immunol 2013; 14: 37.
Kim KS, Jung H, Shin IK, Choi BR, Kim DH . Induction of interleukin-1 beta (IL-1beta) is a critical component of lung inflammation during influenza A (H1N1) virus infection. J Med Virol 2015; 87: 1104–1112.
Poli G, Kinter AL, Fauci AS . Interleukin 1 induces expression of the human immunodeficiency virus alone and in synergy with interleukin 6 in chronically infected U1 cells: inhibition of inductive effects by the interleukin 1 receptor antagonist. Proc Natl Acad Sci USA 1994; 91: 108–112.
Granowitz EV, Saget BM, Wang MZ, Dinarello CA, Skolnik PR . Interleukin 1 induces HIV-1 expression in chronically infected U1 cells: blockade by interleukin 1 receptor antagonist and tumor necrosis factor binding protein type 1. Mol Med 1995; 1: 667–677.
Witkin SS, Linhares IM, Gerber S, Caetano ME, Segurado AC . Interleukin-1 receptor antagonist gene polymorphism and circulating levels of human immunodeficiency virus type 1 RNA in Brazilian women. J Virol 2001; 75: 6242–6244.
Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014; 505: 509–514.
Deeks SG . HIV infection, inflammation, immunosenescence, and aging. Annu Rev Med 2011; 62: 141–155.
Romani L . Immunity to fungal infections. Nat Rev Immunol 2004; 4: 1–23.
Vonk AG, Netea MG, van Krieken JH, Iwakura Y, van der Meer JW, Kullberg BJ . Endogenous interleukin (IL)-1 alpha and IL-1 beta are crucial for host defense against disseminated candidiasis. J Infect Dis 2006; 193: 1419–1426.
Caffrey AK, Lehmann MM, Zickovich JM, Espinosa V, Shepardson KM, Watschke CP et al. IL-1alpha signaling is critical for leukocyte recruitment after pulmonary Aspergillus fumigatus challenge. PLoS Pathog 2015; 11: e1004625.
Isaacs A, Lindenmann J . Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci 1957; 147: 258–267.
Pestka S, Krause CD, Walter MR . Interferons, interferon-like cytokines, and their receptors. Immunol Rev 2004; 202: 8–32.
Gonzalez-Navajas JM, Lee J, David M, Raz E . Immunomodulatory functions of type I interferons. Nat Rev Immunol 2012; 12: 125–135.
Kawai T, Akira S . The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 2010; 11: 373–384.
Watanabe T, Asano N, Fichtner-Feigl S, Gorelick PL, Tsuji Y, Matsumoto Y et al. NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J Clin Invest 2010; 120: 1645–1662.
Pandey AK, Yang Y, Jiang Z, Fortune SM, Coulombe F, Behr MA et al. NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog 2009; 5: e1000500.
Chiu YH, Macmillan JB, Chen ZJ . RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009; 138: 576–591.
Parvatiyar K, Zhang Z, Teles RM, Ouyang S, Jiang Y, Iyer SS et al. The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat Immunol 2012; 13: 1155–1161.
Thompson MR, Sharma S, Atianand M, Jensen SB, Carpenter S, Knipe DM et al. Interferon gamma-inducible protein (IFI) 16 transcriptionally regulates type i interferons and other interferon-stimulated genes and controls the interferon response to both DNA and RNA viruses. J Biol Chem 2014; 289: 23568–23581.
Sun L, Wu J, Du F, Chen X, Chen ZJ . Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013; 339: 786–791.
Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol 2001; 2: 1144–1150.
Colonna M, Krug A, Cella M . Interferon-producing cells: on the front line in immune responses against pathogens. Curr Opin Immunol 2002; 14: 373–379.
Trinchieri G . [Physiology of the immune system]. Minerva Anestesiol 1978; 44: 673–678.
