
Abstract
Background:
Prostate-specific antigen (PSA) screening for prostate cancer results in a large number of unnecessary prostate biopsies. There is a need for specific molecular markers that can be used in combination with PSA to improve the specificity of PSA screening. We examined GADD45a methylation in blood DNA as a molecular marker for prostate cancer diagnosis.
Methods:
The study included 82 men, with PSA levels >4 ng ml−1 and/or abnormal digital rectal exam, who underwent prostate biopsy. We compared GADD45a methylation in DNA from serum and buffy coat in 44 patients (22 prostate cancer and 22 benign). GADD45a methylation in serum DNA was examined in 82 patients (34 cancer and 48 benign).
Results:
There was no significant difference in buffy coat GADD45a methylation between cancer and benign patients. Serum GADD45a methylation was significantly higher in cancer than in benign patients. Classification and regression tree predictive model for prostate cancer including risk groups defined by PSA, free circulating DNA (fcDNA) level and GADD45a methylation yielded specificity of 87.5%, sensitivity of 94.1% and receiver operator characteristic curve area of 0.937.
Conclusions:
Serum GADD45a methylation in combination with PSA and fcDNA level was useful in distinguishing benign from prostate cancer patients.
Similar content being viewed by others

Main
Prostate cancer is the second most diagnosed cancer and the second leading cause of cancer-related death in the United States with estimated 220 800 new cases and 27 450 deaths in 2015 (Siegel et al, 2015). Serum prostate-specific antigen (PSA) levels and digital rectal exam (DRE) are currently used in prostate cancer screening. Although PSA screening is highly sensitive, it lacks specificity and results in a large number of false positives. Only about 25–35% of patients with elevated PSA levels (4.1–9.9 ng ml−1) undergoing prostate biopsy are diagnosed with prostate cancer (Smith et al, 1997) indicating that two-third or more of patients have elevated PSA caused by non-malignant conditions (Partin et al, 1990; Nadler et al, 1995). Widespread PSA screening has led to a large proportion of patients being subjected to the discomfort of prostate biopsies resulting in increased health-care costs. Moreover, PSA screening results in overdetection of indolent prostate cancer on one hand (Etzioni et al, 2002) and missing high-grade prostate cancers in patients with normal PSA levels on the other (Thompson et al, 2004). Hence, there is an unmet need for specific molecular markers that can be used in combination with PSA to improve the specificity of prostate cancer-screening tests.
A blood-based test that can accurately predict the presence of prostate cancer may greatly help prostate cancer screening by eliminating unnecessary prostate biopsies. Free circulating DNA (fcDNA) in serum/plasma has provided an attractive tool in cancer diagnosis due to the non-invasive nature of sample procurement. We have previously demonstrated that serum fcDNA levels increases the specificity of the PSA test in prostate cancer diagnosis (Gordian et al, 2010). Although fcDNA levels may be helpful in prostate cancer diagnosis, it is not presently used in the clinical setting. One of the limitations of using elevated levels of fcDNA as a marker is that it is not specific to prostate cancer and can occur in other cancers as well (Shapiro et al, 1983; Silva et al, 1999; Wu et al, 2002; Sozzi et al, 2003; Taback et al, 2004; Flamini et al, 2006; Ren et al, 2006). Moreover, studies have shown that increasing levels of fcDNA in plasma and serum can be detected in benign pathological conditions as well (Chang et al, 2003; Okkonen et al, 2011).
Because of their chemical and biological stability, DNA methylation-based biomarkers have greater clinical applications in cancer detection, diagnosis and targeted therapies compared with RNA or protein. DNA methylation occurs primarily in CpG dinucleotides and is a common event in cancer. Methylation patterns that are specific to prostate cancer have been previously identified in tumour tissue (Goering et al, 2012; Yang and Park, 2012). However, this is limited by the invasiveness of the sample procurement method and increased costs. DNA methylation patterns in blood of prostate cancer patients and have been shown to be similar to that from tissues (Bastian et al, 2008; Ellinger et al, 2008). GSTP1 is the most studied DNA methylation marker in prostate cancer (Heyn and Esteller, 2012). GSTP1 hypermethylation was identified as an early marker of prostate cancer as it was detected in a large proportion of proliferative inflammatory atrophy lesions (Nakayama et al, 2003). GSTP1 hypermethylation was also detected in various bodily fluids such as urine, serum and ejaculate (Goessl et al, 2000; Reibenwein et al, 2007; Ellinger et al, 2008; Sunami et al, 2009). Other genes that exhibit a high incidence of hypermethylation in prostate cancer include APC, RASSFIA, PTGS2, TIG1 and MDRI (Bastian et al, 2008; Ellinger et al, 2008; Ellinger and Bastian, 2010).
In our previous study on GADD45a methylation in prostate cancer (Ramachandran et al, 2009), we found that methylation of four CpGs at positions −737, −723, −706 and −690 with respect to the transcription start site correlated inversely with GADD45a expression levels in prostate cancer cell lines. The 5′ four-CpG region was also frequently methylated in prostate cancer tissues compared with benign tissues. Methylation of this region correlated inversely with gene expression in prostate cancer and benign prostate tissues. So far, methylation of GADD45a has not been studied as a blood-based biomarker of prostate cancer.
