
Abstract
IL-17 is one of the most potent and most actively investigated proinflammatory cytokines. In this study, we examined the effect of IL-17 on mesenchymal stem cells (MSCs) under the influence of inflammatory cytokines. Ironically, IL-17 dramatically enhanced the immunosuppressive effect of MSCs induced by IFNγ and TNFα, revealing a novel role of IL-17 in immunosuppression. Interestingly, we found that this action of IL-17 was dependent on the promoted expression of a key immune suppressive molecule, inducible nitric oxide synthase (iNOS), in MSCs. In a concanavalin A (ConA)-induced hepatitis mouse model, we found that IL-17 also enhanced the in vivo immunosuppressive effect of MSCs in an iNOS-dependent manner. Moreover, this promoting effect of IL-17 was found to be exerted through enhancing mRNA stability by modulating the protein level of ARE/poly(U)-binding/degradation factor 1 (AUF1), a well-known factor that promotes mRNA decay. In auf1−/− MSCs, IFNγ and TNFα could induce maximal immunosuppressive effect, both in vitro and in vivo, without the need for IL-17. Thus, our studies demonstrated that in the presence of MSCs, IL-17 promotes immunosuppression.
Similar content being viewed by others


Main
Interleukin 17A (hereafter referred to as IL-17) is the first member discovered of the IL-17 cytokine family.1 It is one of the key inflammatory cytokines during immune responses, primarily produced by Th17 cells.2, 3 IL-17 is well-known for its proinflammatory functions in infectious, inflammatory, and autoimmune responses.4, 5, 6 IL-17 signals through a heteromeric receptor complex composed of IL-17RA and IL-17RC.7, 8 Upon IL-17 binding, IL-17RA recruits Act1, a critical downstream mediator of the IL-17-induced signaling process.9, 10 Although much is known about the IL-17-induced signaling pathways, its cellular targets and mode of action during inflammatory processes and autoimmune diseases remain elusive.
Mesenchymal stem cells (MSCs) exist in various tissues, including bone marrow, muscle, teeth, skin, and fat.11 Upon stimulation by cytokines in an inflammatory environment, MSCs can secrete a variety of chemokines, immune-regulatory molecules, and growth factors. These products can further recruit other cells including inflammatory cells, modulate immune responses, participate in tissue repair, and affect pathological progression.12, 13, 14, 15 Although we have reported that among all the cytokines produced by activated T cells, interferon-γ (IFNγ), tumor necrosis factor-α (TNFα), and likely IL-1 are the key initiating cytokines in inducing immunosuppression by MSCs;13 it is not known whether other cytokines accompanying an inflammatory response can also affect the immunosuppressive effects of MSCs. In this study, we found that the otherwise most potent inflammatory cytokine, IL-17, could in fact effectively synergize with IFNγ and TNFα in inducing immunosuppression by promoting inducible nitric oxide synthase (iNOS) expression in MSCs.
Besides transcription, the abundance of mRNAs is also regulated by their decay rate.16 Regulation of mRNA decay is executed through interactions between specific mRNA sequences and the trans-acting factors that bind them.16, 17 The ARE-binding protein ARE/poly(U)-binding/degradation factor 1 (AUF1) is abundant in lymphoid organs,18 regulates various immune-related molecules, including IL-2, GM-CSF, TNFα, iNOS, and IL-6 mRNAs, and thus controls expression of these inflammatory proteins during immune responses.19, 20 In our study, we showed that IL-17 could enhance iNOS mRNA stability through reducing AUF1 protein levels in MSCs treated with IFNγ and TNFα. Thus, AUF1 serves as a control point through which IL-17 must act to exert its immunosuppressive function in MSCs in an inflammatory environment.
Because of their powerful immunomodulatory functions, MSCs are being investigated for their therapeutic potential in many diseases. Although various studies have demonstrated the therapeutic effect of MSCs on chronic diseases, including graft-versus-host disease, Crohn’s disease, liver cirrhosis, systemic lupus erythomatosus, and multiple sclerosis (MS),21, 22, 23, 24, 25 little is known about the effectiveness of MSCs in treating acute inflammatory disorders such as acute liver injury. Here, we showed that MSCs pretreated with inflammatory cytokines together with IL-17 could effectively cure concanavalin A (ConA)-induced liver injury in mice in an iNOS-dependent manner. Consistent with our in vitro findings, auf1−/− MSCs treated with inflammatory cytokines alone demonstrated similar effects and IL-17 supplementation was not needed. We believe that our findings not only revealed a novel role of IL-17 in enhancing immunosuppression, but also provided novel findings of the action of AUF1 in the IL-17 signaling pathway, and better understanding of the therapeutic effects of MSCs.
Results
IL-17 enhances the immunosuppressive effect of MSCs
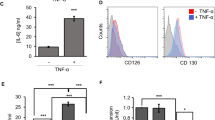
Our previous studies have shown that a combination of IFNγ with TNFα is required to elicit the immunosuppressive effect of MSCs. We examined whether IL-17 could affect IFNγ and TNFα-induced immunosuppression of MSCs. We found that T-cell proliferation was reduced in the presence of IFNγ and TNFα, and surprisingly, this suppression was markedly enhanced by IL-17 (Figure 1a). As IL-17 signals through IL-17RA and IL-17RC, the expression of these receptors in MSCs was determined by quantitative RT-PCR (Figure 1b), in which a mouse macrophage cell line Raw 264.7 was used as a positive control. Cell surface expression of the receptor was also confirmed by flow cytometric analysis (Figure 1c).