Iwasaki A, Medzhitov R . Toll-like receptor control of the adaptive immune responses. Nat Immunol 2004; 5: 987–995.
Dunn GP, Koebel CM, Schreiber RD . Interferons, immunity and cancer immunoediting. Nat Rev Immunol 2006; 6: 836–848.
Ivashkiv LB, Donlin LT . Regulation of type I interferon responses. Nat Rev Immunol 2014; 14: 36–49.
Platanias LC . Mechanisms of type-I-and type-II-interferon-mediated signalling. Nat Rev Immunol 2005; 5: 375–386.
Nguyen KB, Watford WT, Salomon R, Hofmann SR, Pien GC, Morinobu A et al. Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science 2002; 297: 2063–2066.
Ivashkiv LB, Donlin LT . Regulation of type I interferon responses. Nat Rev Immunol 2014; 14: 36–49.
Rauch I, Muller M, Decker T . The regulation of inflammation by interferons and their STATs. JAKSTAT 2013; 2: e23820.
Yan N, Chen ZJ . Intrinsic antiviral immunity. Nat Immunol 2012; 13: 214–222.
Versteeg GA, Garcia-Sastre A . Viral tricks to grid-lock the type I interferon system. Curr Opin Microbiol 2010; 13: 508–516.
McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A . Type I interferons in infectious disease. Nat Rev Immunol 2015; 15: 87–103.
Durbin JE, Fernandez-Sesma A, Lee CK, Rao TD, Frey AB, Moran TM et al. Type I IFN modulates innate and specific antiviral immunity. J Immunol 2000; 164: 4220–4228.
Bogdan C . The function of type I interferons in antimicrobial immunity. Curr Opin Immunol 2000; 12: 419–424.
Majer O, Bourgeois C, Zwolanek F, Lassnig C, Kerjaschki D, Mack M et al. Type I interferons promote fatal immunopathology by regulating inflammatory monocytes and neutrophils during Candida infections. PLoS Pathog 2012; 8: e1002811.
Biondo C, Midiri A, Gambuzza M, Gerace E, Falduto M, Galbo R et al. IFN-alpha/beta signaling is required for polarization of cytokine responses toward a protective type 1 pattern during experimental cryptococcosis. J Immunol 2008; 181: 566–573.
Sionov E, Mayer-Barber KD, Chang YC, Kauffman KD, Eckhaus MA, Salazar AM et al. Type I IFN induction via poly-ICLC protects mice against cryptococcosis. PLoS Pathog 2015; 11: e1005040.
Tandon RN, Feuillette AR, Mahouy G, Badillet G, Friedman RM, Maheshwari RK . Interferon protects mice against an extracellular infection of Aspergillus fumigatus. Ann N Y Acad Sci 1988; 544: 409–411.
Maheshwari RK, Tandon RN, Feuillette AR, Mahouy G, Badillet G, Friedman RM . Interferon inhibits Aspergillus fumigatus growth in mice: an activity against an extracellular infection. J Interferon Res 1988; 8: 35–44.
Inglis DO, Berkes CA, Hocking Murray DR, Sil A . Conidia but not yeast cells of the fungal pathogen Histoplasma capsulatum trigger a type I interferon innate immune response in murine macrophages. Infect Immun 2010; 78: 3871–3882.
Bourgeois C, Majer O, Frohner IE, Lesiak-Markowicz I, Hildering KS, Glaser W et al. Conventional dendritic cells mount a type I IFN response against Candida spp. requiring novel phagosomal TLR7-mediated IFN-beta signaling. J Immunol 2011; 186: 3104–3112.
Lippmann J, Muller HC, Naujoks J, Tabeling C, Shin S, Witzenrath M et al. Dissection of a type I interferon pathway in controlling bacterial intracellular infection in mice. Cell Microbiol 2011; 13: 1668–1682.
de la Maza LM, Peterson EM, Goebel JM, Fennie CW, Czarniecki CW . Interferon-induced inhibition of Chlamydia trachomatis: dissociation from antiviral and antiproliferative effects. Infect Immun 1985; 47: 719–722.