In this study, we examine the usefulness of GADD45a methylation in blood DNA as a marker of prostate cancer alone and in combination with PSA and fcDNA in 82 patients. We also compared GADD45a methylation in matched DNA from serum and buffy coat in 44 patients.
Materials and methods
Study patients
We have previously conducted a prospective study that examined the use of serum fcDNA levels as a biomarker of prostate cancer in a ‘representative’ group of patients that underwent prostate cancer screening (Gordian et al, 2010). This study included 252 men, with PSA levels >4 ng ml−1 and/or abnormal DRE, who underwent prostate biopsy at the Urology Clinic of University of Miami/Jackson Memorial Hospitals between 2004 and 2008 under an institutional review board protocol. Patients with other malignancies and patients not willing to sign an informed consent form were excluded. A racially diverse population comprising of non-Hispanic blacks, Hispanic whites, non-Hispanic whites and Hispanic blacks was included. Baseline demographics, medical history and other pertinent information were obtained from the hospital charts. Levels of fcDNA in serum before prostate biopsy were quantitated by real-time PCR amplification of the GSTP1 gene (Gordian et al, 2010). The present study included a subset of 82 patients with sufficient serum for GADD45a methylation analysis. We first determined whether GADD45a methylation in serum or buffy coat or both correlate with prostate biopsy results and whether there is correlation between GADD45a methylation in serum and buffy coat. For this, we compared GADD45a methylation at CpG 1 site in matched DNA from serum and buffy coat in 22 prostate cancer and 22 benign patients. This assay was done using Ms-SNuPE analysis. Following this, we examined GADD45a methylation at four CpG sites in serum in the larger group of 82 patients (34 cancer and 48 benign) using a more robust pyrosequencing analysis.
Blood collection and DNA isolation
Blood was collected in 10-ml serum plus blood collection tubes (BD Vacutainer, Franklin Lakes, NJ, USA) before patients undergoing prostate biopsy. Serum was separated by centrifugation at 3500 r.p.m. for 15 min at room temperature. DNA was extracted from 2 ml serum using QIAamp UltraSens virus kit (Qiagen, Valencia, CA, USA) following the manufacturer’s protocol and stored at −20 °C until further analysis.
Ms-SNuPE analysis of GADD45a methylation
Bisulphite conversion of the serum DNA was carried out as previously described (Das et al, 2006). After bisulphite treatment, the DNA template was amplified with primer sets: forward: 5′-TTGGGTTGTTAGGGATTTTTATATG-3′; reverse: 5′-AAAATCTTTTCCACAAAAAACAAAA-3′ for the 5′ four-CpG region (Ramachandran et al, 2009). Ms-SNuPE analysis was done with the primer spanning CpG 1: 5′-AAACTAATCTAC-3′. Ms-SNuPE reactions were carried out using 1 μl of the PCR product in a 25 μl reaction mixture containing 1 × PCR buffer (Sigma, St Louis, MO, USA), 1 μ M of Ms-SNuPE primer, 1 μ Ci of 32P-labelled dATP or 32P-labelled dGTP and 1 U of Taq Polymerase (Sigma) (Gonzalgo and Jones, 1997; Ramachandran et al, 2007, 2009). Percent methylation in each CpG was calculated as G/(G+A) × 100.
Pyrosequencing analysis of GADD45a methylation
Methylation levels were quantified using pyrosequencing (EpigenDx, Hopkiton, MA, USA), to examine the methylation status at four CpG sites in the GADD45a 5′-UTR located between –737 and −690 relative to the transcription start site. Bisulphite-treated DNA template was amplified by nested PCR. The outer PCR product of 116 bp was generated using primers, forward: 5′-TTGGGTTGTTAGGGATTTTTATATG-3′ and reverse: 5′-AAAATCTTTTCCACAAAAAACAAAA-3′. This was then purified using the Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI, USA) after agarose gel electrophoresis. The resulting DNA was amplified using forward primer: 5′-TGGGTTGTTAGGGATTTTTATATG-3′ and the biotinylated reverse primer: 5′-AAAATCTTTTCCACAAAAAACA-3′. The biotinylated primer was used to capture one single-stranded DNA template for pyrosequencing (Royo et al, 2007). The PCR products (10 μl) were sequenced using the forward sequencing primer: 5′-GATTTTTATATGTGGTTATT-3′ on the Pyrosequencing PSQ96 HS System (Biotage, Uppsala, Sweden). Loci were analysed as a T/C SNP using QCpG software (Biotage). Percent methylation was calculated as the relative peak height of T vs C at each CpG site. Each plate contained unmethylated, partially methylated and heavily methylated DNAs (SssI-treated DNA) for quality control.
