
Abstract
Although several studies have reported that locally administering oncolytic viruses effectively targets malignancies, the efficacy of systemically administered oncolytic viruses is restricted. Recently, however, it was reported that systemic administration of oncolytic vesicular stomatitis virus adsorbed onto antigen-specific lymphocytes was effective against malignancies. We hypothesized that intravenously administering such virus might have significant potential in treatment of the malignant tumors. We adsorbed oncolytic herpes simplex virus-1 mutant R3616 onto lymphocytes harvested from mice with acquired antitumor immunity. We administered adsorbed R3616 to peritoneally disseminated tumors and analyzed the efficacy of this treatment. Mice administered adsorbed R3616 survived significantly longer than mice administered R3616 adsorbed onto non-specific lymphocytes, or mice administered either virus or tumor antigen-specific lymphocytes alone. In this context, herpes oncolytic virus is a promising treatment not only for primary lesions, but also for multiple metastasizing lesions. This treatment strategy may become one of the most effective methods for systemic virus delivery.
Similar content being viewed by others


Introduction
Colorectal cancer is the most common cancer of the digestive tract.1, 2 To date, all studies on peritoneal carcinomatosis from colorectal cancer have shown that there is a high incidence of abdominal dissemination in patients with a poor prognosis; such patients have a median survival of 5−9 months.2, 3, 4 This observation clearly indicates the importance of treating peritoneal dissemination in colorectal cancer patients.
Oncolytic viral therapy has been developed as a new modality for the treatment of advanced cancers.5 In 1991, Martuza et al.6 demonstrated the effects of herpes simplex virus type-1 (HSV-1) against a brain tumor. Subsequently, both basic science studies and clinical trials have examined various oncolytic viral therapies.7 To date, numerous viruses, including adenovirus, HSV and Newcastle virus, have been used in clinical trials.8, 9, 10, 11, 12, 13, 14, 15
Clinical trials and preclinical studies with adenovirus and HSV have indicated that direct intratumoral injections of oncolytic viruses yield significant therapeutic effects.16 However, in patients with disseminated metastases, the virus must be systemically delivered via an intravenous (i.v.) route to the metastatic sites.17 One preclinical study showed that i.v. or intraportally administering HSV effectively treated multiple liver metastases.5 By contrast, intraperitoneal (i.p.) virus administration was significantly more effective than i.v. administration in mice bearing peritoneal metastases, with no significant differences in peritoneal metastases between the i.v. treated and control groups.18 As viruses adhere nonspecifically to the endothelium and are neutralized by the immune system, viral titers at the target site may be insufficient in cases of i.v. administration.19 Several clinical trials have examined i.v. administered adenoviruses and Newcastle viruses, but the antitumor effects of these therapies are not definitive.8, 10, 11 Therefore, it is important to establish an effective method for systemic delivery of oncolytic viruses.
It has been proposed that cells could function as a ‘Trojan horse’ vehicle for delivery of viruses into tumors, as follows: cells would first be infected with an oncolytic virus in vitro, then systemically injected and subsequently carried to the tumor beds to release the oncolytic virus.20 A previous study examined the effectiveness of loading oncolytic measles viruses onto nonspecific T cells, and showed that this method prevented neutralization by the immune system.21 Another study examined the dissemination and availability of vaccinia virus treatment using cytokine-induced killer cells as a carrier.22 A recent study evaluated the effectiveness of loading oncolytic vesicular stomatitis virus onto antigen-specific T cells, and showed that these T cells were more efficiently delivered to lung tumors in mice than the virus alone.23 Various carriers have been evaluated, and antigen-specific T cells have often been used as a vector carrier.24, 25, 26, 27, 28, 29 Tumor antigen-specific T lymphocytes are a logical carrier for systemic virus delivery.20, 30 Moreover, tumor antigen-specific lymphocytes have been clearly shown to have inherent antitumor effects on their own.30, 31, 32
The antitumor efficacy of HSV adsorbed on the tumor antigen-specific lymphocytes has not been reported. We tested the hypothesis that HSV treated in this manner could be delivered systemically, and investigated the effects of HSV on primary and metastatic tumors.
Materials and methods
Cells and viruses
Vero, African green monkey kidney cells, were obtained from American Type Culture Collection (Manassas, VA). MC26, a mouse colon carcinoma cell line, was obtained from the National Cancer Institute Tumor Repository (Frederick, MD). Vero and MC26 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum and 1% penicillin/streptomycin. The HSV-1 mutant, R3616, was kindly provided by Bernard Roizman (University of Chicago, Chicago, IL). Virus was propagated and titered on Vero cells, aliquoted and stored at −80 °C.