Kazar J, Gillmore JD, Gordon FB . Effect of Interferon and Interferon Inducers on Infections with a Nonviral Intracellular Microorganism, Chlamydia trachomatis. Infect Immun 1971; 3: 825–832.
Walberg K, Baron S, Poast J, Schwartz B, Izotova L, Pestka S et al. Interferon protects mice against inhalation anthrax. J Interferon Cytokine Res 2008; 28: 597–601.
Freudenberg MA, Merlin T, Kalis C, Chvatchko Y, Stubig H, Galanos C . Cutting edge: a murine, IL-12-independent pathway of IFN-gamma induction by gram-negative bacteria based on STAT4 activation by Type I IFN and IL-18 signaling. J Immunol 2002; 169: 1665–1668.
Manca C, Tsenova L, Freeman S, Barczak AK, Tovey M, Murray PJ et al. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J Interferon Cytokine Res 2005; 25: 694–701.
Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM et al. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha /beta. Proc Natl Acad Sci USA 2001; 98: 5752–5757.
Ordway D, Henao-Tamayo M, Harton M, Palanisamy G, Troudt J, Shanley C et al. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J Immunol 2007; 179: 522–531.
Stanley SA, Johndrow JE, Manzanillo P, Cox JS . The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol 2007; 178: 3143–3152.
Antonelli LR, Gigliotti Rothfuchs A, Goncalves R, Roffe E, Cheever AW, Bafica A et al. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J Clin Invest 2010; 120: 1674–1682.
Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 2010; 466: 973–977.
Redford PS, Mayer-Barber KD, McNab FW, Stavropoulos E, Wack A, Sher A et al. Influenza A virus impairs control of Mycobacterium tuberculosis coinfection through a type I interferon receptor-dependent pathway. J Infect Dis 2014; 209: 270–274.
O'Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B et al. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med 2004; 200: 437–445.
Carrero JA, Calderon B, Unanue ER . Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med 2004; 200: 535–540.
Auerbuch V, Brockstedt DG, Meyer-Morse N, O'Riordan M, Portnoy DA . Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med 2004; 200: 527–533.
Martin FJ, Gomez MI, Wetzel DM, Memmi G, O'Seaghdha M, Soong G et al. Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J Clin Invest 2009; 119: 1931–1939.
Merches K, Khairnar V, Knuschke T, Shaabani N, Honke N, Duhan V et al. Virus-induced type I interferon deteriorates control of systemic Pseudomonas aeruginosa infection. Cell Physiol Biochem 2015; 36: 2379–2392.
Henderson C, Goldbach-Mansky R . Monogenic autoinflammatory diseases: new insights into clinical aspects and pathogenesis. Curr Opin Rheumatol 2010; 22: 567–578.
Crow YJ, Manel N . Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol 2015; 15: 429–440.
Huang Y, Blatt LM, Taylor MW . Type 1 interferon as an antiinflammatory agent: inhibition of lipopolysaccharide-induced interleukin-1 beta and induction of interleukin-1 receptor antagonist. J Interferon Cytokine Res 1995; 15: 317–321.
Tilg H, Mier JW, Vogel W, Aulitzky WE, Wiedermann CJ, Vannier E et al. Induction of circulating IL-1 receptor antagonist by IFN treatment. J Immunol 1993; 150: 4687–4692.
Coclet-Ninin J, Dayer JM, Burger D . Interferon-beta not only inhibits interleukin-1beta and tumor necrosis factor-alpha but stimulates interleukin-1 receptor antagonist production in human peripheral blood mononuclear cells. Eur Cytokine Netw 1997; 8: 345–349.