IL-17 enhances the immunosuppressive effect of MSCs. (a) Cloned MSCs were first treated with the indicated combinations of recombinant cytokines IFNγ, TNFα, and IL-17 (2 ng/ml each) for 12 h, then co-cultured with T-cell blasts at a 1 : 20 ratio (MSC: T cells), and proliferation was assessed by 3H-thymidine incorporation after an additional 12 h. (b) The mRNA expression of IL-17 receptor family members in MSCs or cell line Raw 264.7 were examined by quantitative RT-PCR. (c) Surface expression of IL-17RA was detected by flow cytometry with cloned MSCs. (d and e) MSCs were first treated with IFNγ and TNFα, with or without IL-17 (10 ng/ml). IFNγ and TNFα were supplemented at different cytokine concentrations, for 12 h, and then co-cultured with T-cell blasts (d) or T-cell hybridoma A1.1 cells (e) at a ratio of 1 : 20 for 12 h. T-cell proliferation was measured by 3H-thymidine incorporation. (f) MSCs were first treated with IFNγ and TNFα (2 ng/ml) with graded concentrations of IL-17 for 12 h, and then co-cultured with T-cell hybridoma A1.1 cells at a ratio of 1 : 10 for 12 h. T-cell proliferation was measured by 3H-thymidine incorporation. (g) MSCs were co-cultured with fresh C57BL/6 splenocytes activated with anti-CD3 and anti-CD28, or in the presence of antibodies against IL-17A, at a 1 : 20 or 1 : 40 ratio (MSCs: splenocytes), for 48 h, and then cell proliferation was assessed by 3H-thymidine incorporation. (h) MSCs were first treated with IFNγ and TNFα (2 ng/ml), or together with IL-17 for 12 h, and then co-cultured with T-cell hybridoma A1.1 cells at a ratio of 1 : 10, in the presence or absence of L-NMMA, for 12 h. T-cell proliferation was measured by 3H-thymidine incorporation. Proliferation values represent means±S.E.M. of three wells from a representative of three independent experiments
As the concentrations of inflammatory cytokines vary at different stages of an inflammatory response, we examined the concentration dependence of IFNγ and TNFα on IL-17–enhanced immunosuppression. IL-17 was able to enhance the immunosuppressive effect of MSCs on T cells at IFNγ and TNFα concentrations as low as 1–2 ng/ml each (Figures 1d and e). Even at higher concentrations of IFNγ and TNFα (10–20 ng/ml), IL-17 still improved immunosuppression by MSCs though the effect was less pronounced. Nonetheless, in the presence of MSCs, as little as 0.5 ng/ml IL-17 was sufficient to elicit a dramatic decrease in T-cell proliferation (Figure 1f). We also examined the role of T-cell death in this effect of IL-17. However, our results showed that the strongly inhibited T-cell proliferation by MSCs pretreated with IFNγ+TNFα+IL-17 was not due to T-cell death (Supplementary Figure S1).
To verify that IL-17 contributes to the immunosuppressive effect of MSCs during a T-cell immune response, antibody against IL-17 was used to neutralize it in a co-culture system with MSC-activated splenocytes. While proliferation of activated splenocytes was markedly inhibited by MSCs, this inhibition was partially reversed upon addition of antibody against IL-17 (Figure 1g). The optimal effect of anti-IL-17 occurred with a MSC/splenocyte ratio of 1 : 40. At a MSC/splenocyte ratio of 1 : 20, the reversal effect was less pronounced, but still statistically significant. Furthermore, this enhancing effect of IL-17 on immunosuppression of MSCs was completely reversed by iNOS inhibitor, L-NMMA (Figure 1h), suggesting that iNOS has a crucial role in this action of IL-17.
To expand our studies, we compared the function of IL-17 between bone marrow–derived MSCs and adipose-derived MSCs. IL-17 could also dramatically enhance IFNγ and TNFα-induced immunosuppression in adipose-derived MSCs (Supplementary Figure S2A), indicating that this effect of IL-17 may be independent of the source of MSCs. As the self-renewal capacity and plasticity may be altered after several passages, we examined whether the responsiveness of MSCs to IL-17 treatment was compromised as passage number increased. IL-17 still enhanced the immunosuppressive effect of MSCs at higher passage number, even though IFNγ and TNFα can induce modestly more immunosupression in this situation (Supplementary Figure S2B).
IL-17 synergizes with inflammatory cytokines to induce the expression of immune modulatory genes in MSCs
We have shown that IL-17 can enhance the immunosuppressive effect of MSCs. To reveal the mechanism, we initially hypothesized that IL-17 affects the proliferation of MSCs, especially considering that IL-17 has been reported to promote the proliferation of human MSCs.26 However, we found that in the presence of IFNγ and TNFα, the addition of IL-17 modestly inhibited the proliferation of MSCs (Supplementary Figure S2C). Thus, the promoting effect of IL-17 on immunosuppression by MSCs is not through influencing the proliferation of MSCs.
We previously showed that NO and chemokines, acting in concert, are the key molecules mediating the immunosuppressive effects of MSCs. The induction of iNOS and some chemokine genes in MSCs is initiated by IFNγ and TNFα.27 As IL-17 enhanced immunosuppression by MSCs cultured with IFNγ and TNFα (Figures 1d and e), and iNOS was essential for this effect of IL-17 (Figure 1h), we hypothesized that IL-17 synergizes with IFNγ and TNFα to induce the expression of iNOS or chemokines in MSCs. To test this hypothesis, MSCs were cultured with various combinations of IFNγ, TNFα, and/or IL-17, and the expression levels of some key immune modulatory genes were assessed. Compared with MSCs cultured with single or double combinations of cytokines, addition of IFNγ, TNFα, and IL-17 dramatically increased the expression of iNOS mRNA (Figure 2a). Western blotting analysis and assay of nitrate in supernatant also confirmed an increase of iNOS protein (Figure 2b, Supplementary Figure S2D). However, the expression of other chemokines such as CCL2, CCL5, CXCL9, and CXCL10, which have pivotal roles in the immunosuppressive effects of MSCs, were all unaffected by addition of IL-17 (Figure 2c), indicating a specific effect of IL-17 on iNOS expression. The addition of IL-17 can induce MSCs to produce large amounts of NO, which can further inhibit the proliferation of T cells. Excessive NO can also exhibit a cytotoxic effect on MSCs, resulting in suppressed proliferation of IFNγ+TNFα+IL-17 treated MSCs (Supplementary Figure S2C).

IL-17 synergizes with inflammatory cytokines to induce the expression of immune modulatory genes in MSCs. (a and c) MSCs were cultured with different combinations of inflammatory cytokines IFNγ, TNFα, and IL-17 (10 ng/ml) for 12 h, and then cells were harvested for total RNA extraction. The mRNA expression levels of iNOS (a) and chemokines CCL2, CCL5, CXCL9, CXCL10 (c) were detected by quantitative RT-PCR. (b) MSCs were cultured with IFNγ and TNFα, or together with IL-17 (10 ng/ml) for the indicated times, and the protein levels of iNOS were detected by western blotting analysis. (d) MSCs were supplemented with Sup-CD3 or Sup-CD3 pretreated with antibodies against IL-17, and cells were collected for RNA extraction after 12 h (Sup-CD3: supernatant from splenocytes activated by anti-CD3 and anti-CD28). The expression of the iNOS, CCL2, CCL5, CXCL9 and CXCL10 genes was measured by quantitative RT-PCR. (e) MSCs were treated with Sup-CD3 or Sup-CD3 pretreated with antibodies against IL-17, and the protein level of iNOS was assessed by western blotting analysis after 24 h. mRNA expression values are means±S.E.M. of three wells from a representative of three independent experiments. Western blotting data are from a representative of three independent experiments
To verify that the effects on gene expression were due to IL-17, MSCs were cultured with supernatant from anti-CD3 and anti-CD28–activated splenocytes in the presence or absence of neutralizing antibody against IL-17. Addition of anti-IL-17 to supernatant diminished its ability to induce iNOS gene expression without affecting CCL2, CCL5, CXCL9, or CXCL10 (Figures 2d and e). Furthermore, we also verified our findings by blocking signal transduction of IL-17 in MSCs by knocking down Act1, a key component in the signal transduction pathway from the IL-17 receptor. As has been shown in other cells,10 the phosphorylation of IκBα, ERK, p65, and JNK, key molecules in the IL-17 signaling pathway, were impaired in these Act1 knockdown MSCs (Supplementary Figure S3A). As expected, IL-17 cannot upregulate IFNγ and TNFα-induced iNOS expression in Act1 knockdown MSCs (Supplementary Figures S3B, C). At the cellular level, the immunosuppressive effect of IFNγ and TNFα could not be further enhanced by IL-17 in the absence of Act1 (Supplementary Figure S3D).