Statistical analysis
Patient groups (prostate cancer vs benign) were compared with respect to categorical variables using the Fisher’s exact test or the χ2-test. For continuous variable approximately normal distributed, such as age, log2 PSA and log2 fcDNA, patient groups were compared by the two-sample Student‘s t-test. Patient groups were compared with respect to PSA and fcDNA original data using the Wilcoxon–Mann–Whitney two-sample rank-sum test. This test is based on the ranks, as opposed to the actual measurements, and is recommended when data is not approximately normal distributed. Multivariate predictive model for prostate cancer was generated using Classification and Regression Tree (CART) (Breiman et al, 1984) analysis implemented in the R-package RPART (recursive partitioning for classification, regression and survival trees), version 4.1–8 (http://CRAN.R-project.org/package=rpart) (Therneau et al, 2014). In assessing the performance of the decision tree model, we report misclassification error rate considering all data as well as the misclassification error rate obtained by leave-one-out cross-validation. We employed logistic regression analysis to estimate odds ratios (ORs) and corresponding 95% confidence intervals (CIs) associated to individual binary predictors and risks groups identified by the RPART analysis. The Firth’s penalised likelihood approach was employed owing to small sample size and quasi-complete separation of data resulting in sparse data for some risk groups. (Heinze and Schemper, 2002; Heinze, 2006). Statistical analyses were performed using SAS version 9.3 (SAS Institute, Inc., Cary, NC, USA) and R package 3.1.1.
Results
Patient characteristics
Characteristics of patients grouped according to prostate biopsy are reported in Table 1. Of the total 82 patients analysed in this study, 34 were diagnosed with prostate cancer and 48 were benign based on prostate biopsy. The mean age was 64.9 years in prostate cancer and 64.5 in benign patients. The prostate cancer group had a higher percentage of blacks (44%) than the benign group (29%). The majority of blacks (79%) were non-Hispanic and the majority of whites (96%) were Hispanic. Following the exclusion of the two largest PSA values (1981 and 5000 ng ml−1), the data follows normal distribution; mean PSA in the cancer group (mean=34.7 ng ml−1; s.d.=62.6, max =306.4 ng ml−1) was significantly different from that of benign group (mean=7.1 ng ml−1; s.d.=4.1) using t-test (P=0.018). The number of patients with PSA>10 ng ml−1 was greater in prostate cancer (47%) than in benign (27%). The majority of prostate cancer patients (47.1%) had a Gleason score of 6 (3+3).
Correlation of serum and buffy coat GADD45a methylation with biopsy result in 44 matched samples
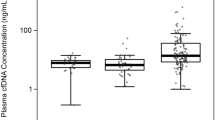
First, we determined whether serum and/or buffy coat GADD45a methylation correlated with prostate biopsy results. For this, we analysed methylation of a CpG site located at −737 (CpG 1) with respect to transcription start site by Ms-SNuPE (Ramachandran et al, 2009) in a subset of 44 patients comprising of 22 prostate cancer and 22 benign patients. Patient groups (prostate cancer vs benign) were compared using the two-sample t-test. Difference in CpG 1 methylation levels in serum between cancer vs benign was statistically significant (P=0.004), with mean levels higher in prostate cancer (33.9%) as compared with benign (14.6%) (Figure 1A). Similar results were obtained using log base 2-transformed data (P=0.027; data not shown). There was no statistically significant difference in methylation between cancer (18.5%) and benign (20.1%) in buffy coat DNA (P=0.686; Figure 1B). This was also true for log base 2-transformed data (P=0.405; data not shown). No significant correlations were seen between serum and buffy coat in both prostate cancer (r=0.047; P=0.836) and benign groups (r=0.273; P=0.219; Figure 1C). As we found low correlation of buffy coat and serum GADD45a methylation levels and also lack of significant difference in buffy coat GADD45a methylation levels between the benign and prostate cancer groups, we decided that further analysis would be limited to serum DNA. Despite high reproducibility and ease of being conducted in the lab, Ms-SNuPE has a few limitations. Only one CpG could be analysed per assay and it also required use of radioisotopes. Pyrosequencing is a more robust high-throughput assay that allows analysis of multiple CpGs at a time. We measured methylation levels in all four CpGs (−737 (CpG 1), −723 (CpG 2), −706 (CpG 3) and −690 (CpG 4) with respect to transcription start site) in serum in this subset of patients. Our results for CPG 1 were similar to that observed by Ms-SNuPE assay. Significantly higher levels of methylation were observed in prostate cancer group compared with that of benign (mean: 19.3% vs 6.4%, P=0.006; Figure 1D). The correlation between serum GADD45a methylation levels measured by Ms-SNuPE and that measured by pyrosequencing were very high in both groups, prostate cancer (r=0.972, P<0.0001) and benign (r=0.776, P<0.0001; Figure 1E).

Methylation in GADD45a CpG 1 in matched serum and buffy coat DNA samples. Box plots of percent GADD45a methylation in serum (A and D) and buffy coat (B) by biopsy result. Low correlation was seen between GADD45a methylation levels in serum and buffy coat by Ms-SNuPE analysis (C). GADD45a serum methylation was higher in cancer as compared with benign patients measured by Ms-SNuPE (A) and pyrosequencing (Pyro) (D). High correlation was observed between serum GADD45a methylation analysed by Ms-SNuPE and pyrosequencing (E).