Cytotoxic assay
Lymphocyte and virus cytotoxicity were determined using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Cells were plated onto 96-well plates at 5 × 103 cells per well and grown for 36 h. Lymphocytes or viruses were added at various multiplicities of infection (MOI) and incubated for 3 or 4 days. The number of surviving cells was quantified by a colorimetric MTT assay, and percent cell survival was calculated compared with control (mock-infected) cells.
Animal studies
BALB/c mice were obtained from Japan CLEA (Hamamatsu, Japan). Animal studies were performed in accordance with the guidelines issued by the Nagoya University Animal Center. Mice were age- and sex-matched. To establish peritoneally disseminated tumors, MC26 cells in 500 μl of DMEM were injected into the abdominal cavity of BALB/c mice. To establish subcutaneous tumors, MC26 cells in 100 μl of DMEM were injected into the flanks of mice. Animals were randomly assigned to different groups before treatment. To study tumor immunity, MC26 cells (1 × 103 plaque-forming units (pfu)) in 500 μl of DMEM were injected i.p. into five BALB/c mice. After 24 h, the mice were treated i.p. with 1 × 106 pfu of R3616 in 500 μl of DMEM. After 56 days, four of the five mice were cured, and were subsequently injected subcutaneously (s.c.) with 1 × 103 pfu MC26 cells in 100 μl of DMEM (thus becoming anti-tumor immunized mice). Control BALB/c mice were also injected with MC26 cells (1 × 103 pfu) in 100 μl of DMEM s.c., and the tumor volumes in both groups were monitored.
To establish tumor antigen-specific lymphocytes (or non-specific lymphocytes), MC26 cells (1 × 105 pfu) and R3616 (1 × 105 pfu) were co-cultured for 24 h; alternatively, MC26 cells (1 × 105 pfu) were frozen and thawed twice. Cells were injected i.p. into five BALB/c mice. After 28 days, lymphocytes were harvested from spleen and thymus as follows: tissues were removed from mice, soaked in DMEM, minced and pushed out the contents to release blood cells. Fragments were settled and the supernatant fluid containing blood cells was obtained. Red blood cells were lysed using ACK (ammonium chloride–potassium) buffer (0.15 M NH4Cl, 1.0 mM KHCO3, 0.1 mM EDTA, pH 7.2−7.4). Lymphocytes of ∼100% of purity were obtained. These cells were used for in vitro assays and in vivo injections.
In vitro loading of lymphocytes with HSV-1
Lymphocytes from mice were infected with R3616 at an MOI of 1 for 2 h at 37 °C. Virus-loaded lymphocytes were washed in phosphate-buffered saline at 4 °C, and then used for in-vivo injections or in-vitro assays. Lymphocytes were mixed with R3616 at an MOI of 1; the ratio of adsorbed R3616 was 20:1 (lymphocyte vs R3616). We checked the adsorbed ratio by titering on Vero cells after freeze/thaw of virus-adsorbed lymphocytes (data not shown).
Statistical analysis
Two-sample unequal variance was analyzed by Student's t-test. Survival was analyzed by the Kaplan−Meier method and log-rank test using the JMP software (SAS Institute, Cary, NC). Statistical significance was defined as P<0.05.
Results
Effects of tumor immunity in vivo
We analyzed the effects of tumor immunity in vivo. Tumor-immunized mice had a significant reduction in tumor growth, compared with the control mice (P=0.0148; Figure 1). We observed that BALB/c mice acquired tumor-specific immunity following i.p. administration of MC26 cells.

Effects of tumor immunity in vivo. MC26 cells (1 × 103) were injected intraperitoneally into five BALB/c mice. After 24 h, the mice were treated with 1 × 106 plaque-forming unit R3616. After 56 days, four of the five mice were cured, and were injected subcutaneously (s.c.) with MC26 cells (1 × 103; group A; N=4). Control BALB/c mice were also injected s.c. with MC26 cells (1 × 103; group B; N=4). Tumor volumes were monitored and calculated based on the formula V=1/2 × (longest dimension) × (width)2 (P=0.0148).
