
Abstract
In liver ischemic preconditioning (IP), stimulation of adenosine A2a receptors (A2aR) prevents ischemia/reperfusion injury by promoting diacylglycerol-mediated activation of protein kinase C (PKC). By concerting diacylglycerol to phosphatidic acid, diacylglycerol kinases (DGKs) act as terminator of diacylglycerol signalling. This study investigates the role of DGK in the development of hepatocyte IP. DGK activity and cell viability were evaluated in isolated rat hepatocytes preconditioned by 10 min hypoxia followed by 10 min re-oxygenation or by the treatment with the A2aR agonist, CGS21680, and subsequently exposed to prolonged hypoxia. We observed that after IP or A2aR activation, a decrease in DGK activity was associated with the onset of hepatocyte tolerance to hypoxia. CGS21680-induced stimulation of A2aR specifically inhibited DGK isoform θ by activating RhoA–GTPase. Consistently, both siRNA-mediated downregulation of DGK θ and hepatocyte pretreatment with the DGK inhibitor R59949 induced cell tolerance to hypoxia. The pharmacological inhibition of DGK was associated with the diacylglycerol-dependent activation of PKC δ and ɛ and of their downstream target p38 MAPK. In conclusion, we unveil a novel signalling pathway contributing to the onset of hepatocyte preconditioning, which through RhoA–GTPase, couples A2aR to the downregulation of DGK. Such an inhibition is essential for the sustained accumulation of diacylglycerol required for triggering PKC-mediated survival signals.
Similar content being viewed by others


Main
Adenosine production and release by stressed cells has been shown to have an important function in modulating tissue damage and repair.1 Among these effects, adenosine is regarded as a key mediator in the development of cell tolerance to ischemia and reperfusion injury triggered by ischemic preconditioning (IP).2, 3, 4 IP has been proposed as a simple procedure to reduce ischemia/reperfusion injury after hepatic resection and orthotopic liver transplantation.5 However, the application of IP in hepatic surgery is limited by the high inter-individual variability of the responses,6 indicating the need for a better inside in the mechanisms involved.
The importance of adenosine in hepatic preconditioning is supported by the observation that targeted deletion of hepatic ecto-5′-nucleotidase blocks adenosine generation during hypoxia and abolishes liver protection by IP.7 The hepatocyte response to adenosine autocrine loop has been characterized to some extent showing that the stimulation of adenosine A2a receptors (A2aR) is coupled to the combined activation of phosphatidylinositol 3-kinase/Akt pathway and the novel protein kinases C (nPKC), namely, PKCδ and PKCɛ that on their turn lead to the stimulation of a network of protein kinases including p38 MAP kinase (p38 MAPK).8, 9 The membrane recruitment and activation of PKCδ and PKCɛ is fully dependent on their direct interaction with diacylglycerol generated by A2aR-induced activation of phospholipase C-γ, through a mechanism requiring both Src and PI3K activities.8, 9 These finding are consistent with central role of nPKCs in the development of the protective action of IP in several tissues.10, 11, 12
An increasing body of evidence suggests that diacylglycerol signalling is also regulated by diacylglycerol kinases (DGKs), which metabolize diacylglycerol to phosphatidic acid.13 DGKs are a family of enzymes encoded by 10 distinct genes, grouped in five structurally related classes.13 Accordingly, the genetic ablation of some DGK isoforms results in the deregulated activation of specific diacylglycerol-mediated signalling pathways.13 These observations suggest that the regulation of DGK activity may provide a strict control of the kinetic as well as of the extent of diacylglycerol signalling. However, direct evidence that extracellular ligands control intracellular diacylglycerol signalling by negative regulating specific DGK isoforms is still missing. Thus, we set to investigate whether negative regulation of a DGK activity might contribute to diacylglycerol-dependent activation of PKCδ/ɛ on adenosine-induced hepatic preconditioning.
Results
Hypoxic preconditioning and activation of adenosine receptors negatively regulate DGKθ in primary hepatocytes
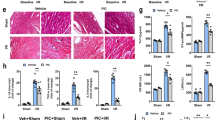
We first investigated whether preconditioning of rat hepatocytes affected the total DGK activity. Figure 1a shows that hepatocyte preconditioning, obtained by the 10 min exposure to hypoxia followed by 10 min re-oxygenation, reduced total DGK activity by about 50%. A similar effect was also induced by hepatocytes treatment with either adenosine (10 μM) or CGS21680 (1 μM), a specific A2aR agonist (Figure 1b and c). Concomitantly, CGS21680 increased at least two-folds the intracellular diacylglycerol concentration (Figure 1d). To identify the DGK isoforms putatively regulated by A2aR, DGK expression was investigated in primary hepatocytes by RT-PCR. DGKζ, θ and δ were the most expressed isoforms, whereas DGKα was expressed at low levels and DGKβ, γ, ɛ, η and ι were not expressed (Figure 2). We obtained no positive signals with several primer pairs for rat DGKκ (not shown). As the suppression of DGK-1, the Caenorhabditis elegans orthologue of DGKθ, positively regulates diacylglycerol in response to the serotonin receptor stimulation,14, 15 we set to investigate the regulation of DGKθ in response to A2aR activation. The DGKθ enzymatic activity measured in immunoprecipitates obtained using two distinct anti-DGKθ antibodies raised against different epitopes was significantly reduced on CGS21680 stimulation (Figure 3a and b), whereas DGKθ protein expression was not affected (Figure 3). DGKθ involvement was further confirmed by the use of a well-characterized anti-DGKθ neutralizing antibody16 that decreased by about 80% the recovered DGKθ activity in immunoprecipitates (Figure 3c). All together, these data indicate that the activation of A2aR provides a signal that negatively regulates DGKθ activity, likely contributing to the sustained accumulation of diacylglycerol.