Serum GADD45a methylation in benign and prostate cancer patients
We examined the pattern of methylation of four CpGs in 34 cancer and 48 benign patients by pyrosequencing analysis. The distributions of serum GADD45a methylation in biopsy-positive and -negative patients were compared using the two-sample Student’s t-test and also using the non-parametric Mann–Whitney–Wilcoxon rank-sum test. In all CpGs, means and medians of GADD45a methylation were significantly higher in cancer than in benign patients. Greater variability of GADD45a methylation was seen among the cancer patients compared with benign patients (Figure 2A). Serum GADD45a methylation measured at each of the four CpGs, and also sum of four CpGs were log base 2-transformed for easy visualisation and to improve the approximation to normal distribution. Log-transformed data by biopsy result were compared using the two-sample Student’s t-test. Mean values at each CpG, and also the sum of four CpGs were significantly higher in cancer than in benign group (Figure 2B). The results are summarised in Table 2.

Percent GADD45a methylation in serum (pyrosequencing) by biopsy result. Jittered box plots of original data (A), and of log base 2-transformed data (B).
Correlation of serum GADD45a methylation with Gleason score in prostate cancer patients
In order to assess the prognostic value of GADD45a methylation in serum, we examined whether the levels of serum GADD45a methylation correlates with Gleason score among the 34 prostate cancer patients. Of these, there were 16 patients with Gleason score 6, 9 with Gleason score 7 and 9 patients with Gleason score 8–10. We did not observe correlation of GADD45a methylation levels at any of the four CpG sites with Gleason score. There was no statistically significant difference between GADD45a means of the three Gleason score groups considering the individual CpG or the sum of four CpGs. ANOVA P-values were 0.320 for CpG 1, 0.380 for CpG 2, 0.500 for CpG 3, 0.785 for CpG 4 and 0.443 for sum of four CpGs (data not shown).
Prostate cancer predictive models based on PSA, fcDNA levels and GADD45a methylation
In order to understand the relationship between potential predictors of prostate cancer and to develop a multivariate predictive model, we employed Classification and Regression Tree (CART) analysis. As shown in Figure 3A, the RPART analysis resulted in classification trees defining risk groups based on cut-off points for PSA, fcDNA and GADD45a methylation (sum of four CpGs). Age, race and ethnicity were also included in the RPART model statement; however, none of these demographic characteristics was predictive to the extent of defining partition groups in the presence of the other variables. The strongest predictor was PSA level 16.9 ng ml−1. All 13 patients with PSA ⩾16.9 ng ml−1 had cancer. Using this PSA cutoff of 16.9 ng ml−1 resulted in 100% specificity, but only 38.2% sensitivity. The next strongest predictor was fcDNA level ⩾188 ng ml−1. Of note, the fcDNA cutoff point by CART analysis is consistent with previous study with 252 patients (Gordian et al, 2010), in which we found that the cutoff point of 180 ng ml−1 best discriminated between cancer and benign patients. Among the 69 patients with PSA <16.9 ng ml−1, 51 had fcDNA level <188, among which 43 were benign and 8 had cancer. Of the 18 patients with fcDNA ⩾188 ng ml−1, 13 had cancer and 5 were benign. The combination of PSA with fcDNA had a specificity of 89.5% and resulted in improvement of sensitivity to 76.5%. The third strongest predictor was GADD45a methylation (sum of four CpGs) at a cutoff point of 89.6. Seven patients had GADD45a methylation ⩾89.6 among those with PSA <16.9 and fcDNA <188; of these six had cancer and one was benign. Of the 44 patients with sum of four CpG methylation level <89.6, 42 were benign and 2 had cancer. The regression tree model misclassified 8 patients (9.8%), 6/48 benign (12.5%) and 2/34 cancer (5.9%), yielding a specificity and sensitivity of 87.5% and 94.1%, respectively. The leave-one-out cross-validation misclassification error was 9.9%.

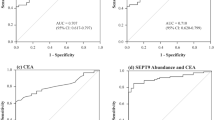
(A) Classification tree based on cut points for PSA, fcDNA and sum of GADD45a four-CpG methylation (CpGs_sum). Age, race and ethnicity were also included in each model statement under RPART but none defined partition. Below each prediction node label (corresponding to majority vote) is shown the number of observations from benign and cancer groups, respectively. The corresponding misclassification rates were 8/82 (9.8%) overall, 6/48 (12.5%) in benign and 2/34 (5.9%) in cancer group. These implied specificity 42/48 (87.5%) and sensitivity 32/34 (94.1%). In addition, the leave-one-out cross-validation misclassification error was 9.9%. (B) Receiver-operating curves for comparison of the classification tree models including PSA, fcDNA and GADD45a methylation (solid and dashed lines) to other fitted models.