IL-17 together with IFNγ and TNFα-pretreated MSCs alleviate ConA-induced liver injury in an iNOS-dependent manner
To investigate the promoting effect of IL-17 on IFNγ and TNFα-induced immunosuppression of MSCs in vivo, we employed the ConA-induced liver injury mouse model. ConA-induced liver injury is a well-established in vivo model of hepatocyte apoptosis induced by acute immune responses, in which T lymphocytes are identified as the major effector cells.28 Wild-type MSCs were pretreated or not with IFNγ and TNFα in the presence or absence of IL-17 for 12 h. After ConA administration for 30 min, these differently pretreated MSCs were intravenously injected into the mice. The immunosuppression mediated by MSCs is exerted locally. Thus, GFP-transgenic MSCs were transfused into mice that received ConA injection 30 min earlier. Localization of administrated MSCs in the damaged liver was indeed observed (Supplementary Figure S4). Interestingly, liver injury was not inhibited in mice administrated with wild-type MSCs or wild-type MSCs pretreated with IFNγ and TNFα. However, MSCs pretreated with IFNγ, TNFα, and IL-17 almost completely inhibited liver damage, exhibited as a dramatic reduction of serum alanine aminotransferase (ALT) level, centrilobular necrosis, and inflammatory infiltration (Figures 3a, c and d). Detailed analyses of immune cells in the liver showed that the infiltration of mononuclear cells (MNCs), CD3+CD4+, and CD3+CD8+ T cells were dramatically decreased in mice administered with wild-type MSCs pretreated with IFNγ and TNFα, together with IL-17 (Figure 3b). However, the ameliorated effects of MSCs pretreated with IFNγ, TNFα, and IL-17 in liver injury was not observed in iNOS−/− MSCs (Figures 3a, c and d), indicating that iNOS is a key mediator in the promoting effect of IL-17 on immunosuppression by MSCs in vivo.

MSCs pretreated with IL-17 together with IFNγ and TNFα alleviate ConA-induced liver injury in an iNOS-dependent manner. Mice were intravenously injected with ConA (15 mg/kg); 30 min later, untreated or cytokine-pretreated wild-type or iNOS−/− MSCs were transfused. After 7.5 h, serum and livers were sampled. (a) Serum levels of ALT were measured. (b) Absolute numbers of MNCs in liver tissues were calculated (n=5 mice per group). Absolute numbers of CD3+CD4+ and CD3+CD8+ T cells were determined by flow cytometry. (c) Hematoxylin & eosin staining of liver sections at 8 h after ConA administration. N: necrosis area. (d) Percentages of necrosis were calculated
IL-17 reverses the suppression of gene expression imposed by RNA-binding protein AUF1
Messenger RNAs encoding iNOS and many cytokines/chemokines are rapidly degraded after interaction with mRNA decay factors, which provides a mechanism to limit the abundance of these molecules during an immune response. Activation of signaling pathways, particularly during immune responses, stabilizes many of these mRNAs to increase their expression. Indeed, a major mechanism by which IL-17 influences the expression of many inflammatory mediator genes is by stabilizing their mRNAs.29, 30 AUF1 has been shown to promote the degradation of iNOS mRNA.20 We thus hypothesized that AUF1 may act to limit the expression of iNOS mRNA and that IL-17 may block this activity of AUF1 (thereby increasing gene expression).
To test this hypothesis, cytokine-induced gene expression was compared between MSCs derived from bone marrow of wild-type and auf1−/− mice.31 Cells were cultured as before with IFNγ and TNFα, with or without IL-17. Addition of IL-17 (together with IFNγ and TNFα) dramatically further increased iNOS mRNA in wild-type MSCs (Figure 4a). In contrast, iNOS mRNA was much higher in auf1−/− MSCs cultured with IFNγ and TNFα; interestingly, addition of IL-17 had little effect (i.e., IL-17 increased iNOS mRNA abundance less than twofold). Likewise, IFNγ and TNFα were sufficient to maximally induce iNOS protein in auf1−/− MSCs without the need for IL-17, in contrast to wild-type MSCs (Figure 4b, compare lanes 7 and 8 with lanes 2 and 5). To verify these findings, we knocked down AUF1 in wild-type MSCs (Figure 4c), and treated these cells with IFNγ and TNFα, with or without IL-17. Consistent with auf1−/− MSCs, IFNγ and TNFα induced maximal iNOS mRNA and protein expression in AUF1-knockdown MSCs (Figures 4d and e), and the enhancing effect of IL-17 was abated. Moreover, IL-17 further increased the suppressive activity of IFNγ and TNFα-stimulated wild-type MSCs on T-cell proliferation; however, IFNγ and TNFα were sufficient to induce maximal immunosuppression in auf1−/− MSCs and IL-17 did not further enhance the inhibitory effect on T-cell proliferation (Figure 4f).

IL-17 reverses the suppression of gene expression imposed by RNA-binding protein AUF1. (a and d) WT MSCs and auf1−/− MSCs (a) or shCTRL MSCs and shAUF1 MSCs (d) were treated with IFNγ and TNFα, or together with IL-17 (all cytokines supplemented at 10 ng/ml) for 12 h, and iNOS mRNA expression was measured by quantitative RT-PCR. (b) WT MSCs were treated with IFNγ and TNFα (10 ng/ml), or together with different concentrations of IL-17; auf1−/− MSCs were treated with IFNγ and TNFα (10 ng/ml), or together with 10 ng/ml of IL-17. After 24 h, cells were harvested for detection of iNOS by western blotting analysis. (c) MSCs were stably transfected with plasmid expressing shRNA against AUF1 or plasmid encoding scrambled sequences (CTRL), and cells were collected for identification of AUF1 by quantitative RT-PCR and Western Blot. These cells are referred to as shAUF1 MSCs or shCTRL MSCs. (e) shCTRL MSCs or shAUF1 MSCs were stimulated with IFNγ and TNFα, or together with IL-17 (all cytokines supplemented at 10ng/ml) for 12 or 24 h. iNOS expression was detected by western blot. (f) WT MSCs or auf1−/− MSCs were first treated with IFNγ and TNFα, with or without IL-17 (10 ng/ml), IFNγ and TNFα were supplemented at different concentrations, for 12 h, and then co-cultured with T-cell hybridoma A1.1 cells at a ratio of 1 : 10 for 12 h. T-cell proliferation was measured by 3H-thymidine incorporation, taking the proliferation level of A1.1 alone as 100%. mRNA expression values are means±S.E.M. of three wells from a representative of three independent experiments. Western blotting data are representative of three independent experiments. Proliferation values are means±S.E.M. of three wells from a representative of three independent experiments
IL-17 enhances gene expression through modulating AUF1-mediated mRNA decay in MSCs
Given the effects of AUF1 and IL-17 on gene expression and immunosupression by MSCs, we considered the possibility that knockout of AUF1 alone would be sufficient to provide the degree of mRNA stabilization that would normally require IL-17. mRNA decay assays were performed with wild-type and auf1−/− MSCs, cultured with IFNγ and TNFα, with or without IL-17. In wild-type MSCs, iNOS mRNA was relatively unstable with a half-life of 4.3±1.4 h; IL-17 led to a twofold stabilization of iNOS mRNA (Figure 5a); P<0.05. The CCL2 and CXCL10 mRNAs, which did not respond to IL-17 (see Figure 2c), were not stabilized by IL-17, as would be expected (Figure 5a; t1/2=∼2 h for both mRNAs, with or without IL-17). In contrast to wild-type MSCs, knockout of AUF1 strongly stabilized iNOS mRNA (Figure 5b; t1/2>10 h). In these auf1−/− MSCs, IL-17 had no effect on the half-life of iNOS mRNA (Figure 5b). We also verified these findings in AUF1-knockdown MSCs (Figure 5c).