Hypoxic preconditioning and adenosine A2a receptor (A2aR) stimulation decrease diacylglycerol kinase (DGK) activity and promote diacylglycerol accumulation in isolated hepatocytes. DGK activity was evaluated in the homogenates of primary rat hepatocytes undergoing preconditioning by exposure to transient hypoxia/reoxygenation (a), 15 min pre-incubation with adenosine (10 μM) (b) or A2aR stimulation with CGS21680 (1 μM) (c). A representative determination of phosphatidic acid (PA) determination is shown together with the quantification of five independent experiments. Statistical significance: *P<0.05; **P<0.01. (d) The quantification of the hepatocyte diacylglycerol (DAG) content after treatment with CGS21680 (1 μM) for 15 or 60 min. The data are mean±S.E.M. of five replicates. Statistical significance *P<0.01

Characterization of the different diacylglycerol kinase (DGK) isoforms expressed in parenchimal liver cells. RT-PCR was used to verify the expression of the different DGKs in the HTC cell line and primary rat hepatocytes. Rat brain was used as positive control. GAPDH was used as loading control. The values above each band represent the densitometric analysis normalized by the value of the brain

Modulation of DGKθ by the adenosineA2a receptor agonist CGS21680. (a, b) DGKθ was immunopurified with two different anti-DGKθ antibodies (Dgkθ-1 and Dgkθ-2) from extracts of hepatocytes treated with CGS21680 (1 μM) for 15 or 30 min. DGKθ enzymatic activity was measured as reported in ‘Materials and methods’ section. A representative experiment of phosphatidic acid (PA) determination is shown together with the quantification of three independent experiments. WB, represents western blot analysis of DGKθ protein in total hepatocyte lysates after 30 min incubation with CGS21680 (1 μM). The data are mean±S.E.M. of five replicates. Statistical significance *P<0.01. (c) DGKθ was immunopurified from hepatocytes and the enzymatic activity was measured in presence of control IgG or DGKθ neutralizing antibodies (DGKθ ab) as reported in ‘Materials and methods’ section. Phosphatidic acid (PA) determination is shown in the lower panel. The values are mean±S.E.M. from a representative experiment performed in triplicate. Statistical significance *P<0.01
RhoA-dependent inhibition of DGKθ mediates A2aR-induced protection of hepatocytes from hypoxia-induced cell death
Biochemical evidence both in neuroblastoma17 and in C. elegans motoneurons14 shows that the enzymatic activity of DGKθ and of its orthologue DGK-1 is strongly inhibited on direct interaction with the small GTPase RhoA.18 Thus, we set to investigate the interaction between active RhoA and DGKθ in PC12 cells,, a rat cell line expressing A2aR19, 20 transiently co-transfected with FLAG-tagged DGKθ and myc-tagged RhoA. Consistently with earlier published data,18 FLAG-DGKθ co-immunoprecipited with constituitvely active myc-RhoA-V14, but not with inactive myc-RhoA-N14 mutants from unstimulated cells (Figure 4). Moreover, FLAG-DGKθ specifically co-precipitated with myc-RhoA in anti-myc immunoprecipitates from CGS21680-stimulated cells, but not from unstimulated cells (Figure 4). In the whole, these data confirmed that indeed DGKθ is recruited in a complex with active RhoA. We then moved to investigate whether RhoA regulated DGKθ activity on A2aR activation in primary rat hepatocytes. To this aim, we first verified whether RhoA was regulated by activation of A2aR. Indeed, on 15 min treatment, CGS21680 increased by 50% RhoA–GTP complex, as measured by pull-down assay with immobilized rothekin-Rho binding domain (Figure 5a and b). Such A2aR-induced activation of RhoA was insensitive to the addition of pertussis toxin (data not shown). Next, to verify whether DGKθ inhibition downstream from A2aR was mediated by RhoA–GTP, primary hepatocytes were treated with the cell permeable analogue of Clostridrium botulinum-derived C3 transferase (1 μM), a highly specific RhoA inhibitor stabilizing RhoA in the inactive GDP-bound form. Inhibition of RhoA entirely reverted CGS21680-induced inhibition of DGKθ (Figure 5c), showing that RhoA–GTP mediated the inhibition of DGKθ triggered on A2aR activation. Moreover, the inhibition of RhoA by C3 tranferase abolished the protection against hypoxic injury in hepatocyte treated with CGS21680 (Figure 5d). In the whole, these data showed that the cytoprotective signalling downstream from A2aR requires the activation of RhoA–GTPase followed by the inhibition of DGKθ on binding of RhoA to its catalytic site.

The adenosineA2a receptor agonist CGS21680 promotes DGKθ association with RhoA/C GTPase. DGKθ/RhoA association was evaluated in PC12 cells transfected with FLAG-DGKθ and either with wild-type myc-RhoA (wt) or constitutive active myc-RhoA-V14 (active) or inactive myc-RhoA-N19 (inactive) mutants. After trasfection, cells were serum starved for 24 h and treated with CGS21680 for 30 min. Cells were then lysed and transfection verified by western blotting and protein association was assessed after immunoprecipitation with anti-myc-specific antibodies using western blotting with anti-FLAG-specific antibodies

CGS21680 promotes the activation of RhoA/C GTPase, which is required for the inhibition of DGKθ and the protection of hepatocytes against hypoxic damage. (a) Primary rat hepatocytes were stimulated with CGS21680 (1 μM) for 30 min and active RhoA/C was purified by pull-down GST-Rhotekin-RBD and visualized by western blotting with RhoA/C antibodies (middle panel). The GST and GST-Rhotekin-RBD loading was verified by western blotting with GST antibodies (lower panel). The RhoA/C content in total cell lysate is also shown (upper panel). (b) Densitometric quantification of RhoA/C activity in four independent experiments. Statistical significance: *P<0.05. (c) Effect of the RhoA inhibitor Clostridrium botulinum-derived C3 transferase (C3) (1 μM), on DGKθ activity in primary rat hepatocytes. The cells were pretreated with C3 for 120 min and then stimulated with CGS21680 (1 μM) for 30 min. The data are mean±S.E.M. of five replicates. Statistical significance *P<0.01 versus control and C3-treated cells. (d) Viability of primary rat hepatocytes incubated for 90 min under normoxic (95% air–5% CO2) or hypoxic (95% N2–5% CO2) condition in the presence or in absence of the adenosine A2a receptor agonist CGS21680 and with or without the RhoA/C inhibitor C3 transferase (C3). CGS21680 (1 μM) was added 15 min before hypoxic exposure, while hepatocytes were pretreated with C3 for 2 h. The results are means of five different experiments +S.D. Statistical significance: *P<0.001 versus control, hypoxia+CGS21680 or C3
To provide further support to the claim that RhoA-mediated inhibition of DGKθ contributed to A2aR-dependent signalling in preconditioning, we downregulated the expression of DGKθ in HTC cells by specific siRNAs. HTC cells are rat hepatocyte-derived cell lines that, similar to primary hepatocytes, undergoe an A2aR-mediated preconditioning.21 Downregulation of DGKθ expression was obtained by transient transfection of two different specific siRNAs or a control siRNA and verified by western blot 48 h after transfection. As shown in Figure 6a, both anti-DGKθ siRNAs specifically downregulated DGKθ and not the other major hepatocytes DGK isoforms (DGKα, δ and ζ). On 60 min exposure to hypoxia, un-transfected cells and cells transfected with control siRNA underwent a significant loss of cell viability (Figure 6b). Conversely, HTC cells transfected with either of the DGKθ-specific siRNAs were fully protected from hypoxic damage, as were the cells pretreated with CGS21680 (Figure 6b). Thus, these data show that specific downregulation of DGKθ isoform in HTC cells provides a sufficient signalling to induce tolerance to hypoxic damage.