Reznikov LL, Puren AJ, Fantuzzi G, Muhl H, Shapiro L, Yoon DY et al. Spontaneous and inducible cytokine responses in healthy humans receiving a single dose of IFN-alpha2b: increased production of interleukin-1 receptor antagonist and suppression of IL-1-induced IL-8. J Interferon Cytokine Res 1998; 18: 897–903.
Billiau A . Anti-inflammatory properties of type I interferons. Antiviral Res 2006; 71: 108–116.
Radwan M, Stiefvater R, Grunert T, Sharif O, Miller I, Marchetti-Deschmann M et al. Tyrosine kinase 2 controls IL-1ss production at the translational level. J Immunol 2010; 185: 3544–3553.
Castiglia V, Piersigilli A, Ebner F, Janos M, Goldmann O, Dambock U et al. Type I interferon signaling prevents IL-1beta-driven lethal systemic hyperinflammation during invasive bacterial infection of soft tissue. Cell Host Microbe 2016; 19: 375–387.
Henry T, Kirimanjeswara GS, Ruby T, Jones JW, Peng K, Perret M et al. Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections. J Immunol 2010; 184: 3755–3767.
Perkins DJ, Rajaiah R, Tennant SM, Ramachandran G, Higginson EE, Dyson TN et al. Salmonella typhimurium co-opts the host type I IFN system to restrict macrophage innate immune transcriptional responses selectively. J Immunol 2015; 195: 2461–2471.
de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE . Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med 1991; 174: 1209–1220.
Aman MJ, Keller U, Derigs G, Mohamadzadeh M, Huber C, Peschel C . Regulation of cytokine expression by interferon-alpha in human bone marrow stromal cells: inhibition of hematopoietic growth factors and induction of interleukin-1 receptor antagonist. Blood 1994; 84: 4142–4150.
Naveau S, Emilie D, Borotto E, Portier A, Lazizi Y, Giraud V et al. Interleukin-1 receptor antagonist plasma concentration is specifically increased by alpha-2A-interferon treatment. J Hepatol 1997; 27: 272–275.
Danis VA, Kulesz AJ, Nelson DS, Brooks PM . Cytokine regulation of human monocyte interleukin-1 (IL-1) production in vitro. Enhancement of IL-1 production by interferon (IFN) gamma, tumour necrosis factor-alpha, IL-2 and IL-1, and inhibition by IFN-alpha. Clin Exp Immunol 1990; 80: 435–443.
Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011; 34: 213–223.
Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O'Garra A . IL-10 inhibits cytokine production by activated macrophages. J Immunol 1991; 147: 3815–3822.
Reboldi A, Dang EV, McDonald JG, Liang G, Russell DW, Cyster JG . Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science 2014; 345: 679–684.
Schindler R, Ghezzi P, Dinarello CA . IL-1 induces IL-1. IV. IFN-gamma suppresses IL-1 but not lipopolysaccharide-induced transcription of IL-1. J Immunol 1990; 144: 2216–2222.
Hara K, Shirasuna K, Usui F, Karasawa T, Mizushina Y, Kimura H et al. Interferon-tau attenuates uptake of nanoparticles and secretion of interleukin-1beta in macrophages. PLoS One 2014; 9: e113974.
Blazek K, Eames HL, Weiss M, Byrne AJ, Perocheau D, Pease JE et al. IFN-lambda resolves inflammation via suppression of neutrophil infiltration and IL-1beta production. J Exp Med 2015; 212: 845–853.
Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol 2010; 11: 395–402.
Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol 2010; 11: 385–393.
Sauer JD, Witte CE, Zemansky J, Hanson B, Lauer P, Portnoy DA . Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 2010; 7: 412–419.
Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O'Rourke K et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci USA 2010; 107: 9771–9776.
Tian Z, Shen X, Feng H, Gao B . IL-1 beta attenuates IFN-alpha beta-induced antiviral activity and STAT1 activation in the liver: involvement of proteasome-dependent pathway. J Immunol 2000; 165: 3959–3965.