IL-17 enhances gene expression by modulating AUF1-mediated mRNA decay in MSCs. (a–c) Wild-type (a) or auf1−/− MSCs (b), shCTRL MSCs or shAUF1 MSCs (c) were treated with IFNγ and TNFα, with or without IL-17 (all cytokines supplemented at 10 ng/ml) for 6 h, and then actinomycin D (5 μg/ml) was added to stop transcription. At the indicated time points, mRNA levels were assayed by quantitative RT-PCR, taking the expression level at the time of actinomycin D addition as 100%. mRNA expression values are means±S.E.M. of three wells from a representative of three independent experiments. (d) Wild-type MSCs were treated with IFNγ and TNFα, with or without IL-17 (all cytokines supplemented at 10 ng/ml). At different time points, cells were collected for analysis of AUF1 protein level by western blot. (e) Wild-type MSCs were treated with IL-17 (10 ng/ml) for different time, and cells were harvested for analysis of interaction of Act1 and AUF1 by immunoprecipitation. Western blotting data is from a representative of two independent experiments
To assess the interaction of IL-17 and AUF1 on the enhancing effect of IL-17 on MSC immunosuppression, the protein level of AUF1 was analyzed in MSCs treated with IFNγ and TNFα, in the presence or absence of IL-17, as a function of time. We found that the addition of IL-17 could reduce AUF1 protein levels in IFNγ+TNFα-treated MSCs (Figure 5d). Therefore, IL-17 enhances iNOS mRNA stability by reducing AUF1 protein levels. Thus, AUF1 is one key factor mediating the enhancing effect of IL-17 on IFNγ and TNFα-induced iNOS expression by MSCs. Further experiments were employed to assess potential interactions between AUF1 and IL-17 downstream molecules. As either knockdown of Act1 or knockout of AUF1 in MSCs abolished the ability of IL-17 to enhance their immunosuppressive effects (Supplementary Figure S3 and Figure 4), we assessed interaction of Act1 and AUF1 in MSCs upon IL-17 stimulation using co-immunoprecipitation. IL-17 stimulation increased the interaction of endogenous Act1 with AUF1 (Figure 5e). These data support the ideas that AUF1 downregulation and Act1–AUF1 interactions are key steps in the enhancing effect of IL-17 on both gene expression and immunosupression in MSCs.
Critical roles of AUF1 in IL-17–enhanced therapeutic effect of MSCs on ConA-induced acute liver injury
Given the crucial role of AUF1 in the enhancing effect of IL-17 on immunosuppression by MSCs in vitro, we next examined the effects of IL-17 on immunosuppression in vivo by wild-type and auf1−/− MSCs. Wild-type and auf1−/− MSCs were first pretreated with or without IFNγ and TNFα, in the presence or absence of IL-17 for 12 h, and then these cells were transfused into mice that received ConA injection 30 min earlier. Consistent with the results in Figure 3, wild-type MSCs pretreated with IFNγ, TNFα, and IL-17 showed the best therapeutic effect on liver injury, in which liver injury was almost completely alleviated with sharply reduced serum ALT activity, liver necrosis, and lymphocyte infiltration (Figures 6a, c and d). However, maximal therapeutic effects on ConA-induced liver injury were also achieved with auf1−/− MSCs pretreated with only IFNγ and TNFα, without the need for IL-17 (Figures 6a, c and d). Infiltration of MNCs and CD3+CD4+ and CD3+CD8+ T cells in liver was also dramatically decreased in mice administered with wild-type MSCs pretreated with IFNγ, TNFα, and IL-17, or in mice administered auf1−/− MSCs pretreated with IFNγ and TNFα, either with or without IL-17 (Figure 6b). The percentages of CD3+CD4+ T cells, CD3+CD8+ T cells, Th1, Th17, and Treg subpopulations were analyzed in liver-infiltrated cells among the different groups. However, no significant changes were observed among them (Supplementary Figures S5A, B and C). Thus, the therapeutic effect of MSCs pretreated with IFNγ, TNFα, and IL-17 on ConA-induced liver injury was mainly dependent on their robust suppression of T-cell proliferation, but not by affecting differentiation of T-cell subpopulations.

Critical roles of AUF1 in IL-17–enhanced therapeutic effect of MSCs on ConA-induced acute liver injury. Mice were intravenously injected with ConA (15 mg/kg); 30 min later, untreated or cytokine-pretreated wild-type or auf1−/− MSCs were transfused. After 7.5 h, serum and livers were sampled. (a) Serum levels of ALT were measured. (b) Absolute numbers of MNCs in liver tissues were calculated (n=3–5 mice per group). Absolute numbers of CD3+CD4+ and CD3+CD8+ T cells were determined by flow cytometry. (c) Hematoxilin & eosin staining of liver sections at 8 h after ConA administration. N: necrosis area. (d) Percentages of necrosis were calculated
Discussion
The interactions between MSCs and the inflammatory environment have been actively studied in the last few years in both in vitro and in vivo models. We have previously elucidated critical roles of NO and chemokines in the immunosuppressive effect of MSCs, with the co-presence of IFNγ and TNFα, or IL-1 as a requisite.13 As IL-17 is one of the most potent inflammatory cytokines and is often elevated in the serum and tissues of patients with inflammatory or autoimmune diseases,32 it is important to examine the influence of this key inflammatory cytokine in modulating the immunosuppressive properties of MSCs. In addition, to elicit the immunosuppressive property of MSCs, critical levels of inflammatory cytokines are required.33 It is possible that the presence of IL-17 can further change the dynamics of the interaction between MSCs and inflammatory cytokines. In our study, we found that IL-17 could enhance the immunosuppressive function of MSCs, even in the presence of low doses of inflammatory cytokines IFNγ and TNFα.