siRNAs DGKθ downregulation in HTC cells protects against cell killing by hypoxia as the adenosine A2a receptor stimulation. (b) HTC cells were trasfected with control siRNA or two different DGKθ siRNAs (siRNA1 and siRNA2) and submitted to 60 min incubation under either normoxic or hypoxic conditions. When indicated, HTC were pretreated 15 min with A2aR agonist CGS21680 (1 μM). Results are mean of three different experiments. Statistical significance: *P<0.05 versus hypoxia. (a) The capacity of the different siRNA to specifically block DGKθ expression was assessed by western blot (WB) using antibodies against different DGK isoenzymes. Equal amounts of proteins were assessed by using actin as loading control. One experiment representative of three
Pharmacological inhibition of DGK is sufficient to activate signals leading to hepatocyte resistance to hypoxic damage
To identify the key role played by the negative regulation of DGKθ in mediating adenosine-induced preconditioning, led us investigate whether direct modulation of diacylglycerol turnover through pharmacological inhibition of DGK could induce hepatocytes resistance to hypoxic damage. The DGK inhibitor R59949 is known to preferentially inhibit DGKα, β, γ and δ isoforms,22 whereas DGKζ, one of the major DGK isoforms expressed in hepatocytes, is resistant to R5994922 and no information is available on the R59949 sensitivity to other DGK isoforms. Herein, we show that R59949 at 10 μM, but not at lower concentrations, effectively inhibited hepatocyte DGKθ activity as assayed ‘in vitro’ in anti-DGKθ immunoprecipitates (Figure 7a). Accordingly, in the same cells, 10 μM R59949 induced a sustained increase in intracellular diacylglycerol concentration (Figure 7b). Pretreatment with R59949 also protected primary hepatocytes from hypoxia-induced cell death (Figure 7c), whereas it did not affect viability of control cells. Moreover, in primary hepatocytes as well as in HTC cells, R59949 increased PKCδ and PKCɛ activities in both membrane and cytosolic fractions (Figure 8a). The extent of both diacylglycerol accumulation (Figure 7b) and enzymatic activation of PKCδ and PKCɛ was highly similar to that induced by CGS21680-mediated activation of A2aR (Figure 8a). Moreover, neither pharmacological inhibition of DGK nor A2aR stimulation promoted the enzymatic activities of PKCα, β1 and β2, which are regulated by both calcium and diacylglycerol (Figure 8a). We showed earlier that PKCδ/ɛ-dependent phosphorylation of p38 MAPK is required for A2aR-induced protection from hypoxic damage.7 Consistently, R59949-mediated inhibition of DGK stimulated the dual phosphorylation Thr180/Tyr182 of p38 MAPK (Figure 8b). The effect induced by DGK inhibition was comparable to that obtained by the activation of A2aR and was prevented by the PKC inhibitor chelerythrine (50 μM) (Figure 8b). Moreover, the inhibition of either PKC or p38 MAPK kinase with, respectively, chelerythrine or SB203580 (5 μM) abolished R59949-induced protective effects against hypoxic hepatocyte death (Figure 7c). All together, these findings indicate that the pharmacological inhibition of DGK provides sufficient diacylglycerol accumulation to trigger PKC-mediated signals leading to resistance to hypoxic damage.