Next, we employed univariate and multivariate logistic regression analysis to estimate ORs and corresponding 95% CIs associated to risks groups identified by the RPART analysis. (Table 3). Univariate logistic models were computed for examining the effect of individual variables: PSA >16.9 vs ⩽16.9 ng ml−1, fcDNA >188 vs ⩽188 ng ml−1 and GADD45a methylation >89.6 vs ⩽89.6, on the risk of prostate cancer in comparison with benign patients. Odds ratios for PSA, fcDNA or GADD45a methylation comparing prostate cancer vs benign patients based on various fitted logistic models are reported in Table 3. Univariate analysis including only PSA >16.9 vs ⩽16.9 ng ml−1 yielded an OR of 60.9 (95% CI: 3.11, >999; P=0.007; AUC=0.691), model including only fcDNA >188 vs ⩽188 ng/ml yielded an OR of 8.6 (95% CI: 2.7, 27.0; P=0.0002; AUC=0.698), and model including only GADD45a methylation >89.6 vs ⩽89.6 yielded an OR of 29.1 (95% CI: 3.57, 237.1; P=0.002; AUC=0.681). Figure 3B shows the ROC curves for various fitted models. There were significant improvements (P<0.001) in prediction comparing models including risk groups vs model including PSA+fcDNA or model including GADD45a alone. Receiver-operating characteristic AUCs for the multivariate models defined by PSA, fcDNA levels and GADD45a methylation derived from regression trees analysis without and with adjustment for race, ethnicity and age were 0.937 and 0.952, respectively, and these were significantly higher than those for the univariate models and the model including PSA+fcDNA (AUC=0.746). Thus, the composite models including these three markers were effective in discriminating biopsy positive from biopsy negative patients.
Discussion
The present study included 82 subjects who were part of our previous study that examined the usefulness of fcDNA as a biomarker of prostate cancer (Gordian et al, 2010). These subjects had sufficient stored serum for methylation analysis. We first determined whether GADD45a methylation in serum or buffy coat or both correlate with prostate biopsy results and whether there is correlation between GADD45a methylation in serum and buffy coat. For this, we compared GADD45a methylation in matched DNA from serum and buffy coat in 22 prostate cancer and 22 benign patients. This assay was done using Ms-SNuPE analysis (Ramachandran et al, 2009). We found statistically significant difference in serum methylation between cancer and benign groups. However, we did not observe statistically significant difference in buffy coat methylation between cancer and benign groups. There was no significant correlation between serum and buffy coat in both prostate cancer and benign groups. Hence, we proceeded to study serum GADD45a methylation in 82 patients comprising of 34 cancer and 48 benign patients using a more robust pyrosequencing analysis.
Although there has been a lot of interest in using DNA derived from peripheral blood as biomarkers for prostate cancer diagnosis and prognosis, the relative significance of serum vs buffy coat DNA is currently unknown. Several studies have examined DNA methylation in buffy coat as a biomarker of various cancers including prostate cancer (Goessl et al, 2000; Wong et al, 2000; Suga et al, 2008; Marsit et al, 2011; You et al, 2012). The advantages of using buffy coat include recovery of large amounts of DNA as compared with serum or plasma, and the ability to store for prolonged periods of time (Mychaleckyj et al, 2011). Buffy coat DNA is comprised of cell-bound DNA including blood cell DNA as well as DNA from circulating tumour cells. On the other hand, the source of serum/plasma fcDNA is not clear. Despite several possible sources such as apoptosis/necrosis of tumour cells, blood cells, lysed circulating tumour cells and active release of fcDNA by living cells (van der Vaart and Pretorius, 2008), tumour DNA constitutes a large proportion of fcDNA in cancer patients.
There are conflicting reports in literature regarding the correlation of buffy coat and serum DNA methylation with tumour. Goessl et al (2000) examined GSTP1 methylation in prostate tissue and compared with bodily fluids such as serum, plasma, buffy coat and ejaculate. They found higher correlation of GSTP1 methylation in serum and plasma with that of prostate tissue compared with that of buffy coat. Contrary to this, Wong et al (2000) examined p15 methylation in tumours, plasma, serum and buffy coat samples from hepatocellular carcinoma patients and found that buffy coat p15 methylation showed a higher correlation with that of tumour DNA compared with serum and plasma. We found that GADD45a methylation in serum fcDNA but not buffy coat DNA distinguishes between benign and prostate cancer patients.
The first study of non-invasive methylation biomarker of prostate cancer was the analysis of GSTP1 promoter methylation in prostate cancer and benign patients. GSTP1 promoter hypermethylation was observed in 72% of prostate cancer patients, but none of the patients with benign prostatic hyperplasia (BPH) (Goessl et al, 2000, 2001). The usefulness of hypermethylation of other genes including RASSF1A, RARβ2, MDR1, EDNRB, TIG1 and Reprimo in serum as a diagnostic marker of prostate cancer has also been shown (Ellinger et al, 2008; Hoque, 2009). A pooled analysis of about 900 prostate cancer cases in several studies that examined the presence of GSTP1 hypermethylation in fcDNA showed that the assay specificity was high (>93%), but the sensitivity was varying (ranging from 11 to 100%). Moreover, it was found that the sensitivity was increased when methylation of multiple genes was analysed (Ellinger and Bastian, 2010). To our knowledge, ours is the first study to examine the role of GADD45a methylation in early detection of prostate cancer. The cutoff level of 89.6 for sum of four CpG methylation could distinguish between biopsy-positive and biopsy-negative patients with a high specificity of 97.9% and moderate sensitivity of 38.2% (AUC=0.681). There are only a limited number of reports on quantitative analysis of serum DNA methylation in prostate cancer. Our results are comparable to one such study that showed that GSTP1 hypermethylation could discriminate prostate cancer and BPH patients at a high specificity of 92.9% and sensitivity of 42.3% (AUC=0.684; Ellinger et al, 2008).