The identification of the role of IL-17 in enhancing immunosuppression by MSCs is a novel concept. Since its discovery, IL-17 has been widely reported to exacerbate disease progress in multiple autoimmune diseases, including rheumatoid arthritis, MS, and inflammatory bowel disease, in which the IL-17 level is dramatically elevated.32 In addition, disease progression slows when IL-17 is genetically ablated or IL-17 blocking antibody is administered.34 Thus, it is well accepted that IL-17 is critical in promoting inflammation and autoimmunity. However, IL-17 not always promotes immune responses, as it has been reported that IL-17 has a protective function in gut inflammatory disorders. Genetic ablation or neutralization of IL-17 can actually aggravate disease progress in dextran-sulphate-sodium-induced colitis in mice.35, 36 However, the molecular mechanisms underlining this apparently opposite effect of IL-17 is not known. Our study shows that IL-17 can enhance the immunosuppressive property of MSCs, and it is possible that MSCs may not suppress immune responses effectively without IL-17. Thus, these findings provide novel information for a better understanding of the pathophysiological roles of IL-17. We also examined the function of human IL-17 in human MSCs. As inhibition on T-cell responses by human MSCs is induced by IFNγ,37 we tested if human IL-17 could affect IFNγ-induced immunosuppression in human MSCs. We co-cultured human MSCs with anti-CD3 and anti-CD28–activated human peripheral blood mononuclear cells (PBMCs), in the presence of IFNγ, or together with IL-17, and detected the proliferation of PBMCs. Our results showed that the addition of IL-17 could weakly promote IFNγ-induced immunosuppression by human MSCs, although not as significant as in the mouse system (data not shown). More detailed investigations are necessary to understand the role of various cytokine combinations in the context of IL-17-regulated immunosuppression of human MSCs.
ConA-induced liver injury in mice is a well-established model for investigating the pathophysiological process of autoimmune or viral fulminant hepatitis, in which acute immune responses have a pivotal role in mediating liver damage.28 It is well established that suppression of immune responses can dramatically attenuate ConA-induced liver injury.38 Adipose tissue–derived stromal cells have been shown to reduce liver damage in this model.39, 40 In the present investigation, we used bone marrow–derived MSCs and investigated the role of IL-17 in modulating MSC-mediated therapy of liver injury. We found that in this acute inflammatory disease model, MSCs only marginally affected disease progression. We reasoned that the immunosuppressive capacity of MSCs requires stimulation by inflammatory cytokines. Although many cytokines can be produced after ConA administration in vivo, these cytokines remain at high levels for only a short time28 and thus may not be able to endow MSCs with immunosuppressive capacity when administered at a later time. To circumvent this scenario, we pretreated MSCs with inflammatory cytokines IFNγ and TNFα for 12 h. Clearly, the therapeutic effect was improved to some extent (see Figures 3 and 6). Interestingly, when we pretreated MSCs with inflammatory cytokines IFNγ and TNFα together with IL-17, there was a dramatic enhancement of the therapeutic effect. This enhancement is likely related to a robust induction of iNOS by the combination with IL-17 (Figure 2), as iNOS−/− MSCs didn’t present any therapeutic effect on this liver injury model. Thus, this experiment revealed a novel in vivo role of IL-17 in enhancing immunosuppression.
AUF1 is a well-known RNA-binding protein that promotes degradation of ARE-containing mRNAs encoding numerous cytokines, cell cycle regulators, and other inflammatory molecules.20, 41, 42 Indeed, IL-17 induces cytokine/chemokine gene expression via mRNA stabilization.29 Knockout of AUF1 in MSCs led to induction and stabilization of iNOS and IL-6 mRNAs in MSCs treated with IFNγ and TNFα (Figure 4 and data not shown), indicating that AUF1 critically controls expression of these genes in MSCs. Moreover, AUF1 knockout eliminated the requirement for IL-17 to modulate gene expression, mRNA stabilization, and immunosuppression by MSCs. Further investigations revealed AUF1–Act1 interactions and the action of IL-17 on AUF1 in MSCs. We found that addition of IL-17 reduced AUF1 protein levels in IFNγ+TNFα treated MSCs, and this effect led to stabilization of iNOS and IL-6 mRNAs ( Figure 5 and data not shown). Our results also showed that Act1, a crucial adaptor protein that binds the IL-17 receptor, interacted with AUF1 upon IL-17 stimulation of MSCs. In addition, IL-17 signal transduction was defective in MSCs in the absence of AUF1 (data not shown). These data suggest that AUF1 must have dual roles in the enhancing effect of IL-17: AUF1 is required to initiate IL-17 signaling; AUF1 also serves as a target of IL-17 and is downregulated by IL-17 in MSCs treated with IFNγ and TNFα. Thus, we have uncovered novel roles of AUF1 in the enhancing effect of IL-17 on immunosuppression.
In conclusion, we have demonstrated that IL-17 can enhance immunosuppression in the presence of MSCs. This novel effect of IL-17 is exerted in part by reversing the suppression of gene expression normally conferred by mRNA decay factor AUF1. It would be of great interest to uncover the detailed molecular mechanisms underlying the control of AUF1 by IL-17 in MSCs. In addition, much needs to be learned about the kinetics of IL-17 production during various inflammation processes and how such kinetics affects the therapeutic effect of MSCs. Nevertheless, we believe that further investigations in this direction will lead to better application of MSCs in clinical settings.
Materials and Methods
Reagents and mice
Recombinant mouse IFNγ, TNFα, IL-17A, and antibodies against IL-17A were from eBiosciences (La Jolla, CA, USA). Recombinant mouse IL-2 was from R&D Systems (Minneapolis, MN, USA). Antibodies against β-actin, GAPDH, iNOS, p65, p-IκBα, p-p65, p-JNK, and p-ERK1/2 were from Cell Signaling Technology (Danvers, MA, USA). Antibody against Act1 was from Santa Cruz Biotechnology (Dallas, TX, USA). L-NMMA, propidium iodide, and actinomycin D were purchased from Sigma-Aldrich (St Louis, MO, USA). Concanavaline A (ConA) was from Vector Labs (Burlingame, CA, USA).
C57BL/6 mice were purchased from the Shanghai Laboratory Animal Center of the Chinese Academy of Sciences (Shanghai, China) and maintained under specific pathogen-free conditions in the vivarium of Shanghai Jiao Tong University School of Medicine with water and food provided ad libitum. All animal protocols are approved by our Institutional Animal Care and Use Committee.