The inhibition of DGK promotes diacylglycerol accumulation and protection against hypoxic injury in primary rat hepatocytes. (a) DGKθ activity was evaluated after immunopurification with specific antibodies from extracts of hepatocytes incubation with increasing concentration of the DGK inhibitor R59949. The values are mean±S.E.M. from a representative experiment performed in quadruplicate. (b) Quantification of diacylglycerol content of primary rat hepatocytes after treatment with R59949 (10 μM) for 15 or 60 min. The data are mean±S.E.M. of four replicates. Statistical significance: *P< 0.05. (c) Viability of primary rat hepatocytes incubated 90 min under normoxic or hypoxic conditions in presence or absence of R59949 (10 μM). The effects of inhibiting PKC with cheletrytrine (50 μM) or p38MAPK with SB203580 (2 μM) are also shown. The results are means of four different experiments +S.D. Statistical significance: **P<0.003 versus control, hypoxia+R59949, SB203580, R59949 or chelerythrine

DGK inhibition induces the activation of PKCδ/ɛ and p38 MAPK in both primary hepatocytes and HTC cells. (a) The activity of different PKC isoforms was measured in the immunoprecipitated fractions obtained from primary hepatocytes or HTC cells incubated 30 min with R59940 (10 μM) or CGS21680 (1 μM). The results are expressed as percentage of the values in the controls and are means of three different experiments +S.D. Statistical significance: *P< 0.05 versus controls. (b) The activation of p38 MAPK was estimated by western blotting using antibodies against dual (Thr180/Tyr182) p38 MAPK after 45 min incubation with R59940 or CGS21680. In some experiments, cells were pretreated with the PKC inhibitor cheletrytrine (50 μM). The relative intensity of phosphorylated and non-phosphorylated bands was measured by videodensitometry and the results were expressed as ratios after normalization at one of the controls. A representative experiment is shown together with the quantification of five independent experiments. Statistical significance: *P<0.005 versus control or cells treated with R59940+cheletrytrine or CGS21680+cheletrytrine
Discussion
It is increasingly clear that the development of IP in many tissues, including the liver, requires the activation of a complex network of signals comprising cell-surface receptors, redox signals and diverse arrays of protein kinases such as reperfusion injury salvage kinases that rapidly re-modulate several cell functions to better cope with energy loss, oxidative stress and ion unbalances associated with hypoxia and reperfusion.3, 4, 10, 23, 24 Among these protein kinases, PKCδ and PKCɛ have a key function by coupling adenosine receptors to downstream stress-activated protein kinases Erk1/2 and p38 MAPKs.3, 4, 24 Moreover, translocation of PKCɛ to the mitochondria has been implicated in preventing mitochondrial permeability transition on myocardial reperfusion.25 PKCδ and PKCɛ membrane recruitment and activation are fully dependent on their direct interaction with diacylglycerol, generated by adenosine-induced activation of phospholipase C-γ and diacylglycerol analogues fully mimic the activation of the signals that onset IP.8, 9, 26 However, the accumulation of cellular diacylglycerol is dependent also on the rate of its rapid metabolism to phosphatidic acid by DGKs.13 Recent data indicate that the genetic deletion of specific DGK isoforms results in a sustained diacylglycerol signalling in different cell systems and tissues.13 Moreover, the role of DGKs as the terminator of diacylglycerol-mediated signals has been recently underscored by the finding that several DGK isoforms are recruited to the activated muscarinic receptors, in which they rapidly metabolize diacylglycerol similarly to the action of phosphodiesterase towards cAMP.27 However, so far, no direct biochemical evidence is available for a receptor-mediated negative regulation of any DGK isoform. In this study, we show that in hepatocytes both hypoxic preconditioning and A2aR stimulation lead to a strong decrease in total DGK activity that contributes to diacylglycerol accumulation. In particular, we show that enzymatic activity of DGKθ, that is, along DGKζ the most abundant isoform in hepatocytes, is strongly inhibited on A2aR activation. This provides the first demonstration of a negative regulation of a DGK isoform by extracellular ligands. However, we cannot rule out that other DGK isoforms beside DGKθ may be inhibited on hepatocyte A2aR activation. Nevertheless, the demonstration that specific genetic downregulation of DGKθ expression is sufficient to protect HTC hepatoma cells from hypoxic damage, indicates that the negative regulation of the DGKθ isoform may be sufficient to trigger the onset of cytoprotective signals on A2aR activation by hypoxic preconditioning.
The localization and function of DGKθ have been reported earlier to be regulated by several G-protein coupled receptors, although the molecular mechanisms underlying such a regulation have not been fully investigated. For instance, in vascular smooth muscle cells purinergic receptors promote PKC-mediated recruitment of DGKθ to the plasma membrane, whereas norepinephrine stimulates DGKθ activity at lipid rafts.28, 29, 30, 31 Herein, we show that on ligand stimulation of A2aR, RhoA is activated, binds and inhibits DGKθ . Such an Rho-A signalling is required for A2aR-induced inhibition of DGKθ as well as for the development of cytoprotection. These findings are consistent with the evidence that both DGKθ and its C. elegans orthologue, DGK-1 are inhibited on the direct binding of active RhoA to their catalytic domain.18, 32 Moreover, in motoneurons of C. elegans, the activation of the RhoA orthologue, Rho-1, leads to diacylglcyerol accumulation, which then allows the recruitment and activation of PKC-1 and UNC-13 proteins.33 Thus, our finding that in hepatocytes RhoA-mediated inhibition of DGKθ triggers cytoprotective signalling downstream from A2a receptors, unveils the general relevance of a novel Rho-mediated signalling pathway linking G-protein coupled receptors to the negative regulation of DGKθ activity. Such a novel pathway is conserved from C. elegans to different mammalian tissues and may enable G-protein coupled receptors to regulate the extent and the kinetics of diacylglycerol-mediated signalling, by fine-tuning the enzymatic activity of DGKθ. This is also the first evidence for a direct activation of RhoA by A2aR, albeit in hepatic stellate cells A2aR has been reported recently to mediate RhoA inhibition through the cAMP-mediated activation of protein kinase A.34 It is known that G-protein coupled receptors activate RhoA–GTPases, by coupling the pertussis toxin insensitive Gα12/13 or Gαq/11 proteins to, respectively, p115 or Trio RhoGEFs.35 Moreover, previous evidence indicates that A2aR couples to Gα12/13 in endothelial cells.36 On the other hand, A2aR-induced activation of hepatocyte RhoA is pertussis toxin insensitive, suggesting that it may be mediated through either Gα12/13 or Gαq/1137 and ruling out the involvement of inhibitory G (Gαi)-proteins, which instead transduce A2aR-induced Src-mediated signalling to PI3-kinase and PLC-γ.9 These findings indicate that A2aRs by interacting with, respectively, Gαi or Gα12/13(q/11) GTPases promote both the PLC-γ-mediated diacylglycerol synthesis and, at the same time, inhibit diacylglycerol catabolism by DGK (Figure 9). Thus, the accumulation of diacylglycerol required for preconditioning-induced PKCδ/ɛ activation results from the fine-tuning of the enzymes responsible for both diacylglycerol generation and catabolism. Such an interpretation is consistent with a growing body of evidence suggesting that the temporal and spatial regulation of intracellular second messengers depends on the simultaneous reciprocal regulation of both activating and de-activating enzymes, such as adenylate cyclase and phosphodiesterase for cAMP, and PI3-kinase and PTEN for PI(3,4,5)P3.38, 39 Moreover, these observations underscore the relevance of negative regulators for the transduction of the complex signals required for the onset of the cytoprotective machinery in IP. In this latter respect, a decline in the activity of PTEN is evident in parallel with PI3K activation during brain and myocardium preconditioning.40, 41 By unveiling that the RhoA-mediated-negative regulation of DGKθ is essential for A2aR-dependent hepatocyte preconditioning, these results suggest that the pharmacological inhibition of DGK may likely trigger cytoprotection. Indeed, the treatment of primary hepatocytes with the DGK inhibitor R59949 is sufficient to induce adequate diacylglycerol accumulation to activate diacylglycerol-dependent nPKCs, fully protecting hepatocytes from hypoxic damage. On pharmacological inhibition of DGK, adequate diacylglycerol levels likely build from the unstimulated basal phospholipids turnover, even in absence of a receptor-mediated activation of PLC-γ. At the concentrations used in this study, R59949 inhibits several DGK isoforms including DGKθ, DGKδ and DGKα, but does not affect DGKζ, which is along with DGKθ the most abundant DGK isoform expressed in hepatocytes. Thus, the inhibition of DGKθ may be largely responsible for R59949-induced protection, although we cannot rule out the involvement of other DGKs, such as DGKδ and DGKα.