Cytotoxicity of tumor-specific lymphocytes in vitro
We evaluated the cytotoxicity of tumor-specific lymphocytes against MC26 cells in vitro. MC26 cells (1 × 105 pfu) and R3616 (1 × 105 pfu) were co-cultured for 24 h (group A). Group B consisted of MC26 cells (1 × 105 pfu) that were frozen and thawed twice, and injected i.p. with 500 μl of DMEM into BALB/c mice. After 28 days, lymphocytes were harvested from groups A, B and C (control group; mice injected with an equivalent amount of phosphate-buffered saline). MC26 cells were treated with each type of lymphocytes (from group A, B or C) at a ratio of 100:1 (lymphocytes:MC26 cells). The number of surviving cells was quantified after 72 h. Lymphocytes from group B were significantly more cytotoxic than those from group C (P<0.0001). In addition, lymphocytes from group A were significantly more cytotoxic than those from group B (P=0.0012). We observed that lymphocytes from tumor antigen-immunized mice were effective against tumor cells in vitro. Moreover, lymphocytes from MC26 and R3616-injected mice were more effective than lymphocytes from MC26-injected mice (Figure 2).

Cytotoxicity of tumor antigen-specific lymphocytes against MC26 cells in vitro. MC26 cells (1 × 105) and 1 × 105 plaque-forming unit R3616 were co-cultured for 24 h (group A), or MC26 cells (1 × 105; group B) were frozen and thawed twice. Cells were injected intraperitoneally into five BALB/c mice. After 28 days, lymphocytes were harvested from groups A, B and C (control group; phosphate-buffered saline-injected mice). MC26 cells were treated with each type of lymphocytes (group A, B, and C lymphocytes) at a ratio of 100:1 (lymphocytes:MC26 cells). Surviving cells were quantified after 72 h. Group B lymphocytes were significantly more cytotoxic than group C lymphocytes (P<0.0001). In addition, group A lymphocytes were significantly more cytotoxic than group B lymphocytes (P=0.0012).
Cytotoxicity of R3616 adsorbed onto tumor antigen-specific lymphocytes in vitro
We evaluated the cytotoxicity of lymphocyte-adsorbed R3616 against MC26 cells in vitro. MC26 cells were treated with R3616 that had been adsorbed onto tumor antigen-specific or non-specific lymphocytes at several ratios (lymphocytes:MC26 cells; 0.05:1, 0.5:1, 5:1). The number of surviving cells was determined after 4 days. Lymphocytes were infected with R3616 at an MOI of 1, and the ratio of adsorbed R3616 was 20:1 (lymphocyte vs R3616). At a lymphocyte:MC26 cell ratio of 0.05:1, there were no significant differences between R3616 adsorbed onto tumor antigen-specific lymphocytes and R3616 adsorbed onto non-specific lymphocytes (P=0.5652). However, at lymphocyte:MC26 cell ratios of 0.5:1 and 5:1, R3616 adsorbed onto tumor antigen-specific lymphocytes was significantly more cytotoxic than R3616 adsorbed onto non-specific lymphocytes (P=0.0405, P<0.0001; Figure 3). We observed that R3616 adsorbed onto tumor antigen-specific lymphocytes was more effective against tumors than R3616 adsorbed onto non-specific lymphocytes in vitro.

Cytotoxicity of R3616 adsorbed onto tumor antigen-specific lymphocytes, against MC26 cells in vitro. Non-frozen 1 × 105 MC26 cells (group A) or phosphate-buffered saline (PBS; group B) were injected intraperitoneally into 5 BALB/c mice for each groups. After 28 days, lymphocytes were harvested from each group, and R3616 were adsorbed onto them at a multiplicities of infection (MOI)=1 for 2 h at 37 °C. The virus-loaded lymphocytes were washed in PBS at 4 °C. MC26 cells were treated with R3616-loaded tumor antigen-specific or control lymphocytes at several different ratios (lymphocytes:MC26 cells=0.05:1, 0.5:1 or 5:1). Surviving cells were quantified after 4 days (when R3616 was added to lymphocytes at MOI=1, the ratio of lymphocyte to R3616 was 20:1). At a lymphocyte:MC26 cell ratio of 0.05:1, there were no significant differences between R3616 adsorbed onto tumor antigen-specific lymphocytes and R3616 adsorbed onto non-specific lymphocytes (P=0.5652). However, at ratios of 0.5:1 and 5:1, R3616 adsorbed onto tumor antigen-specific lymphocytes was significantly more cytotoxic than R3616 adsorbed onto non-specific lymphocytes (P=0.0405, P<0.0001, respectively).