Kohase M, Zhang YH, Lin JX, Yamazaki S, Sehgal PB, Vilcek J . Interleukin-1 can inhibit interferon-beta synthesis and its antiviral action: comparison with tumor necrosis factor. J Interferon Res 1988; 8: 559–570.
Coulombe F, Jaworska J, Verway M, Tzelepis F, Massoud A, Gillard J et al. Targeted prostaglandin E2 inhibition enhances antiviral immunity through induction of type I interferon and apoptosis in macrophages. Immunity 2014; 40: 554–568.
Xu XJ, Reichner JS, Mastrofrancesco B, Henry WL Jr, Albina JE . Prostaglandin E2 suppresses lipopolysaccharide-stimulated IFN-beta production. J Immunol 2008; 180: 2125–2131.
McNab FW, Ewbank J, Howes A, Moreira-Teixeira L, Martirosyan A, Ghilardi N et al. Type I IFN induces IL-10 production in an IL-27-independent manner and blocks responsiveness to IFN-gamma for production of IL-12 and bacterial killing in Mycobacterium tuberculosis-infected macrophages. J Immunol 2014; 193: 3600–3612.
Koo IC, Wang C, Raghavan S, Morisaki JH, Cox JS, Brown EJ . ESX-1-dependent cytolysis in lysosome secretion and inflammasome activation during mycobacterial infection. Cell Microbiol 2008; 10: 1866–1878.
Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J et al. Mycobacterium tuberculosis differentially activates cGAS- and inflammasome-dependent intracellular immune responses through ESX-1. Cell Host Microbe 2015; 17: 799–810.
Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J et al. The cytosolic sensor cGAS detects Mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe 2015; 17: 811–819.
Collins AC, Cai H, Li T, Franco LH, Li XD, Nair VR et al. Cyclic GMP-AMP synthase is an innate immune DNA sensor for Mycobacterium tuberculosis. Cell Host Microbe 2015; 17: 820–828.
Dey B, Dey RJ, Cheung LS, Pokkali S, Guo H, Lee JH et al. A bacterial cyclic dinucleotide activates the cytosolic surveillance pathway and mediates innate resistance to tuberculosis. Nat Med 2015; 21: 401–406.
Snell LM, Brooks DG . New insights into type I interferon and the immunopathogenesis of persistent viral infections. Curr Opin Immunol 2015; 34: 91–98.
Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013; 340: 202–207.
Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013; 340: 207–211.
Ng CT, Sullivan BM, Teijaro JR, Lee AM, Welch M, Rice S et al. Blockade of interferon Beta, but not interferon alpha, signaling controls persistent viral infection. Cell Host Microbe 2015; 17: 653–661.
Tanabe M, Matsumoto T, Shibuya K, Tateda K, Miyazaki S, Nakane A et al. Compensatory response of IL-1 gene knockout mice after pulmonary infection with Klebsiella pneumoniae. J Med Microbiol 2005; 54: 7–13.
Parker D, Planet PJ, Soong G, Narechania A, Prince A . Induction of type I interferon signaling determines the relative pathogenicity of Staphylococcus aureus strains. PLoS Pathog 2014; 10: e1003951.
Kafka D, Ling E, Feldman G, Benharroch D, Voronov E, Givon-Lavi N et al. Contribution of IL-1 to resistance to Streptococcus pneumoniae infection. Int Immunol 2008; 20: 1139–1146.
Biondo C, Mancuso G, Midiri A, Signorino G, Domina M, Lanza Cariccio V et al. Essential role of interleukin-1 signaling in host defenses against group B streptococcus. MBio 2014; 5: e01428–01414.
Zwijnenburg PJ, van der Poll T, Florquin S, Roord JJ, Van Furth AM . IL-1 receptor type 1 gene-deficient mice demonstrate an impaired host defense against pneumococcal meningitis. J Immunol 2003; 170: 4724–4730.
Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R et al. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol 2007; 178: 3126–3133.
Gratz N, Hartweger H, Matt U, Kratochvill F, Janos M, Sigel S et al. Type I interferon production induced by Streptococcus pyogenes-derived nucleic acids is required for host protection. PLoS Pathog 2011; 7: e1001345.
Feng CG, Scanga CA, Collazo-Custodio CM, Cheever AW, Hieny S, Caspar P et al. Mice lacking myeloid differentiation factor 88 display profound defects in host resistance and immune responses to Mycobacterium avium infection not exhibited by Toll-like receptor 2 (TLR2)- and TLR4-deficient animals. J Immunol 2003; 171: 4758–4764.
Denis M . Recombinant murine beta interferon enhances resistance of mice to systemic Mycobacterium avium infection. Infect Immun 1991; 59: 1857–1859.
Rogers HW, Tripp CS, Schreiber RD, Unanue ER . Endogenous IL-1 is required for neutrophil recruitment and macrophage activation during murine listeriosis. J Immunol 1994; 153: 2093–2101.
Rogers HW, Sheehan KC, Brunt LM, Dower SK, Unanue ER, Schreiber RD . Interleukin 1 participates in the development of anti-Listeria responses in normal and SCID mice. Proc Natl Acad Sci USA 1992; 89: 1011–1015.
Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S . Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol 2012; 13: 954–962.
Raupach B, Peuschel SK, Monack DM, Zychlinsky A . Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect Immun 2006; 74: 4922–4926.
del Barrio L, Sahoo M, Lantier L, Reynolds JM, Ceballos-Olvera I, Re F . Production of anti-LPS IgM by B1a B cells depends on IL-1beta and is protective against lung infection with Francisella tularensis LVS. PLoS Pathog 2015; 11: e1004706.
Kang TJ, Basu S, Zhang L, Thomas KE, Vogel SN, Baillie L et al. Bacillus anthracis spores and lethal toxin induce IL-1beta via functionally distinct signaling pathways. Eur J Immunol 2008; 38: 1574–1584.
Wang JP, Lee CK, Akalin A, Finberg RW, Levitz SM . Contributions of the MyD88-dependent receptors IL-18R, IL-1R, and TLR9 to host defenses following pulmonary challenge with Cryptococcus neoformans. PLoS One 2011; 6: e26232.
Bellocchio S, Montagnoli C, Bozza S, Gaziano R, Rossi G, Mambula SS et al. The contribution of the Toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. J Immunol 2004; 172: 3059–3069.
Hung CY, Jimenez-Alzate Mdel P, Gonzalez A, Wuthrich M, Klein BS, Cole GT . Interleukin-1 receptor but not Toll-like receptor 2 is essential for MyD88-dependent Th17 immunity to Coccidioides infection. Infect Immun 2014; 82: 2106–2114.
Deepe GS Jr, McGuinness M . Interleukin-1 and host control of pulmonary histoplasmosis. J Infect Dis 2006; 194: 855–864.
Voronov E, Dotan S, Gayvoronsky L, White RM, Cohen I, Krelin Y et al. IL-1-induced inflammation promotes development of leishmaniasis in susceptible BALB/c mice. Int Immunol 2010; 22: 245–257.
Kostka SL, Knop J, Konur A, Udey MC, von Stebut E . Distinct roles for IL-1 receptor type I signaling in early versus established Leishmania major infections. J Invest Dermatol 2006; 126: 1582–1589.
Charmoy M, Hurrell BP, Romano A, Lee SH, Ribeiro-Gomes F, Riteau N et al. The Nlrp3 inflammasome, IL-1beta, and neutrophil recruitment are required for susceptibility to a nonhealing strain of Leishmania major in C57BL/6 mice. Eur J Immunol 2016; 46: 897–911.