Proposed role of DGKθ inhibition and phospholypase C activation in the recruitment of protein PKC-dependent survival pathways responsible for inducing the resistance to hypoxia of preconditioned hepatocytes. During hepatocytes preconditioning, adenosine A2 receptor (A2aR) stimulation induces RhoA/C activation and an RhoA/C-dependent inhibition of DGKθ activity. The consequent decrease in diacylglycerol (DAG) metabolism to phosphatidic acid (PA) allows a stable increase in DAG intracellular level that maintains the activation of the DAG-dependent cytoprotective signals
In conclusion, the data presented characterize a novel signalling pathway in the mechanisms by which adenosine promotes hepatic preconditioning involving DGKθ downregulation. These findings indicate DGK modulation as a potential target of new pharmacological strategies aimed to induce tolerance to ischemia/reperfusion injury in organs such as the liver, the myocardium and the kidney, which relay on nPCK activity for the development of the cytoprotective effects of preconditioning.
Materials and Methods
Hepatocyte preparation and treatments
Primary hepatocytes were isolated by collagenase (Sigma, Milan, Italy) liver perfusion of fed male Wistar rats (180–250 g weight) (Harlan, San Pietro al Natisone, Udine, Italy).8 The use and care of the animals were approved by the Italian Ministry of Health. Hepatocytes were suspended in Krebs-Henseleit-Hepes medium (final cell density of 106/ml) and preconditioned by exposure to 10 min of hypoxia (95% N2–5% CO2) followed by 10 min re-oxygenation (95% air–5% CO2) or by 15 min treatment with CGS21680 (Sigma) as described earlier.8 Hypoxic damage was evaluated by exposing hepatocytes to 90 min hypoxia immediately after preconditioning procedures. Control cells were maintained under normoxic atmosphere (95% air–5% CO2). The inhibitors SB203580, chelerythrine (Sigma) and the cell-permeable C. boltulinum-derived C3 transferase (Citoskeleton, Denver, CO, USA) were added, respectively, 5, 15 and 120 min before preconditioning. R59949 (Sigma) was added 15 min before hypoxic exposure.
Cell cultures and treatments
HTC rat hepatoma cell line was obtained from the European Collection of Cell Cultures and cultured in DMEM-HAM F12 medium containing 10% foetal calf serum and 1% penicillin/streptomycin, 1% glutamine. The preconditioning procedures and treatments were the same as those used for primary hepatocytes. PC12 cell line was kind gift from Prof. C Isidoro (University of East Piedmont, Novara, Italy). Cells were cultured on collagen-coated dishes in RPMI medium containing 15% foetal calf serum and 1% penicillin/streptomicin, 1% glutamine. PC12 cells were transfected using lipofectamine (Invitrogen, San Giuliano, Milan, Italy) with either FLAG-tagged DGKθ and the myc-tagged forms of wild-type RhoA or constitutively active mutant V14-RhoA or inactive mutant RhoA-N1918, 32 using. FLAG-DGKθ/myc-RhoA association was evaluated in 106 transfected PC12 cells serum starved for 24 h and treated with CGS21680 for 30 min. After treatments, the cells were lysed and immunoprecipitated with anti-myc-specific antibodies (Santa Crutz Biotechnologies, Santa Crutz, CA, USA) and protein G Sepharose beads. The immunoprecipitates were analysed with anti-FLAG-specific antibodies (Sigma).
Assay of total and isoform-specific DGK activity
For total DGK assay, 106 hepatocytes were homogenated in 1 ml of a buffer containing 25 mM Hepes pH 8, 10% glycerol, 150 mM NaCl, 5 mM EDTA, 2 mM EGTA, 1 mM ZnCl2, 50 mM ammonium molibdate, 10 mM NaF, 1 mM sodium orthovanadate and Sigma protease inhibitor cocktail and spinned at 500 × g for 15 min. The enzyme activity was assayed in 25 μl of the homogenate by measuring phosphatidic acid formation during 5 min incubation at 30°C in the presence of 1 mg/ml diolein, 5 mM ATP, 3 μCi/ml [γ32P]-ATP (GE Healthcare, Cologno Monzese, Milan, Italy), 10 mM MgCl2, 1 mM ZnCl2, 1 mM EGTA in 25 mM Hepes pH 8, final reaction volume 50 μl. Reaction was stopped with 200 μl of 1 M HCl and lipids were extracted with 200 μl chloroform/methanol (1 : 1). Phosphatidic acid was separated by thin layer chromatography (TLC) in chloroform/methanol/water/25% ammonium hydroxide (60 : 47 : 11 : 4) using TLC plates coated earlier with potassium oxalate 1.3%, EDTA 5 mM/methanol (3 : 2) and desiccated. [γ32P]-phosphatidic acid formation was identified by comigration with non-radioactive phosphatidic acid formation standards stained by exposure to iodine vapours. Radioactive signals were detected and quantified by GS-250 Molecular Imager and Phosphor Analyst Software (Bio-Rad, Milan, Italy).
The specific DGKθ activity was evaluated by the same method after DGKθ immunoprecipitation with protein A Sepharose and two anti-DGKθ-specific antibodies recognizing different DGKθ epitopes (Santa Cruz Biotechnologies) according to Houssa et al.18 DGKθ western blotting was performed using 25 μg of hepatocyte proteins and a specific anti-DGKθ antibody (BD Biosciences, San Jose, CA, USA) . To verify the effects of neutralizing antibodies on DGK activity, control IgG (Santa Cruz Biotechnologies, Milan, Italy) and anti-DGKθ inhibitory IgG (50 μg/ml; BD Biosciences) were added to the samples 30 min before the assay. The results were acquired and quantified with GS-250 Molecular Imager and Phosphor Analyst Software (Bio-Rad).