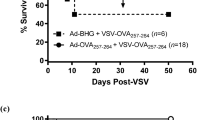
Survival of mice bearing peritoneal metastases after i.v. administering R3616 adsorbed onto tumor antigen-specific lymphocytes
We analyzed survival of mice bearing MC26 peritoneal metastases following i.v. administration (tail-vein injection) of R3616 adsorbed onto tumor antigen-specific lymphocytes. MC26 cells (1 × 104) in 500 μl of DMEM were injected i.p. into BALB/c mice. After 24 h, the mice were treated i.v. with 1 × 106 tumor antigen-specific lymphocytes in 500 μl of DMEM (group 1; L(MC26)), 1 × 106 non-specific lymphocytes in 500 μl of DMEM (group 2; L(control)), 1 × 106 pfu R3616 adsorbed onto 1 × 106 tumor antigen-specific lymphocytes in 500 μl of DMEM (group 3; L(MC26)+R3616), 1 × 106 pfu R3616 adsorbed onto 1 × 106 non-specific lymphocytes in 500 μl of DMEM (group 4; L(control)+R3616), 500 μl of phosphate-buffered saline (group 5), or 1 × 106 pfu R3616 in 500 μl of DMEM (group 6). The ratio of adsorbed R3616 was 20:1 (lymphocytes vs R3616). There was no death at early stage after administration, but all mice died after tumor grew. No deaths were related with severe side effects caused by treatments. There were no significant statistical differences in the survival rate among groups 1, 2, 5 and 6, even though tumor antigen-specific lymphocytes and the viruses alone showed tendency to expand the survival rate. Group 3 mice survived significantly longer than group 1 mice (P=0.0055). In addition, although all mice in group 1 died within 60 days, 40% of the mice in group 3 were cured. Furthermore, group 3 mice survived significantly longer than group 4 mice (P=0.0005). As the ratio of adsorbed R3616 was 20:1 (lymphocyte vs R3616), mice in groups 3 and 6 were injected with only 5 × 104 pfu R3616 and 1 × 106 pfu R3616, respectively. Nevertheless, group 3 mice survived significantly longer than group 6 mice (P=0.0064). Although all of the mice in group 6 died within 54 days, 40% of the mice in group 3 were cured (Figure 4). These findings clearly showed that R3616 adsorbed onto tumor-specific lymphocytes have significant antitumor effects in vivo.

Survival rate of mice bearing MC26 peritoneal metastases after intravenous (i.v.) administration of R3616 adsorbed onto tumor-specific lymphocytes. MC26 cells (1 × 104) were injected intraperitoneally into BALB/c mice (N=10 per group). After 24 h, the mice were treated i.v. with 1 × 106 tumor antigen-specific lymphocytes (group 1; L (MC26)), 1 × 106 non-specific lymphocytes (group 2; L (control)), 1 × 106 plaque-forming unit (pfu) R3616 for adsorption onto 1 × 106 tumor antigen-specific lymphocytes (group 3; L (MC26)+R3616), 1 × 106 pfu R3616 for adsorption onto 1 × 106 non-specific lymphocytes (group 4; L (control)+R3616), phosphate buffered saline (group 5), or 1 × 106 pfu R3616 (group 6). There were no significant statistical differences in survival between groups 1, 2, 5 and 6. Tumor antigen-specific lymphocytes and virus alone showed a tendency to increase survival, but did not exhibit significant efficacy against tumors in our animal studies. Group 3 mice survived significantly longer than other groups.
Discussion
Here we showed that i.v. administration of oncolytic herpes virus adsorbed onto tumor antigen-specific lymphocytes is an effective antitumor treatment. In the past, oncolytic viruses have been mainly introduced into tumors by direct injection, but this therapeutic method is insufficient to treat multiple systemically disseminated tumors. Although a method to systemically deliver oncolytic viruses is needed, administering the virus alone i.v. does not provide sufficient antitumor efficacy in this model. However, i.v. administration of HSV adsorbed onto tumor antigen-specific lymphocytes is a promising treatment for malignancies. It has been proposed that this method would inhibit both virus neutralization by the host immune response and nonspecific virus adhesion to the host cells.33, 34 In contrast to liposome-conjugated tumor-specific antibodies, tumor antigen-specific lymphocytes themselves have antitumor efficacy via mechanisms related to lymphocyte activation, including perforin, granzyme and Fas ligand apoptosis. Moreover, lymphocytes can specifically target virus to the tumor site. This new strategy is more effective than either tumor antigen-specific lymphocytes alone or virus alone.