Cells
MSCs were generated using our previously described protocol.13 Briefly, tibia and femur bone marrow of wild-type, iNOS−/−, or auf1−/− mice was harvested. Cells were cultured in DMEM medium supplemented with 10% FBS, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (complete medium, all from Invitrogen, Carlsbad, CA, USA). All nonadherent cells were removed after 24 h (hr), and adherent cells were maintained. Medium was changed every 2–3 days. To obtain MSC clones, cells at confluence were harvested and seeded into 96-well plates by limited dilution. Individual clones were then picked and expanded. These MSCs were capable of differentiating into adipocytes and osteocytes under the respective differentiation conditions. Cells were used before the 20th passage.
The generation of T-cell blasts was done as previously described.13, 43
Proliferation assay
To assay cell proliferation, 0.5 μCi of 3H-thymidine (Tdr, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, China) was added to each well of 96-well plates 6 h before termination of the cultures by freezing. Plates were then thawed, cells were harvested, and incorporated 3H-Tdr was assessed with a Wallac Microbeta scintillation counter (Perkin-Elmer, Waltham, MA, USA).
DNA content analysis
DNA content analysis was done as previously described.44 Briefly, A1.1 cells were co-cultured with MSCs for 12 h, harvested, and washed once with PBS. The cells were then resuspended in staining buffer (PBS supplemented with 50 μg/ml propidium iodide, 0.2% saponin, 10 μg/ml RNase A). After 30 min incubation in dark, samples were analyzed by flow cytometry on a FACS Calibur flow cytometer (Becton Dickinson, San Jose, CA, USA).
RNA isolation and gene expression assay
Total RNA was isolated with the RNAprep Pure Cell/Bacteria Kit (TIANGEN Biotech, Beijing, China). First-strand cDNA synthesis was performed with a cDNA synthesis kit (TIANGEN Biotech). The levels of mRNAs were measured by quantitative RT-PCR (7900 HT; Applied Biosystems, Foster City, CA, USA) with SYBR Green Master Mix (TaKaRa Biotech, Dalian, China) and normalized to the level of β-actin mRNA. Sequences of forward and reverse primer pairs are as follows:
AUF1: forward 5′-CAGCAGAGTGGTTATGGGAAAGTATCC-3′ and reverse
5′-GACAGGAGCCACCTTCAAATGAATC-3′;
β-actin: forward 5′-CCACGAGCGGTTCCGATG-3′ and reverse
5′-GCCACAGGATTCCATACCCA-3′;
CCL2: forward 5′-TCTCTCTTCCTCCACCACCATG-3′ and reverse
5′-GCGTTAACTGCATCTGGCTGA-3′;
CCL5: forward 5′-TTTCTACACCAGCAGCAAGTGC-3′ and reverse
5′-CCTTCGTGTGACAAACACGAC-3′;
CXCL9: forward 5′-AGTGTGGAGTTCGAGGAACCCT-3′ and reverse
5′-TGCAGGAGCATCGTGCATT-3′;
CXCL10: forward 5′-TAGCTCAGGCTCGTCAGTTCT-3′ and reverse
5′-GATGGTGGTTAAGTTCGTGCT-3′.
IL-6: forward 5′-GAGGATACCACTCCCAACAGACC-3′ and reverse
5′-AAGTGCATCATCGTTGTTCATACA-3′;
IL-17RA: forward 5′-AGTGTTTCCTCTACCCAGCAC-3′ and reverse
5′-GAAAACCGCCACCGCTTAC-3′;
IL-17RC: forward 5′-GCTGCCTGATGGTGACAATGT-3′ and reverse
5′-TGGACGCAGGTACAGTAAGAAG-3′;
iNOS: forward 5′-CAGCTGGGCTGTACAAACCTT-3′ and reverse
5′-CATTGGAAGTGAAGCGTTTCG-3′.
Messenger RNA decay assays
Messenger RNA decay assays were performed essentially as described.45, 46, 47 Briefly, MSCs were incubated with cytokine combinations for 6 h. Actinomycin D (Act.D) was added into the medium at a final concentration of 5 μg/ml to stop transcription. At various time points after addition of Act.D, cells were harvested for the extraction of total RNA. Levels of iNOS, IL-6, CCL2, and CXCL10 mRNAs were measured at each time point by quantitative RT-PCR. Percentages of mRNA remaining at each time point were plotted versus time after Act.D addition. First-order decay constants, k, were determined by nonlinear regression. The associated mRNA half-lives, t1/2, were calculated with the equation t1/2=ln2/k.
RNA interference
Plasmids bearing shRNA targeting AUF1 mRNA (5′-GTTGTAGACTGCACTCTGA-3′) or Act1 mRNA (5′-GACAGTGATGATAATCGTA-3′) were constructed by the insertion of annealed oligonucleotides into the Hind III and BamH I sites of pSILENCER 2.0-U6 (Ambion, Austin, TX, USA). Plasmids bearing scrambled sequences (shCTRL), shRNA targeting AUF1 mRNA (shAUF1) or Act1 mRNA (shAct1) were introduced into MSCs (1.5 × 106 cells) using Amaxa Nucleofector, with program U-023, and the Nucleofector kit V (Amaxa, Cologne, Germany). Cells were seeded in DMEM complete medium in six-well plates. After 2–3 days, the medium was changed, and stably transfected clones were picked and expanded using DMEM complete medium containing 250 μg/ml hygromycin B (Invitrogen) for 3–4 weeks.
Western blotting analysis
Cells were washed twice with ice-cold PBS, harvested and lysed in the RIPA buffer (Millipore, Temecular, CA, USA) containing a cocktail of protease inhibitors (Roche, Natley, NJ, USA) and PMSF (Sigma) for 30 min on ice. Lysates were clarified by centrifugation at 16 000 × g for 15 min. Protein concentration of the supernatant was determined by the Bradford assay (Bio-Rad, Hercules, CA, USA). Protein samples were diluted in 5 × SDS loading buffer and fractionated in a 10% SDS–polyacrylamide gel. Proteins were electroblotted onto a nitrocellulose membrane (Whatman Inc., Clifton, NJ, USA) and incubated for 1 h in 5% nonfat dry milk dissolved in TBST (150 mM NaCl, 50 mM Tris-HCl, pH 7.5, 0.05% Tween 20) at room temperature. The blotting membranes were incubated with primary antibodies overnight at 4 °C, extensively washed in TBST, incubated with hP-conjugated secondary antibody (Cell Signaling) for 1.5 h at room temperature, and washed again with TBST. The blotting membranes were developed with chemiluminescent reagents (Millipore, Billerica, MA, USA) according to the instructions provide by the manufacturer.
Immunoprecipitation
Cells were lysed in RIPA buffer (Millipore, Temecular, CA, USA) containing a cocktail of protease inhibitors (Roche) and PMSF (Sigma) for 30 min on ice. Lysates were clarified by centrifugation at 16 000 × g for 15 min. Protein concentration of the supernatant was determined by the Bradford assay (Bio-Rad). After precleared with protein G sepharose beads, cell extracts were incubated with 20 μl protein G sepharose beads in the presence of 1 μg Act1 antibody or nonspecific IgG overnight at 4 °C. After washing four times with RIPA buffer, beads were suspended in 20 μl 2 × SDS loading buffer, boiled at 100 °C for 10 min, and analyzed by western blot.