RhoA/C–GTP pull-down assay
RhoA/C–GTP pull-down assays were performed according to Ren et al.42 Briefly, 106 hepatocytes were washed in ice-cold PBS and lysed with 1 ml GST-fish buffer (50 mM Tris–HCl, pH 7.2, 1% Triton X-100, 5% sodium deoxicolate, 0.1% SDS, 500 mM NaCl, 10 mM MgCl2, supplemented with fresh 1 mM Na3VO4, protease inhibitors, and 1 mM dithiothreitol); 5% of each sample was directly denatured in Laemmli buffer for whole cell lysate proteins analysis. The remaining clarified lysates were incubated for 60 min with purified GST6-Rhotekin-RBD at 4°C, pre-coupled to Glutathione–Sepharose beads (GE Healthcare, Cologno Monzese, Milan, Italy). After four washes with 1 ml of Tris buffer (1% Triton X-100, 150 mM NaCl, 10 mM MgCl2, protease inhibitors) samples were re-suspended in Laemmli buffer, heat denatured and separated by SDS-PAGE in a 12% polyacrylamide gel. RhoA/C was visualized by western blotting and quantified using Versadoc with Quantity One software (Bio-Rad). RhoA/C activation was calculated as GTP–Rho ratio to Rho in total cell lysate.
RT-PCR determination of rat DGK isoforms
RNA was extracted from 106 hepatocytes or HTC cells using charge switch total RNA cell kit (Invitrogen, San Giuliano, MI, Italy). Reference RNA was extracted from 200 μg rat brain using Tri reagent (Applied Biosystems, Austin, TX, USA). RNA aliquots (200 ng) were retrotranscribed using the RT kit (Applied Biosystems, Austin, TX, USA). One-tenth of the cDNA obtained was amplified using Go-Taq Master Mix (Promega, Milan, Italy). The primers used for the DNA amplification of specific DGK isoforms were
DGKα LEFT PRIMER GAGAAGGAAGCGTTGACAGC RIGHT PRIMER TGAGGCTGATCGCAGAGATA DGKβ LEFT PRIMER CCCCTCAGAATTTTCCCAAC RIGHT PRIMER TGCAAGCACACCATTACCAT DGKγ LEFT PRIMER CACACTCGGCTGAAAGATGA RIGHT PRIMER TTGAGGCTTCCACCTTCATT DGKδ LEFT PRIMER AGTGGGCAAGGCTTATGAGA RIGHT PRIMER GGGTTGGACAAAGAGGATGA DGKɛ LEFT PRIMER GTAATCTGCAGGCAGCAGTG RIGHT PRIMER TCATGCACTCATCGTGGACT DGKζ LEFT PRIMER GGCTTCCAGCAGAAGTTCAC RIGHT PRIMER AGCATGAAGCAGGACACCTT DGKη LEFT PRIMER TTCAGAAGGCTGATGCTGTG RIGHT PRIMER CACATTAAACTGGGCCACCT DGKθ LEFT PRIMER TGTCCCATGAGAAGTGCCTA RIGHT PRIMER GAACCAAAGCAGTGGGCTAC DGKι LEFT PRIMER AACTTTCCGGAAGCAGGTCT RIGHT PRIMER CCACAGGAAGACCAAGAGGA GAPDH LEFT PRIMER ATGACTCTACCCACGGCAAG RIGHT PRIMER GATCTCGCTCCTGGAAGATG
The PCR conditions were the following: 95°C 2′, 30 cycles 95°C 45′′, 60°C 45′′, 72°C 45′′, 72°C 5′. The amplified products were resolved by 2% agarose gel electrophoresis, stained with ethidium bromide and documented with GelDoc system (Bio-Rad). Amplified products match the expected molecular weight.
RNA interference experiments
siRNAs against rat DGKθ were purcased by Sigma-Genosis (Milan, Italy). Sequences were as follows:
siRNA1 sense GUGUACAUUUGGACGUCUAdTdT siRNA1antisense UAGACGUCCAAAUGUACACdTdT siRNA2 sense GUCUUAUCUUCAUUAACAUdTdT siRNA2antisense AUGUUAAUGAAGAUAAGACdTdT.
An siRNA with no matching sequences in the rat genome (Ambion, Austin, TX, USA) was used as negative control. HTC cells were transfected using Lipofectamine 2000 (Invitrogen) according to manufacturer recommendations. Transfection efficiency was analysed after 72 h using BLOCK-iT Fluorescent Oligo (Invitrogen) procedure according to the producer instruction The transfection efficiency of HTC cells with the different siRNAs was more than 75%.
Diacylglycerol quantification
Hepatocyte content of diacylglycerol was evaluated in the lipids extracted from 2 × 106 hepatocytes as described by Bligh and Dyer.43 Briefly, hepatocyte content of diacylglycerol was evaluated by extracting 2 × 106 hepatocyte pellets with 0.750 ml CHCl3/CH3OH (1 : 2 v/v) and the phases were separated by the addition of 0.25 ml CHCl3 and 0.25 ml H2O followed by centrifugation at 1000 × g for 5 min. The lower phase was dried, re-dissolved in 50 μl CHCl3 and spotted on a silica 60 TLC plate (Sigma). Diacylglycerol was eluted with diethylether/heptane/acetic acid (75 : 25 : 1 v : v : v) mixture, the plates were dried and stained with 0.003% Coomassie brilliant blue in 30% CH3OH and 100 mM NaCl for 30 min. The plates were de-stained for 5 min in dyefree solution and the bands were acquired with Versadoc system. The band density was calculated with Quantity One software (Bio-Rad). The amount of diacylglycerol was calculated using a standard curve of dioleyl-diacylglycerol (Sigma).