Oncolytic viral therapy has been heralded as a new treatment modality for advanced cancers.5 Various oncolytic viral therapy studies have been performed, ranging from basic research to clinical trials.7 In addition, several HSV mutants, including HF10, G207, 1716 and NV1020, have demonstrated both safety and efficacy in clinical trials.7, 12, 13, 14, 15
Patients with disseminated metastases of cancer frequently have other systemic metastases in tissues including the liver and lungs, and it is very important to deliver the viruses systemically to these metastatic sites.17 However, Kulu et al.18 reported that i.p. administration was significantly more effective than i.v. administration in mice bearing peritoneal metastases, and that there was no significant difference between the i.v.-treated and control groups. In our animal studies, there was also no significant difference in survival between mice that were i.v.-administered HSV and those left untreated (Figure 4). Viruses can adhere nonspecifically to the endothelium and are neutralized by the immune system, making it difficult to achieve sufficient viral titers at the target site.19 Although clinical trials have examined the efficacy of i.v. administration of adenoviruses and Newcastle viruses, the antitumor effects of these therapies are insufficient.8, 10, 11 As the virus can be neutralized and nonspecifically adhere to host normal cells, the amount of i.v.-administered virus reaching the tumor site is insufficient. Various methods have been examined to increase the efficiency of systemic virus delivery. It has been reported that systemic i.v. delivery of HSV that is encapsulated within liposomes effectively treats multiple liver metastases in the presence of pre-existing viral neutralizing antibodies, and that this method limits viral neutralization.35 The microenvironment of endothelial cells affects viral adhesion, and combination therapy has been used to try to control these effects. A combination of virus and anti-vascular endothelial growth factor resulted in the most dramatic tumor suppression when the anti-vascular endothelial growth factor was administered after the virus i.v.36
There is an emerging hypothesis that the host cells can serve as a ‘Trojan horse’ delivery vehicle; oncolytic virus can adhere to lymphocytes in vitro, and then, these cells can be systemically injected into blood vessels. Once in the bloodstream, these cells carry the virus to tumor beds.20 Various carriers have been evaluated and antigen-specific T cells have often been used.24, 25, 26, 27, 28, 29 Oncolytic viruses adsorbed onto lymphocytes are systemically delivered and released at the tumor site, a protease-rich environment; there, the oncolytic virus spreads and replicates in the tumor cells.27, 33, 37 This strategy is called the ‘hitch-hiking method’. Tumor-specific T lymphocytes are a logical carrier for systemic virus delivery.20, 30 Moreover, tumor-specific lymphocytes themselves have antitumor effects.30, 31, 32 We expect that this new strategy will be extremely effective for systemic delivery when using tumor antigen-specific lymphocytes.
We analyzed the effects of tumor immunity in vivo. Surviving mice, which were treated with HSV against loaded tumors, were rechallenged s.c. with the same tumor cells; the tumor growth in these mice was significantly suppressed (Figure 1). This outcome is assumed to be related to acquired antitumor immunity.
We evaluated the cytotoxicity of tumor-specific lymphocytes against the tumor cells in vitro. Lymphocytes from tumor antigen-acquired mice were significantly more cytotoxic than lymphocytes from control mice. In addition, lymphocytes from co-cultured tumor with R3616-injected mice were significantly more cytotoxic than lymphocytes from tumor-injected mice (Figure 2). We showed that tumor-specific lymphocytes from tumor antigen-injected mice are efficacious as an antitumor immunotherapy, but the antitumor efficacy of lymphocytes from the mice after tumor antigen injected with HSV was even higher. Viral infections induce strong cytotoxic T-cell responses and systemic immune responses against tumors.38 Antigens are likely to be produced when the tumor cells are destroyed during inflammation, when virally encoded Toll-like receptor ligands stimulate dendritic cells to present tumor antigens and elicit effective T-cell responses.39 To generate cancer vaccines, many different strategies have been evaluated in clinical trials, including adjuvant peptides, inactivated cancer cells, DNA, and even mRNA.40, 41 These vaccination strategies can elicit host immune responses, but these low-level responses often have limited efficacy. Although such treatment strategies have been shown to clearly stabilize disease, there have been very few examples of tumor regression. As many tumor antigens are autoantigens, immunological tolerance is a very challenging problem, and it is difficult to generate effective immune responses against these targets.
There were no significant differences in our in-vivo studies among mice that were injected i.v. with non-specific lymphocytes, mice that were injected i.v, with tumor antigen-specific lymphocytes, and mice injected i.v. with phosphate-buffered saline (Figure 4). Tumor antigen-specific lymphocytes had no efficacy against tumors in vivo. Although some effect was observed in vitro, tumor antigen-specific lymphocytes alone were not significantly effective against tumors in our animal studies.