Prognostic value of DNA methylation markers in fcDNA has been previously reported. Reibenwein et al (2007) showed correlation between GSTP1 methylation and Gleason score. Sunami et al (2009) reported that hypermethylation of RASSF1A, RARβ2 and GSTP1 correlated with Gleason score and serum PSA. However, in the study by Ellinger et al (2008), there was no correlation between methylation of GSTP1, TIG1, PTGS2 and Reprimo either in combination or individually with the pathological stage or grade. Among the 34 biopsy-positive patients, we did not find correlation between GADD45a methylation levels and Gleason score. This lack of correlation may be related to the limited sample size.
Prostate cancer screening has remained controversial as it has been associated with a large risk of overdetection (Andriole et al, 2009; Schroder et al, 2009). A large majority of the prostate biopsies performed for increased PSA alone are negative. There have been attempts to reduce the number of unnecessary biopsies by using PSA derivatives as markers. However, many of these markers have not gained acceptance owing to the lack of sensitivity or specificity or both. Garzotto et al (2005) used CART analysis to develop a prostate biopsy algorithm in patients with elevated PSA/abnormal DRE or both. Their CART model included PSA, PSA density, hypoechoic lesions, age and prostate volume. This model had a specificity of 31.3%, sensitivity of 96.6% and AUC of 0.74. In our previous study we found that inclusion of fcDNA and the interaction of fcDNA and PSA to a model with PSA, age and race significantly improved predictivity compared with model without fcDNA yielding a sensitivity and specificity of 95.5% and 33.1%, respectively, AUC 0.742, positive predictive value of 43.8 and negative predictive value of 93.1 (Gordian et al, 2010). To our knowledge, there are no reports on CART models that have included molecular markers for the prediction of prostate cancer. In the present study, the CART model had sensitivity and specificity of 94.1% and 87.5%, respectively, and the AUC was 0.937, indicating high prediction accuracy (positive predictive value=84.2% and negative predictive value=95.5%). We show that if we apply this model including PSA, fcDNA and GADD45a methylation in our patient group, we would eliminate the need for biopsy in 53.6% patients (44 out of 82), as a consequence of our estimated high specificity of 87.5%. Furthermore, only 2 out 34 true positives were missed (false negative rate of 5.9%). However, a major limitation of this study is the small sample size. Owing to the lack of sample availability, this study only included a subset of patients of the larger group that was representative of prostate cancer screening setting. Thus, the results of this study need to be validated in a larger prospective group of patients that undergo prostate biopsy after prostate cancer screening.
Change history
28 July 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Andriole GL, Crawford ED, Grubb RL 3rd, Buys SS, Chia D, Church TR, Fouad MN, Gelmann EP, Kvale PA, Reding DJ, Weissfeld JL, Yokochi LA, O'Brien B, Clapp JD, Rathmell JM, Riley TL, Hayes RB, Kramer BS, Izmirlian G, Miller AB, Pinsky PF, Prorok PC, Gohagan JK, Berg CD (2009) Mortality results from a randomized prostate-cancer screening trial. N Engl J Med 360 (13): 1310–1319.
Bastian PJ, Palapattu GS, Yegnasubramanian S, Rogers CG, Lin X, Mangold LA, Trock B, Eisenberger MA, Partin AW, Nelson WG (2008) CpG island hypermethylation profile in the serum of men with clinically localized and hormone refractory metastatic prostate cancer. J Urol 179 (2): 529–534.
Breiman L, Freidman J, Olshen R, Stone C (1984) Classification and Regression Trees. Wadsworth: Belmont, CA, USA.
Chang CP, Chia RH, Wu TL, Tsao KC, Sun CF, Wu JT (2003) Elevated cell-free serum DNA detected in patients with myocardial infarction. Clin Chim Acta 327 (1–2): 95–101.
Das PM, Ramachandran K, Vanwert J, Ferdinand L, Gopisetty G, Reis IM, Singal R (2006) Methylation mediated silencing of TMS1/ASC gene in prostate cancer. Mol Cancer 5: 28.
Ellinger J, Bastian PJ (2010) Cell-free DNA: a novel biomarker for patients with prostate cancer? Open Prost Cancer J 3: 57–62.
Ellinger J, Haan K, Heukamp LC, Kahl P, Buttner R, Muller SC, von Ruecker A, Bastian PJ (2008) CpG island hypermethylation in cell-free serum DNA identifies patients with localized prostate cancer. Prostate 68 (1): 42–49.