Assay of PKCs activities
PKC isoforms were immunoprecipitated with rabbit polyclonal α, β, β1, δ and ɛ anti-antibodies (Santa Cruz Biotechnology) and protein A Sepharose. After three washes in a buffer containing 10 mmol/l Tris–HCl, 150 mmol/l NaCl, 10 mmol/l MgCl2 and 0.5 mmol/l dithiotreitol, PKC activity was assayed by using 10 g of histone H1 as substrate and a reaction mixture containing 0.1 mmol/l ATP, 2 μCi/sample[32P]ATP (GE Healthcare), 1 g of phophatidylserine and 0.4 μg of diacylglycerol as reported earlier.8
Analysis of p38 MAPK phosphorylation state
The phosphorylation state of p38 MAPK was assessed by using dual (Thr180/Tyr182) phosphorylated p38 MAPK and total p38 MAPK antibodies (PhosphoPlus p38 MAPK antibody kit, New England BioLabs, Beverly, MA, USA).
Determination of cell viability
Cell viability was estimated by microscope counting the hepatocyte excluding Trypan blue and by the determination of nuclear fluorescence staining with propidium iodide.8
Data analysis and statistical calculations
The data were expressed as mean±S.D or ±S.E.M. Statistical analysis was performed by Instat 3 statistical software (GraphPad Software Inc., San Diego, CA, USA) using one-way ANOVA test with Bonferroni's correction for multiple comparisons when more than two groups were analysed. Distribution normality of all groups was preliminary verified by Kolmogorov and Smirnov test. Significance was taken at 5% level.
Abbreviations
- A2aR:
-
adenosine A2a receptors
- PKC:
-
protein kinase C
- DGK:
-
diacylglycerol kinase
- PA:
-
phosphatidic acid
- DAG:
-
diacylglycerol
- IP:
-
ischemic preconditioning
- PI3K:
-
phosphatidylinositol 3-kinase
- PLC:
-
phospholipase C
References
Fredholm BB . Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ 2007; 14: 1315–1323.
Peralta C, Hotter G, Closa D, Prats N, Xaus C, Gelpi E et al. The protective role of adenosine in inducing nitric oxide synthesis in rat liver ischemia preconditioning is mediated by the activation of adenosine A2 receptors. Hepatology 1999; 29: 126–132.
Carini R, Albano E . Recent insights on the mechanisms of liver preconditioning. Gastroenterology 2003; 125: 1480–1491.
Cohen MV, Downey JM . Adenosine: trigger and mediator of cardioprotection. Basic Res Cardiol 2008; 103: 203–215.
Banga NR, Homer-Vanniasinkam S, Graham A, Al-Mukhtar A, White SA, Prasad KR . Ischaemic preconditioning in transplantation and major resection of the liver. Br J Surg 2005; 92: 528–538.
DeOliveira ML, Graf R, Clavien PA . Ischemic preconditioning: promises from the laboratory to patients, sustained or disillusioned? Am J Transplant 2008; 8: 489–491.
Hart ML, Much C, Gorzolla IC, Schittenhelm J, Kloor D, Stahl GL et al. Extracellular adenosine production by ecto-5′- nucleotidase protects during murine hepatic ischemic preconditioning. Gastroenterology 2008; 135: 1739–1750.
Carini R, De Cesaris MG, Splendore R, Vay D, Domenicotti C, Nitti MP et al. Signal pathway involved in the development of hypoxic preconditioning in rat hepatocytes. Hepatology 2001; 33: 131–139.
Carini R, Grazia De Cesaris M, Splendore R, Baldanzi G, Nitti MP, Alchera E et al. Role of phosphatidylinositol 3-kinase in the development of hepatocyte preconditioning. Gastroenterology 2004; 127: 914–923.
Hausenloy DJ, Yellon DM . Reperfusion injury salvage kinase signalling: taking a RISK for cardioprotection. Heart Fail Rev 2007; 12: 217–234.
Bright R, Mochly-Rosen D . The role of protein kinase C in cerebral ischemic and reperfusion injury. Stroke 2005; 36: 2781–2790.
Um JW, Matthews JB, Song JC, Mun EC . Role of protein kinase C in intestinal ischemic preconditioning. J Surg Res 2005; 124: 289–296.
Mérida I, Avila-Flores A, Merino E . Diacylglycerol kinases: at the hub of cell signalling. Biochem J 2008; 409: 1–18.
McMullan R, Hiley E, Morrison P, Nurrish SJ . Rho is a presynaptic activator of neurotransmitter release at pre-existing synapses in C. elegans. Genes Dev 2006; 20: 65–76.
Nurrish S, Ségalat L, Kaplan JM . Serotonin inhibition of synaptic transmission: Galpha(0) decreases the abundance of UNC-13 at release sites. Neuron 1999; 24: 231–242.
Tabellini G, Billi AM, Falà F, Cappellini A, Evagelisti C, Manzoli L et al. Nuclear diacylglycerol kinase-theta is activated in response to nerve growth factor stimulation of PC12 cells. Cell Signal 2004; 16: 1263–1271.
Pacini L, Limatola C, Frati L, Luly P, Spinedi A . Muscarinic stimulation of SK-N-BE(2) human neuroblastoma cells elicits phosphoinositide and phosphatidylcholine hydrolysis: relationship to diacylglycerol and phosphatidic acid accumulation. Biochem J 1993; 289: 269–275.
Houssa B, de Widt J, Kranenburg O, Moolenaar WH, van Blitterswijk WJ . Diacylglycerol kinase theta binds to and is negatively regulated by active RhoA. J Biol Chem 1999; 274: 6820–6822.
Mori Y, Higuchi M, Masuyama N, Gotoh Y . Adenosine A2A receptor facilitates calcium-dependent protein secretion through the activation of protein kinase A and phosphatidylinositol-3 kinase in PC12 cells. Cell Struct Funct 2004; 29: 101–110.
Huang NK, Lin YW, Huang CL, Messing RO, Chern Y . Activation of protein kinase A and atypical protein kinase C by A(2A) adenosine receptors antagonizes apoptosis due to serum deprivation in PC12 cells. J Biol Chem 2001; 276: 13838–13846.
Alchera E, Tacchini L, Imarisio C, Dal Ponte C, De Ponti C, Gammella E et al. Adenosine-dependent activation of hypoxia-inducible factor-1 induces late preconditioning in liver cells. Hepatology 2008; 48: 230–239.