Mattner J, Wandersee-Steinhauser A, Pahl A, Rollinghoff M, Majeau GR, Hochman PS et al. Protection against progressive leishmaniasis by IFN-beta. J Immunol 2004; 172: 7574–7582.
Mattner J, Schindler H, Diefenbach A, Rollinghoff M, Gresser I, Bogdan C . Regulation of type 2 nitric oxide synthase by type 1 interferons in macrophages infected with Leishmania major. Eur J Immunol 2000; 30: 2257–2267.
Diefenbach A, Schindler H, Donhauser N, Lorenz E, Laskay T, MacMicking J et al. Type 1 interferon (IFNalpha/beta) and type 2 nitric oxide synthase regulate the innate immune response to a protozoan parasite. Immunity 1998; 8: 77–87.
Curfs JH, van der Meer JW, Sauerwein RW, Eling WM . Low dosages of interleukin 1 protect mice against lethal cerebral malaria. J Exp Med 1990; 172: 1287–1291.
Voisine C, Mastelic B, Sponaas AM, Langhorne J . Classical CD11c+ dendritic cells, not plasmacytoid dendritic cells, induce T cell responses to Plasmodium chabaudi malaria. Int J Parasitol 2010; 40: 711–719.
Vigario AM, Belnoue E, Gruner AC, Mauduit M, Kayibanda M, Deschemin JC et al. Recombinant human IFN-alpha inhibits cerebral malaria and reduces parasite burden in mice. J Immunol 2007; 178: 6416–6425.
Vigario AM, Belnoue E, Cumano A, Marussig M, Miltgen F, Landau I et al. Inhibition of Plasmodium yoelii blood-stage malaria by interferon alpha through the inhibition of the production of its target cell, the reticulocyte. Blood 2001; 97: 3966–3971.
Liehl P, Zuzarte-Luis V, Chan J, Zillinger T, Baptista F, Carapau D et al. Host-cell sensors for Plasmodium activate innate immunity against liver-stage infection. Nat Med 2014; 20: 47–53.
Haque A, Best SE, Ammerdorffer A, Desbarrieres L, de Oca MM, Amante FH et al. Type I interferons suppress CD4(+) T-cell-dependent parasite control during blood-stage Plasmodium infection. Eur J Immunol 2011; 41: 2688–2698.
Lopez R, Demick KP, Mansfield JM, Paulnock DM . Type I IFNs play a role in early resistance, but subsequent susceptibility, to the African trypanosomes. J Immunol 2008; 181: 4908–4917.
Koga R, Hamano S, Kuwata H, Atarashi K, Ogawa M, Hisaeda H et al. TLR-dependent induction of IFN-beta mediates host defense against Trypanosoma cruzi. J Immunol 2006; 177: 7059–7066.
Costa VM, Torres KC, Mendonca RZ, Gresser I, Gollob KJ, Abrahamsohn IA et al. IFNs stimulate nitric oxide production and resistance to Trypanosoma cruzi infection. J Immunol 2006; 177: 3193–3200.
Chessler AD, Caradonna KL, Da'dara A, Burleigh BA . Type I interferons increase host susceptibility to Trypanosoma cruzi infection. Infect Immun 2011; 79: 2112–2119.
Shim YR, Lee HK . Caspase-1 independent viral clearance and adaptive immunity against mucosal respiratory syncytial virus infection. Immune Netw 2015; 15: 73–82.
Goritzka M, Makris S, Kausar F, Durant LR, Pereira C, Kumagai Y et al. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J Exp Med 2015; 212: 699–714.
Kamperschroer C, Quinn DG . The role of proinflammatory cytokines in wasting disease during lymphocytic choriomeningitis virus infection. J Immunol 2002; 169: 340–349.
Joeckel LT, Wallich R, Metkar SS, Froelich CJ, Simon MM, Borner C . Interleukin-1R signaling is essential for induction of proapoptotic CD8 T cells, viral clearance, and pathology during lymphocytic choriomeningitis virus infection in mice. J Virol 2012; 86: 8713–8719.