Etzioni R, Penson DF, Legler JM, di Tommaso D, Boer R, Gann PH, Feuer EJ (2002) Overdiagnosis due to prostate-specific antigen screening: lessons from U.S. prostate cancer incidence trends. J Natl Cancer Inst 94 (13): 981–990.
Flamini E, Mercatali L, Nanni O, Calistri D, Nunziatini R, Zoli W, Rosetti P, Gardini N, Lattuneddu A, Verdecchia GM, Amadori D (2006) Free DNA and carcinoembryonic antigen serum levels: an important combination for diagnosis of colorectal cancer. Clin Cancer Res 12 (23): 6985–6988.
Garzotto M, Beer TM, Hudson RG, Peters L, Hsieh YC, Barrera E, Klein T, Mori M (2005) Improved detection of prostate cancer using classification and regression tree analysis. J Clin Oncol 23 (19): 4322–4329.
Goering W, Kloth M, Schulz WA (2012) DNA methylation changes in prostate cancer. Methods Mol Biol 863: 47–66.
Goessl C, Krause H, Muller M, Heicappell R, Schrader M, Sachsinger J, Miller K (2000) Fluorescent methylation-specific polymerase chain reaction for DNA-based detection of prostate cancer in bodily fluids. Cancer Res 60 (21): 5941–5945.
Goessl C, Muller M, Heicappell R, Krause H, Miller K (2001) DNA-based detection of prostate cancer in blood, urine, and ejaculates. Ann N Y Acad Sci 945: 51–58.
Gonzalgo ML, Jones PA (1997) Rapid quantitation of methylation differences at specific sites using methylation-sensitive single nucleotide primer extension (Ms-SNuPE). Nucleic Acids Res 25 (12): 2529–2531.
Gordian E, Ramachandran K, Reis IM, Manoharan M, Soloway MS, Singal R (2010) Serum free circulating DNA is a useful biomarker to distinguish benign versus malignant prostate disease. Cancer Epidemiol Biomarkers Prev 19 (8): 1984–1991.
Heinze G (2006) A comparative investigation of methods for logistic regression with separated or nearly separated data. Stat Med 25 (24): 4216–4226.
Heinze G, Schemper M (2002) A solution to the problem of separation in logistic regression. Stat Med 21 (16): 2409–2419.
Heyn H, Esteller M (2012) DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet 13 (10): 679–692.
Hoque MO (2009) DNA methylation changes in prostate cancer: current developments and future clinical implementation. Expert Rev Mol Diagn 9 (3): 243–257.
Marsit CJ, Koestler DC, Christensen BC, Karagas MR, Houseman EA, Kelsey KT (2011) DNA methylation array analysis identifies profiles of blood-derived DNA methylation associated with bladder cancer. J Clin Oncol 29 (9): 1133–1139.
Mychaleckyj JC, Farber EA, Chmielewski J, Artale J, Light LS, Bowden DW, Hou X, Marcovina SM (2011) Buffy coat specimens remain viable as a DNA source for highly multiplexed genome-wide genetic tests after long term storage. J Transl Med 9: 91.
Nadler RB, Humphrey PA, Smith DS, Catalona WJ, Ratliff TL (1995) Effect of inflammation and benign prostatic hyperplasia on elevated serum prostate specific antigen levels. J Urol 154 (2 Pt 1): 407–413.
Nakayama M, Bennett CJ, Hicks JL, Epstein JI, Platz EA, Nelson WG, De Marzo AM (2003) Hypermethylation of the human glutathione S-transferase-pi gene (GSTP1) CpG island is present in a subset of proliferative inflammatory atrophy lesions but not in normal or hyperplastic epithelium of the prostate: a detailed study using laser-capture microdissection. Am J Pathol 163 (3): 923–933.
Okkonen M, Lakkisto P, Korhonen AM, Parviai-nen I, Reinikainen M, Varpula T, Pettila V FINNALI Study Group (2011) Plasma cell-free DNA in patients needing mechanical ventilation. Crit Care 15 (4): R196.
Partin AW, Carter HB, Chan DW, Epstein JI, Oesterling JE, Rock RC, Weber JP, Walsh PC (1990) Prostate specific antigen in the staging of localized prostate cancer: influence of tumor differentiation, tumor volume and benign hyperplasia. J Urol 143 (4): 747–752.
Ramachandran K, Gopisetty G, Gordian E, Navarro L, Hader C, Reis IM, Schulz WA, Singal R (2009) Methylation-mediated repression of GADD45alpha in prostate cancer and its role as a potential therapeutic target. Cancer Res 69 (4): 1527–1535.
Ramachandran K, van Wert J, Gopisetty G, Singal R (2007) Developmentally regulated demethylase activity targeting the betaA-globin gene in primary avian erythroid cells. Biochemistry 46 (11): 3416–3422.