Jiang Y, Sakane F, Kanoh H, Walsh JP . Selectivity of the diacylglycerol kinase inhibitor 3--1piperidinyl)ethyl]-2,3-dihydro-2-thioxo4(1 H) quinazolinone (R59949) among diacylglycerol kinase subtypes. Biochem Pharmacol 2000; 59: 763–767.
Murphy E, Steenbergen C . Mechanisms underlying acute protection from cardiac ischemiareperfusion injury. Physiol Rev 2008; 88: 581–609.
Hausenloy DJ, Yellon DM . Preconditioning and postconditioning: united at reperfusion. Pharmacol Ther 2007; 116: 173–191.
Jabůrek M, Costa AD, Burton JR, Costa CL, Garlid KD . Mitochondrial PKC epsilon and mitochondrial ATPsensitive K+ channel copurify and coreconstitute to form a functioning signaling module in proteoliposomes. Circ Res 2006; 99: 878–883.
Galiñanes M, Goss MW, McGill CJ, Hearse DJ, Brooks G . Diacylglycerol-induced protection against injury during ischemia and reperfusion in the rat heart: comparative studies with ischemic preconditioning. Int J Cardiol 1998; 65: 129–138.
Nelson CD, Perry SJ, Regier DS, Prescott SM, Topham MK, Lefkowitz RJ . Targeting of diacylglycerol degradation to M1 muscarinic receptors by beta-arrestins. Science 2007; 315: 663–666.
van Baal J, de Widt J, Divecha N, van Blitterswijk WJ . Translocation of diacylglycerol kinase theta from cytosol to plasma membrane in response to activation of G protein-coupled receptors and protein kinase C. J Biol Chem 2005; 280: 9870–9878.
Clarke CJ, Ohanian V, Ohanian J . Norepinephrine and endothelin activate diacylglycerol kinases in caveolae/rafts of rat mesenteric arteries: agonist-specific role of PI3-kinase. Am J Physiol Heart Circ Physiol 2007; 292: H2248–H2256.
Bregoli L, Baldassare JJ, Raben DM . Nuclear diacylglycerol kinase-theta is activated in response to alpha-thrombin. J Biol Chem 2001; 276: 23288–23295.
Tu-Sekine B, Ostroski M, Raben DM . Modulation of diacylglycerol kinase theta activity by alpha-thrombin and phospholipids. Biochemistry 2007; 46: 924–932.
Los AP, van Baal J, de Widt J, Divecha N, van Blitterswijk WJ . Structure-activity relationship of diacylglycerol kinase theta. Biochim Biophys Acta 2004; 1636: 169–174.
McMullan R, Nurrish SJ . Rho deep in thought. Genes Dev 2007; 21: 2677–2682.
Sohail MA, Hashmi AZ, Hakim W, Watanabe A, Zipprich A, Groszmann RJ et al. Adenosine induces loss of actin stress fibers and inhibits contraction in hepatic stellate cells via Rho inhibition. Hepatology 2009; 49: 185–194.
Sah VP, Seasholtz TM, Sagi SA, Brown JH . The role of Rho in G protein-coupled receptor signal transduction. Annu Rev Pharmacol Toxicol 2000; 40: 459–489.
Sexl V, Mancusi G, Höller C, Gloria-Maercker E, Schütz W, Freissmuth M . Stimulation of the mitogen-activated protein kinase via the A2A-adenosine receptor in primary human endothelial cells. J Biol Chem 1997; 272: 5792–5799.
Hermans E . Biochemical and pharmacological control of the multiplicity of coupling at Gprotein- coupled receptors. Pharmacol Ther 2003; 99: 25–44.
Houslay MD, Morris NJ, Savage A, Marker A, Bushfield M . Regulation of hepatocyte adenylate cyclase by amylin and CGRP: a single receptor displaying apparent negative cooperatively towards CGRP and simple saturation kinetics for amylin, a requirement for phosphodiesterase inhibition to observe elevated hepatocyte cyclic AMP levels and the phosphorylation of Gi-2. J Cell Biochem 1994; 55: 66–82.
Gericke A, Munson M, Ross AH . Regulation of the PTEN phosphatase. Gene 2006; 374: 1–9.
Cai Z, Semenza G . PTEN activity is modulated during ischemia and reperfusion: involvement in the induction and decay of preconditioning. Circ Res 2005; 97: 1351–1359.
Omori N, Jin G, Li F, Zhang WR, Wang SJ, Hamakawa Y et al. Enhanced phosphorylation of PTEN in rat brain after transient middle cerebral occlusion. Brain Res 2002; 954: 317–322.
Ren XD, Schwartz MA . Determination of GTP loading on Rho. Methods Enzymol 2000; 325: 264–272.
Bligh EG, Dyer WJ . A rapid method of total lipid extraction and purification. Can J Biochem Physiol 1959; 37: 911–917.
Acknowledgements
This work has been supported by the Regional Government of Piedmont (Carini, Fondi Ricerca Sanitaria Finalizzata, 2006, 2007, 2009; Graziani Fondi Ricerca Sanitaria Finalizzata, 2008) by the University ‘Amedeo Avogadro’, by MIUR (Graziani PRIN 2007TE8NFY) and AIRC (Graziani).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by J Cidlowski
Rights and permissions
About this article
Cite this article
Baldanzi, G., Alchera, E., Imarisio, C. et al. Negative regulation of diacylglycerol kinase θ mediates adenosine-dependent hepatocyte preconditioning. Cell Death Differ 17, 1059–1068 (2010). https://doi.org/10.1038/cdd.2009.210
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2009.210
Keywords
This article is cited by
-
Establishment of a DGKθ Endogenous Promoter Luciferase Reporter HepG2 Cell Line for Studying the Transcriptional Regulation of DGKθ Gene
Applied Biochemistry and Biotechnology (2019)
-
A new hypothesis for Parkinson’s disease pathogenesis: GTPase-p38 MAPK signaling and autophagy as convergence points of etiology and genomics
Molecular Neurodegeneration (2018)
-
The expression of diacylglycerol kinase theta during the organogenesis of mouse embryos
BMC Developmental Biology (2013)