We showed that lymphocytes harvested from mice that received virus-adherent lymphocytes i.p. had stronger antigen-recognition capacity and higher cytotoxicity than lymphocytes harvested from mice that received tumor cells alone i.p. (Figure 2). In our in-vitro studies, we found that a high density of antigen-specific lymphocytes is important for effective results (Figure 3). These results indicate that it may be difficult to obtain a high density of these lymphocytes in humans, and that the maximum effects of lymphocyte therapy have not yet been reached in clinical studies. This may be one reason why it is difficult to obtain sufficient antitumor effects with lymphocyte therapy alone. Thus, new therapeutic methods are greatly needed.
We analyzed the survival rate of mice bearing peritoneal metastases after i.v. injection of HSV that was adherent to tumor antigen-specific lymphocytes. This group survived significantly longer than groups that were i.v. injected with HSV-adherent non-specific lymphocytes, tumor antigen-specific lymphocytes alone, or HSV alone. Although all of the mice that received tumor antigen-specific lymphocytes alone and all of the mice in the HSV-treated group died, 4 of 10 mice in the group treated with HSV-adherent tumor antigen-specific lymphocytes were cured (Figure 4).
Interferon (IFN) is a key cytokine in antivirus and anticancer immunity, and acts at the site of tumor-surrounding tissue. Loading lymphocytes with virus induces proinflammatory cytokines that are associated with antiviral responses (IFN-α/β) and T-cell effector cytolytic functions (IFN-γ); IFN is associated with increased cytolytic functions and more effective immune delivery to tumors.23, 42 Infecting tumors with viruses induces antitumor immunity through tyrosinase-related protein 2 and interleukin 12.43 Moreover, indirect tumor cell killing is induced by the activation of cytokine production and host antitumor immunity.23, 42 Virus-adherent lymphocytes become a stimulus for IFN secretion and lead to suppression of tumor growth.
On another front, tumor cytolysis by oncolytic virus releases weak tumor antigens, which are recognized by the host immune system, and can induce immune responses.43, 44 It is thought that long-term antitumor immune memory is initiated by this response.45 Therefore, although we administered HSV-adherent tumor antigen-specific lymphocytes only once, 40% of the treated mice were completely cured. In an immune-competent mouse, the quantity of virus that reaches and infects tumor cells is typically limited, due to virus neutralization and nonspecific adhesion to host cells.23
Pre-existing immunity against the virus affects the balance between antiviral immunity and antitumor immunity.45 Therefore, in the future, it will be necessary to evaluate the efficacy of multiple administration and treatments in the presence of pre-existing virus-neutralizing antibodies. It is possible that tumor antigen-specific lymphocytes inhibit virus neutralization and nonspecific adhesion to host cells, and can effectively deliver oncolytic viruses to the tumor site.
Furthermore, we should point out existing possibility that the adsorption of HSV onto lymphocytes improves the cytolytic functions of the lymphocytes themselves (perforin, granzyme and Fas-ligand apoptosis), resulting in a more efficient antitumor immune response. This type of phenomenon, as opposed to greater efficiency of virus delivery, may be the primary explanation for the outcomes we observed. It will be necessary to evaluate the significance of this potentially beneficial phenomenon in future studies.
We believe that oncolytic HSV-adherent tumor antigen-specific lymphocytes are a promising new treatment method, not only for primary lesions, but also multiple metastatic lesions. This strategy may become one of the most effective methods for systemic virus delivery.
References
Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ . Cancer statistics, 2009. CA Cancer J Clin 2009; 59: 225–249.
Koppe MJ, Boerman OC, Oyen WJ, Bleichrodt RP . Peritoneal carcinomatosis of colorectal origin: incidence and current treatment strategies. Ann Surg 2006; 243: 212–222.
Kerscher A, Esquivel J . Current status and future directions: management of colon cancer with peritoneal dissemination. Future Oncol 2005; 4: 671–679.
Gómez Portilla A, Cendoya I, López de Tejada I, Olabarría I, Martínez de Lecea C, Magrach L et al. Peritoneal carcinomatosis of colorectal origin. Current treatment. Review and update]. Rev Esp Enferm Dig 2005; 97: 716–737.
Nomura N, Kasuya H, Watanabe I, Shikano T, Shirota T, Misawa M et al. Considerations for intravascular administration of oncolytic herpes virus for the treatment of multiple liver metastases. Cancer Chemother Pharmacol 2009; 63: 321–330.
Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM . Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991; 252: 854–856.
Kasuya H, Takeda S, Shimoyama S, Shikano T, Nomura N, Kanazumi N et al. Oncolytic virus therapy--foreword. Curr Cancer Drug Targets 2007; 7: 123–125.