Mordstein M, Kochs G, Dumoutier L, Renauld JC, Paludan SR, Klucher K et al. Interferon-lambda contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog 2008; 4: e1000151.
Crotta S, Davidson S, Mahlakoiv T, Desmet CJ, Buckwalter MR, Albert ML et al. Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog 2013; 9: e1003773.
Tanaka J, Ozaki H, Yasuda J, Horai R, Tagawa Y, Asano M et al. Lipopolysaccharide-induced HIV-1 expression in transgenic mice is mediated by tumor necrosis factor-alpha and interleukin-1, but not by interferon-gamma nor interleukin-6. AIDS 2000; 14: 1299–1307.
He H, Sharer LR, Chao W, Gu CJ, Borjabad A, Hadas E et al. Enhanced human immunodeficiency virus Type 1 expression and neuropathogenesis in knockout mice lacking Type I interferon responses. J Neuropathol Exp Neurol 2014; 73: 59–71.
Torres R, Macdonald L, Croll SD, Reinhardt J, Dore A, Stevens S et al. Hyperalgesia, synovitis and multiple biomarkers of inflammation are suppressed by interleukin 1 inhibition in a novel animal model of gouty arthritis. Ann Rheum Dis 2009; 68: 1602–1608.
Guo B, Chang EY, Cheng G . The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J Clin Invest 2008; 118: 1680–1690.
Teige I, Treschow A, Teige A, Mattsson R, Navikas V, Leanderson T et al. IFN-beta gene deletion leads to augmented and chronic demyelinating experimental autoimmune encephalomyelitis. J Immunol 2003; 170: 4776–4784.
Lauwerys BR, Ducreux J, Houssiau FA . Type I interferon blockade in systemic lupus erythematosus: where do we stand? Rheumatology (Oxford) 2014; 53: 1369–1376.
Crow MK . Advances in understanding the role of type I interferons in systemic lupus erythematosus. Curr Opin Rheumatol 2014; 26: 467–474.
Amezcua-Guerra LM, Ferrusquia-Toriz D, Castillo-Martinez D, Marquez-Velasco R, Chavez-Rueda AK, Bojalil R . Limited effectiveness for the therapeutic blockade of interferon alpha in systemic lupus erythematosus: a possible role for type III interferons. Rheumatology (Oxford) 2015; 54: 203–205.
Nacionales DC, Kelly-Scumpia KM, Lee PY, Weinstein JS, Lyons R, Sobel E et al. Deficiency of the type I interferon receptor protects mice from experimental lupus. Arthritis Rheum 2007; 56: 3770–3783.
Acknowledgements
We thank Drs. Mahtab Moayeri, Dan Barber and Alan Sher for comments and critical reading of the article. This work was supported by the Intramural Research Program of the NIH, National Institute of Allergy and Infectious Diseases.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Mayer-Barber, K., Yan, B. Clash of the Cytokine Titans: counter-regulation of interleukin-1 and type I interferon-mediated inflammatory responses. Cell Mol Immunol 14, 22–35 (2017). https://doi.org/10.1038/cmi.2016.25
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2016.25
Keywords
This article is cited by
-
cGAS-STING, inflammasomes and pyroptosis: an overview of crosstalk mechanism of activation and regulation
Cell Communication and Signaling (2024)
-
The crucial regulatory role of type I interferon in inflammatory diseases
Cell & Bioscience (2023)
-
Mutated Toll-like receptor 9 increases Alzheimer’s disease risk by compromising innate immunity protection
Molecular Psychiatry (2023)
-
Immunogenicity and protective activity of mRNA vaccine candidates against yellow fever virus in animal models
npj Vaccines (2023)
-
CRISPRi screens in human iPSC-derived astrocytes elucidate regulators of distinct inflammatory reactive states
Nature Neuroscience (2022)