ConA-induced liver injury in mice
C57BL/6 mice (8-10 week old) were intravenously injected with ConA in PBS at 15 mg/kg to induce acute inflammatory liver injury. MSCs (5 × 105) derived from wild-type mice, iNOS−/− mice or auf1−/− mice were treated with or without IFNγ and TNFα in the presence or absence of IL-17 (10 ng/ml for each cytokine) for 12 h, and then intravenously administrated into mice that have been treated with ConA for 30 min. Mice were killed and serum and liver tissues were sampled after another 7.5 h. Serum ALT activity was determined by an ALT detection kit (Yihua Medical Scientific, Shanghai, China). Formalin-fixed liver histological sections were stained with hematoxylin & eosin.
Localization of MSCs in mouse liver
Mice were transfused with GFP-transgenic mouse MSCs 30 min after ConA injection. After another 7.5 h, livers were harvested. Frozen sections were stained with anti-GFP antibody (Abcam, New Territories, Hong Kong) and labeled with Alexa Fluor 594 conjugated donkey anti-rabbit secondary antibody (Invitrogen) and DAPI (Sigma-Aldrich) to reveal localization of MSCs.
Isolation of liver MNCs and flow cytometric analysis
Liver mononuclear cells (MNCs) were purified by a 40%/70% percoll gradient48 and stained with anti-CD3-PE, anti-CD4-PerCP/Cy5.5, anti-CD8a-APC, or anti-CD25-FITC (eBiosciences) for 30 min at 4 °C in staining buffer (PBS, 3% FCS). For detection of intracellular proteins, cells were first stimulated with PMA (Sigma) and inomycin (Sigma) for 4 h and stopped by addition of Brefeldin A (Sigma). After cell surface staining, cells were fixed and permeabilized (eBiosciences), and then stained respectively with anti-IFNγ-FITC, anti-IL-17 A-PE, or anti-Foxp3-PE. For detection of surface expression of IL-17RA in cloned MSCs, cells were stained with anti-IL-17RA-PE (eBiosciences). All samples were analyzed by flow cytometry on a FACS Calibur flow cytometer (Becton Dickinson).
Statistical analysis
Nonlinear regression and statistical analyses were performed with PRISM v5 software (GraphPad Software, Inc., La Jolla, CA, USA). Comparisons between samples were performed with the unpaired t-test. Differences with P<0.05 were considered significant (*, P<0.05; **, P<0.01; ***, P<0.001); N.S., not significant.
Abbreviations
- Act.D:
-
actinomycin D
- ALT:
-
alanine aminotransferase
- AUF1:
-
ARE/poly(U)-binding/degradation factor 1
- ConA:
-
concanavalin A
- IFNγ:
-
interferon-γ
- IL-17:
-
interleukin-17
- iNOS:
-
inducible nitric oxide synthase
- MNCs:
-
mononuclear cells
- MSCs:
-
mesenchymal stem cells
- TNFα:
-
tumor necrosis factor-α
References
Rouvier E, Luciani MF, Mattei MG, Denizot F, Golstein P . CTLA-8 cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J Immunol 1993; 150: 5445–5456.
Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 2005; 6: 1123–1132.
Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 2005; 6: 1133–1141.
Kang Z, Altuntas CZ, Gulen MF, Liu C, Giltiay N, Qin H et al. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity 2010; 32: 414–425.
Chabaud M, Lubberts E, Joosten L, van Den Berg W, Miossec P . IL-17 derived from juxta-articular bone and synovium contributes to joint degradation in rheumatoid arthritis. Arthritis Res 2001; 3: 168–177.
Yang B, Wang Y, Zhao C, Yan W, Che H, Shen C et al. Increased Th17 cells and interleukin-17 contribute to immune activation and disease aggravation in patients with chronic hepatitis B virus infection. Immunol Lett 2013; 149: 41–49.
Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR et al. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity 1995; 3: 811–821.
Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J et al. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol 2006; 177: 36–39.
Chang SH, Park H, Dong C . Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem 2006; 281: 35603–35607.
Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol 2007; 8: 247–256.
Rosenbaum AJ, Grande DA, Dines JS . The use of mesenchymal stem cells in tissue engineering: a global assessment. Organogenesis 2008; 4: 23–27.
Xu G, Zhang L, Ren G, Yuan Z, Zhang Y, Zhao RC et al. Immunosuppressive properties of cloned bone marrow mesenchymal stem cells. Cell Res 2007; 17: 240–248.
Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell 2008; 2: 141–150.
Shi Y, Hu G, Su J, Li W, Chen Q, Shou P et al. Mesenchymal stem cells: a new strategy for immunosuppression and tissue repair. Cell Res 2010; 20: 510–518.
Shi Y, Su J, Roberts AI, Shou P, Rabson AB, Ren G . How mesenchymal stem cells interact with tissue immune responses. Trends Immunol 2012; 33: 136–143.
Wu X, Brewer G . The regulation of mRNA stability in mammalian cells: 2.0. Gene 2012; 500: 10–21.
Garneau NL, Wilusz J, Wilusz CJ . The highways and byways of mRNA decay. Nat Rev Mol Cell Biol 2007; 8: 113–126.
Lu JY, Schneider RJ . Tissue distribution of AU-rich mRNA-binding proteins involved in regulation of mRNA decay. J Biol Chem 2004; 279: 12974–12979.
Gratacos FM, Brewer G . The role of AUF1 in regulated mRNA decay. Wiley Interdiscip Rev RNA 2011; 1: 457–473.
Pautz A, Linker K, Altenhofer S, Heil S, Schmidt N, Art J et al. Similar regulation of human inducible nitric-oxide synthase expression by different isoforms of the RNA-binding protein AUF1. J Biol Chem 2009; 284: 2755–2766.
Ali G, Mohsin S, Khan M, Nasir GA, Shams S, Khan SN et al. Nitric oxide augments mesenchymal stem cell ability to repair liver fibrosis. J Transl Med 2012; 10: 75.
Dalal J, Gandy K, Domen J . Role of mesenchymal stem cell therapy in Crohn’s disease. Pediatr Res 2012; 71 (4 Pt 2): 445–451.
Le Blanc K, Frassoni F, Ball L, Locatelli F, Roelofs H, Lewis I et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet 2008; 371: 1579–1586.
Lee H, Park JB, Lee S, Baek S, Kim H, Kim SJ . Intra-osseous injection of donor mesenchymal stem cell (MSC) into the bone marrow in living donor kidney transplantation; a pilot study. J Transl Med 2013; 11: 96.
Ren G, Chen X, Dong F, Li W, Ren X, Zhang Y et al. Concise review: mesenchymal stem cells and translational medicine: emerging issues. Stem Cells Transl Med 2012; 1: 51–58.