Hamid O, Varterasian ML, Wadler S . Phase II trial of intravenous CI-1042 in patients with metastatic colorectal cancer. J Clin Oncol 2003; 21: 1498–1504.
Makower D, Rozenblit A, Kaufman H . Phase II clinical trial of intralesional administration of the oncolytic adenovirus ONYX-015 in patients with hepatobiliary tumors with correlative p53 studies. Clin Cancer Res 2003; 9: 693–702.
Pecora AL, Rizvi N, Cohen GI, Meropol NJ, Sterman D, Marshall JL et al. Phase I trial of intravenous administration of PV701, an oncolytic virus, in patients with advanced solid cancers. J Clin Oncol 2002; 1: 2251–2266.
Lorence RM, Pecora AL, Major PP, Hotte SJ, Laurie SA, Roberts MS et al. Overview of phase I studies of intravenous administration of PV701, an oncolytic virus. Curr Opin Mol Ther 2003; 5: 618–624.
Nakao A, Kimata H, Imai T, Kikumori T, Teshigahara O, Nagasaka T et al. Intratumoral injection of herpes simplex virus HF10 in recurrent breast cancer. Ann Oncol 2004; 15: 988–989.
Markert JM, Medlock MD, Rabkin SD . Conditionally replicating herpes simplex virus mutant, G207, for the treatment of malignant glioma: results of a phase I trial. Gene Ther 2000; 7: 867–874.
Harrow S, Papanastassiou V, Harland J, Mabbs R, Petty R, Fraser M et al. HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Ther 2004; 11: 1648–1658.
Fong Y, Kim T, Bhargava A, Schwartz L, Brown K, Brody L et al. A herpes oncolytic virus can be delivered via the vasculature to produce biologic changes in human colorectal cancer. Mol Ther 2008; 17: 389–394.
Tilstone C . Virotherapy paves the way for new cancer treatment. Lancet Oncol 2004; 5: 136.
Deng W, Jia J . Endothelial progenitor cells as cellular vehicles to deliver oncolytic virus therapies to metastatic tumors: the ‘Trojan horse’ approach. Med Hypotheses 2007; 70: 842–844.
Kulu Y, Dorfman JD, Kuruppu D, Fuchs BC, Goodwin JM, Fujii T et al. Comparison of intravenous versus intraperitoneal administration of oncolytic herpes simplex virus 1 for peritoneal carcinomatosis in mice. Cancer Gene Ther 2008; 16: 291–297.
Fisher K . Striking out at disseminated metastases: the systemic delivery of oncolytic viruses. Curr Opin Mol Ther 2006; 8: 301–313.
Power AT, Bell JC . Cell-based delivery of oncolytic viruses: a new strategic alliance for a biological strike against cancer. Mol Ther 2007; 15: 660–665.
Ong HT, Hasegawa K, Dietz AB, Russell SJ, Peng KW . Evaluation of T cells as carriers for systemic measles virotherapy in the presence of antiviral antibodies. Gene Ther 2007; 14: 324–333.
Thorne SH, Negrin RS, Contag CH . Synergistic antitumor effects of immune cell-viral biotherapy. Science 2006; 311: 1780–1784.
Qiao J, Wang H, Kottke T, Diaz RM, Willmon C, Hudacek A et al. Loading of oncolytic vesicular stomatitis virus onto antigen-specific T cells enhances the efficacy of adoptive T-cell therapy of tumors. Gene Ther 2008; 15: 604–616.
Yotnda P, Savoldo B, Charlet-Berguerand N, Rooney C, Brenner M . Targeted delivery of adenoviral vectors by cytotoxic T cells. Blood 2004; 104: 2272–2280.
Crittenden M, Gough M, Chester J, Kottke T, Thompson J, Ruchatz A et al. Pharmacologically regulated production of targeted retrovirus from T cells for systemic antitumor gene therapy. Cancer Res 2003; 63: 3173–3180.
Chester J, Ruchatz A, Gough M, Crittenden M, Chong H, Cosset FL et al. Tumor antigen-specific induction of transcriptionally targeted retroviral vectors from chimeric immune receptor-modified T cells. Nat Biotechnol 2002; 20: 256–263.
Cole C, Qiao J, Kottke T, Diaz RM, Ahmed A, Sanchez-Perez L et al. Tumor-targeted, systemic delivery of therapeutic viral vectors using hitchhiking on antigen-specific T cells. Nat Med 2005; 11: 1073–1081.
Kottke T, Qiao J, Diaz RM, Ahmed A, Vroman B, Thompson J et al. The perforin-dependent immunological synapse allows T-cell activation-dependent tumor targeting by MLV vector particles. Gene Ther 2006; 13: 1166–1177.