Reibenwein J, Pils D, Horak P, Tomicek B, Goldner G, Worel N, Elandt K, Krainer M (2007) Promoter hypermethylation of GSTP1, AR, and 14-3-3sigma in serum of prostate cancer patients and its clinical relevance. Prostate 67 (4): 427–432.
Ren N, Qin LX, Tu H, Liu YK, Zhang BH, Tang ZY (2006) The prognostic value of circulating plasma DNA level and its allelic imbalance on chromosome 8p in patients with hepatocellular carcinoma. J Cancer Res Clin Oncol 132 (6): 399–407.
Royo JL, Hidalgo M, Ruiz A (2007) Pyrosequencing protocol using a universal biotinylated primer for mutation detection and SNP genotyping. Nat Protoc 2 (7): 1734–1739.
Schroder FH, Hugosson J, Roobol MJ, Tammela TL, Ciatto S, Nelen V, Kwiatkowski M, Lujan M, Lilja H, Zappa M, Denis LJ, Recker F, Berenguer A, Maattanen L, Bangma CH, Aus G, Villers A, Rebillard X, van der Kwast T, Blijenberg BG, Moss SM, de Koning HJ, Auvinen A (2009) Screening and prostate-cancer mortality in a randomized European study. N Engl J Med 360 (13): 1320–1328.
Shapiro B, Chakrabarty M, Cohn EM, Leon SA (1983) Determination of circulating DNA levels in patients with benign or malignant gastrointestinal disease. Cancer 51 (11): 2116–2120.
Siegel RL, Miller KD, Jemal A (2015) Cancer statistics, 2015. CA Cancer J Clin 65 (1): 5–29.
Silva JM, Dominguez G, Garcia JM, Gonzalez R, Villanueva MJ, Navarro F, Provencio M, San Martin S, Espana P, Bonilla F (1999) Presence of tumor DNA in plasma of breast cancer patients: clinicopathological correlations. Cancer Res 59 (13): 3251–3256.
Smith DS, Humphrey PA, Catalona WJ (1997) The early detection of prostate carcinoma with prostate specific antigen: the Washington University experience. Cancer 80 (9): 1852–1856.
Sozzi G, Conte D, Leon M, Ciricione R, Roz L, Ratcliffe C, Roz E, Cirenei N, Bellomi M, Pelosi G, Pierotti MA, Pastorino U (2003) Quantification of free circulating DNA as a diagnostic marker in lung cancer. J Clin Oncol 21 (21): 3902–3908.
Suga Y, Miyajima K, Oikawa T, Maeda J, Usuda J, Kajiwara N, Ohira T, Uchida O, Tsuboi M, Hirano T, Kato H, Ikeda N (2008) Quantitative p16 and ESR1 methylation in the peripheral blood of patients with non-small cell lung cancer. Oncol Rep 20 (5): 1137–1142.
Sunami E, Shinozaki M, Higano CS, Wollman R, Dorff TB, Tucker SJ, Martinez SR, Mizuno R, Singer FR, Hoon DS (2009) Multimarker circulating DNA assay for assessing blood of prostate cancer patients. Clin Chem 55 (3): 559–567.
Taback B, O'Day SJ, Hoon DS (2004) Quantification of circulating DNA in the plasma and serum of cancer patients. Ann N Y Acad Sci 1022: 17–24.
Therneau T, Atkinson B, Ripley B (2014) rpart: Recursive Partitioning and Regression Trees p R package version 4.1-9 Available from http://CRAN.R-project.org/package=rpart.
Thompson IM, Pauler DK, Goodman PJ, Tangen CM, Lucia MS, Parnes HL, Minasian LM, Ford LG, Lippman SM, Crawford ED, Crowley JJ, Coltman CA Jr (2004) Prevalence of prostate cancer among men with a prostate-specific antigen level <or =4.0 ng per milliliter. N Engl J Med 350 (22): 2239–2246.
van der Vaart M, Pretorius PJ (2008) Circulating DNA. Its origin and fluctuation. Ann N Y Acad Sci 1137: 18–26.
Wong IH, Lo YM, Yeo W, Lau WY, Johnson PJ (2000) Frequent p15 promoter methylation in tumor and peripheral blood from hepatocellular carcinoma patients. Clin Cancer Res 6 (9): 3516–3521.
Wu TL, Zhang D, Chia JH, Tsao KH, Sun CF, Wu JT (2002) Cell-free DNA: measurement in various carcinomas and establishment of normal reference range. Clin Chim Acta 321 (1–2): 77–87.
Yang M, Park JY (2012) DNA methylation in promoter region as biomarkers in prostate cancer. Methods Mol Biol 863: 67–109.
You YJ, Chen YP, Zheng XX, Meltzer SJ, Zhang H (2012) Aberrant methylation of the PTPRO gene in peripheral blood as a potential biomarker in esophageal squamous cell carcinoma patients. Cancer Lett 315 (2): 138–144.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Reis, I., Ramachandran, K., Speer, C. et al. Serum GADD45a methylation is a useful biomarker to distinguish benign vs malignant prostate disease. Br J Cancer 113, 460–468 (2015). https://doi.org/10.1038/bjc.2015.240
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.240