Huang H, Kim HJ, Chang EJ, Lee ZH, Hwang SJ, Kim HM et al. IL-17 stimulates the proliferation and differentiation of human mesenchymal stem cells: implications for bone remodeling. Cell Death Differ 2009; 16: 1332–1343.
Xu G, Zhang Y, Zhang L, Roberts AI, Shi Y . C/EBPbeta mediates synergistic upregulation of gene expression by interferon-gamma and tumor necrosis factor-alpha in bone marrow-derived mesenchymal stem cells. Stem Cells 2009; 27: 942–948.
Wang HX, Liu M, Weng SY, Li JJ, Xie C, He HL et al. Immune mechanisms of Concanavalin A model of autoimmune hepatitis. World J Gastroenterol 2012; 18: 119–125.
Hartupee J, Liu C, Novotny M, Li X, Hamilton T . IL-17 enhances chemokine gene expression through mRNA stabilization. J Immunol 2007; 179: 4135–4141.
Faour WH, Mancini A, He QW, Di Battista JA . T-cell-derived interleukin-17 regulates the level and stability of cyclooxygenase-2 (COX-2) mRNA through restricted activation of the p38 mitogen-activated protein kinase cascade: role of distal sequences in the 3'-untranslated region of COX-2 mRNA. J Biol Chem 2003; 278: 26897–26907.
Lu JY, Sadri N, Schneider RJ . Endotoxic shock in AUF1 knockout mice mediated by failure to degrade proinflammatory cytokine mRNAs. Genes Dev 2006; 20: 3174–3184.
Pappu R, Ramirez-Carrozzi V, Sambandam A . The interleukin-17 cytokine family: critical players in host defence and inflammatory diseases. Immunology 2011; 134: 8–16.
Li W, Ren G, Huang Y, Su J, Han Y, Li J et al. Mesenchymal stem cells: a double-edged sword in regulating immune responses. Cell Death Differ 2012; 19: 1505–1513.
van den Berg WB, Miossec P . IL-17 as a future therapeutic target for rheumatoid arthritis. Nat Rev Rheumatol 2009; 5: 549–553.
Ogawa A, Andoh A, Araki Y, Bamba T, Fujiyama Y . Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin Immunol 2004; 110: 55–62.
O’Connor W Jr., Kamanaka M, Booth CJ, Town T, Nakae S, Iwakura Y et al. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol 2009; 10: 603–609.
Ren G, Su J, Zhang L, Zhao X, Ling W, L'Huillie A et al. Species variation in the mechanisms of mesenchymal stem cell-mediated immunosuppression. Stem Cells 2009; 27: 1954–1962.
Lohse AW, Dienes HP, Meyer zum Buschenfelde KH . Suppression of murine experimental autoimmune hepatitis by T-cell vaccination or immunosuppression. Hepatology 1998; 27: 1536–1543.
Kubo N, Narumi S, Kijima H, Mizukami H, Yagihashi S, Hakamada K et al. Efficacy of adipose tissue-derived mesenchymal stem cells for fulminant hepatitis in mice induced by concanavalin A. J Gastroenterol Hepatol 2011; 27: 165–172.
Higashimoto M, Sakai Y, Takamura M, Usui S, Nasti A, Yoshida K et al. Adipose tissue derived stromal stem cell therapy in murine ConA-derived hepatitis is dependent on myeloid-lineage and CD4(+) T-cell suppression. Eur J Immunol 2013; 43: 2956–2968.
Paschoud S, Dogar AM, Kuntz C, Grisoni-Neupert B, Richman L, Kuhn LC . Destabilization of interleukin-6 mRNA requires a putative RNA stem-loop structure, an AU-rich element, and the RNA-binding protein AUF1. Mol Cell Biol 2006; 26: 8228–8241.
Sirenko OI, Lofquist AK, DeMaria CT, Morris JS, Brewer G, Haskill JS . Adhesion-dependent regulation of an A+U-rich element-binding activity associated with AUF1. Mol Cell Biol 1997; 17: 3898–3906.
Devadas S, Das J, Liu C, Zhang L, Roberts AI, Pan Z et al. Granzyme B is critical for T cell receptor-induced cell death of type 2 helper T cells. Immunity 2006; 25: 237–247.
Wang R, Shi YF . A simplified protocol for apoptosis assay by DNA content analysis. Biotechniques 2002 (Suppl): 88–91.
Ysla RM, Wilson GM, Brewer G Chapter 3. Assays of adenylate uridylate-rich element-mediated mRNA decay in cells. Methods Enzymol 2008; 449: 47–71.
Livak KJ, Schmittgen TD . Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25: 402–408.
Schmittgen TD, Zakrajsek BA, Mills AG, Gorn V, Singer MJ, Reed MW . Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Anal Biochem 2000; 285: 194–204.
Lee WY, Salmi M, Kelly MM, Jalkanen S, Kubes P . Therapeutic advantage of anti-VAP-1 over anti-alpha4 integrin antibody in concanavalin a-induced hepatitis. Hepatology 2013; 58: 1413–1423.
Acknowledgements
This work was supported by grants from the Ministry of Science and Technology of China (2010CB945600 and 2011DFA30630), Scientific Innovation Project of the Chinese Academy of Science (XDA 01040107 and XDA 01040110), the Programs of National Natural Science of China (81330046, 81273316), the External Cooperation Program of BIC, Chinese Academy of Sciences (GJHZ201307), Shanghai Municipal Key Projects of Basic Research (12JC1409200), Shanghai Municipal Natural Science Foundation (12ZR1452600), the Knowledge Innovation Program of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (2012KIP202).
Author contributions
XH, YW, GB, and YFS designed research; XH, QY, LL, CX, CZ, and YH performed research and collected data; XH, YS, GB, YW, XC, ML, WC, KC, and QC analyzed data; GX, YZ, JZ, and RJS provided studied material; and XH, YW, YQ, GB, and YS wrote the paper.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by R De Maria
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Han, X., Yang, Q., Lin, L. et al. Interleukin-17 enhances immunosuppression by mesenchymal stem cells. Cell Death Differ 21, 1758–1768 (2014). https://doi.org/10.1038/cdd.2014.85
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2014.85
This article is cited by
-
Adipose-derived mesenchymal stem cells from obese mice prevent body weight gain and hyperglycemia
Stem Cell Research & Therapy (2021)
-
Human Wharton's jelly-derived mesenchymal stem cells alleviate concanavalin A-induced fulminant hepatitis by repressing NF-κB signaling and glycolysis
Stem Cell Research & Therapy (2021)
-
Inflammation-induced inhibition of chaperone-mediated autophagy maintains the immunosuppressive function of murine mesenchymal stromal cells
Cellular & Molecular Immunology (2021)
-
High-mobility group box 1 protein antagonizes the immunosuppressive capacity and therapeutic effect of mesenchymal stem cells in acute kidney injury
Journal of Translational Medicine (2020)
-
Interleukin-17 promotes nitric oxide-dependent expression of PD-L1 in mesenchymal stem cells
Cell & Bioscience (2020)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}