Thanarajasingam U, Sanz L, Diaz R, Qiao J, Sanchez-Perez L, Kottke T et al. Delivery of CCL21 to metastatic disease improves the efficacy of adoptive T-cell therapy. Cancer Res 2007; 67: 300–308.
Dudley ME, Rosenberg SA . Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer 2003; 3: 666–675.
Chang AE, Shu S . Current status of adoptive immunotherapy of cancer. Crit Rev Oncol Hematol 1996; 22: 213–228.
Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002; 298: 850–854.
Pizzato M, Blair ED, Fling M, Kopf J, Tomassetti A, Weiss RA et al. Evidence for nonspecific adsorption of targeted retrovirus vector particles to cells. Gene Ther 2001; 8: 1088–1096.
Harrington K, Alvarez-Vallina L, Crittenden M, Gough M, Chong H, Diaz RM et al. Cells as vehicles for cancer gene therapy: the missing link between targeted vectors and systemic delivery? Hum Gene Ther 2002; 13: 1263–1280.
Shikano T, Kasuya H, Sahin TT, Nomura N, Kanzaki A, Misawa M et al. High Therapeutic Potential for Systemic Delivery of a Liposome-conjugated Herpes Simplex Virus. Curr Cancer Drug Targets 2011; 11: 111–122.
Eshun FK, Currier MA, Gillespie RA, Fitzpatrick JL, Baird WH, Cripe TP . VEGF blockade decreases the tumor uptake of systemic oncolytic herpes virus but enhances therapeutic efficacy when given after virotherapy. Gene Ther 2010; 17: 922–929.
Lichty BD, Stojdl DF, Taylor RA, Miller L, Frenkel I, Atkins H et al. Vesicular stomatitis virus: a potential therapeutic virus for the treatment of hematologic malignancy. Hum Gene Ther 2004; 15: 821–831.
Watanabe D, Goshima F, Mori I, Tamada Y, Matsumoto Y, Nishiyama Y . Oncolytic virotherapy for malignant melanoma with herpes simplex virus type 1 mutant HF10. J Dermatol Sci 2008; 50: 185–196.
Lapteva N, Aldrich M, Rollins L, Ren W, Goltsova T, Chen SY et al. Attraction and activation of dendritic cells at the site of tumor elicits potent antitumor immunity. Mol Ther 2009; 17: 1626–1636.
Fishman M . A changing world for DCvax: a PSMA loaded autologous dendritic cell vaccine for prostate cancer. Expert Opin Biol Ther 2009; 9: 1565–1575.
Miyazawa M, Ohsawa R, Tsunoda T, Hirono S, Kawai M, Tani M et al. Phase I clinical trial using peptide vaccine for human vascular endothelial growth factor receptor 2 in combination with gemcitabine for patients with advanced pancreatic cancer. Cancer Sci 2010; 101: 433–439.
Diaz RM, Galivo F, Kottke T, Wongthida P, Qiao J, Thompson J et al. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res 2007; 67: 2840–2848.
Prestwich RJ, Errington F, Ilett EJ, Morgan RS, Scott KJ, Kottke T et al. Tumor infection by oncolytic reovirus primes adaptive antitumor immunity. Clin Cancer Res 2008; 14: 7358–7366.
Li H, Zhang X . Oncolytic HSV as a vector in cancer immunotherapy. Methods Mol Biol 2010; 651: 279–290.
Ilett EJ, Prestwich RJ, Kottke T, Errington F, Thompson JM, Harrington KJ et al. Dendritic cells and T cells deliver oncolytic reovirus for tumour killing despite pre-existing anti-viral immunity. Gene Ther 2009; 16: 689–699.
Acknowledgements
This work was supported by 2009 Grants-in-Aid for Scientific Research in Japan, and by the Nitto Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Kanzaki, A., Kasuya, H., Yamamura, K. et al. Antitumor efficacy of oncolytic herpes simplex virus adsorbed onto antigen-specific lymphocytes. Cancer Gene Ther 19, 292–298 (2012). https://doi.org/10.1038/cgt.2011.91
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cgt.2011.91
Keywords
This article is cited by
-
Immuno-Oncolytic Viruses: Emerging Options in the Treatment of Colorectal Cancer
Molecular Diagnosis & Therapy (2021)
-
Oncolytic herpes simplex virus and immunotherapy
BMC Immunology (2018)
-
Advance in herpes simplex viruses for cancer therapy
Science China Life Sciences (2013)
