
Abstract
Background:
O6-Methylguanine-DNA methyltransferase (MGMT) loss of expression has been suggested to be predictive of response to temozolomide in neuroendocrine tumours (NETs), but so far, only limited data are available. We evaluated the prognostic and predictive value of MGMT status, assessed by two molecular methods and immunohistochemistry, in a large series of NETs of different origins.
Methods:
A total of 107 patients, including 53 treated by alkylants (temozolomide, dacarbazine or streptozotocin), were retrospectively studied. In each case, we used methyl-specific PCR (MS-PCR) and pyrosequencing for evaluation of promoter methylation and immunohistochemistry for evaluation of protein status.
Results:
MGMT promoter methylation was detected in 12 out of 99 (12%) interpretable cases by MS-PCR and in 24 out of 99 (24%) by pyrosequencing. O6-Methylguanine-DNA methyltransferase loss of expression was observed in 29 out of 89 (33%) interpretable cases. Status of MGMT was not correlated with overall survival (OS) from diagnosis. Progression-free survival and OS from first alkylant use (temozolomide, dacarbazine and streptozotocin) were higher in patients with MGMT protein loss (respectively, 20.2 vs 7.6 months, P<0.001 and 105 vs 34 months, P=0.006) or MGMT promoter methylation assessed by pyrosequencing (respectively, 26.4 vs 10.8 months, P=0.002 and 77 vs 43 months, P=0.026).
Conclusions:
Our results suggest that MGMT status is associated with response to alkylant-based chemotherapy in NETs.
Similar content being viewed by others


Main
Neuroendocrine tumours (NETs) are rare, but their incidence (5.25/100 000 in 2004 in the United States) is rising and their prevalence (35/100 000) is high (Modlin et al, 2008; Yao et al, 2008). As a whole, more than 30% NETs are diagnosed at advanced non-resectable stages, but much higher figures can be achieved for some primary sites, such as the small intestine (Modlin et al, 2008; Yao et al, 2008). Even with the recent availability of targeted therapies, such as sunitinib and everolimus, cytotoxic chemotherapy remains the backbone of systemic treatment (Weatherstone and Meyer, 2012), at least for advanced pancreatic NETs (Delaunoit et al, 2004; Kulke et al, 2010; Auernhammer and Goke, 2011; Walter et al, 2012). Most current chemotherapy regimens are based on alkylating agents, such as streptozotocin (Delaunoit et al, 2004), dacarbazine (Bajetta et al, 2002; Walter et al, 2010) and temozolomide (Kulke et al, 2006; Ekeblad et al, 2007; Strosberg et al, 2011; Chan et al, 2012; Koumarianou et al, 2012; Chan et al, 2013), used alone or in combination with other cytotoxic agents or targeted therapies.
One of the mechanisms of cytotoxicity by alkylating agents is the induction of DNA methylation at O6-guanine sites. Such lesions can be specifically restored by the DNA repair enzyme O6-methylguanine-DNA methyltransferase (MGMT; Christmann et al, 2011): any reduction in MGMT activity may therefore result in an increased sensitivity of tumour cells to alkylating-induced DNA damage and may potentiate the clinical effects of alkylating agents. One of the mechanisms involved in reduced MGMT activity in tumour tissue is the silencing of the corresponding gene through promoter methylation, which has been demonstrated to be a frequent event in many tumour types (Christmann et al, 2011; Wick et al, 2014). For clinical purposes, it has therefore been proposed to study either MGMT protein expression or MGMT promoter methylation in order to predict sensitivity to alkylating agents. However, MGMT promoter methylation has been shown to be neither constantly nor necessarily associated with reduced MGMT protein expression: most studies showed only a poor correlation between the two parameters (Karayan-Tapon et al, 2010; Christmann et al, 2011; Quillien et al, 2012). Nevertheless, it has been shown, first in gliomas (Hegi et al, 2005; Karayan-Tapon et al, 2010; Weller et al, 2010) and more recently in other tumours, such as melanoma (Naumann et al, 2009; Schraml et al, 2012), that MGMT promoter methylation in tumour tissue is predictive of the response to alkylating agents, especially temozolomide, and may prove to be a more reliable predictive biomarker than loss of MGMT expression assessed by immunohistochemistry (Quillien et al, 2012).
Despite the large use of alkylating agents in NETs, comparatively little information is available about MGMT status in these tumours and on its prognostic and predictive relevance. Several studies have shown that MGMT promoter methylation can be detected in a significant proportion of NETs, but little clinical information and no follow-up were available in most of these reports (Toyooka et al, 2001; Chan et al, 2003; House et al, 2003; Liu et al, 2005; Arnold et al, 2007; Welin et al, 2011), except in the most recent one (Schmitt et al, 2014), which found a positive correlation between MGMT promoter methylation and response to temozolomide in 10 patients with pancreatic NETs. Three previous studies attempted to correlate MGMT protein expression assessed by immunohistochemistry with response to temozolomide, but resulted in conflicting results (Ekeblad et al, 2007; Kulke et al, 2009, Schmitt et al, 2014): only one (Kulke et al, 2009) found a positive correlation between loss of MGMT protein and response to temozolomide, but in a short series of pancreatic NETs.
Our aims were therefore: (a) to evaluate the performances in NETs of methods largely used in other tumour types, especially gliomas, for evaluating MGMT status: we combined here two different molecular techniques to study MGMT promoter methylation (methyl-specific PCR (MS-PCR) and pyrosequencing) along with immunohistochemistry to assess MGMT protein expression, (b) to evaluate the prognostic relevance of MGMT status in NETs and its predictive value, not only for temozolomide but also for other alkylating agents, such as streptozotocin and dacarbazine, and (c) to determine whether, in NETs as in other cancers, the presence of MGMT promoter methylation is associated with a so-called hypermethylated phenotype. For this purpose, we retrospectively analyse a large series of 107 patients with NETs of various origins, with complete follow-up in our institution, including 53 patients treated by one or more alkylating agents.
Patients and Methods
Patients
For the predictive study, we retrospectively included all patients having started at least one cycle of chemotherapy containing an alkylating agent (streptozotocin, temozolomide or dacarbazine) for an advanced well-differentiated NET in our institution between January 1982 and December 2012. For the prognostic study, we included an equivalent number of patients referred to our institution between 1997 and 2007, having not received any alkylating agent. Inclusion criteria were: (a) informed consent to clinical research studies; (b) complete follow-up in our institution and (c) availability of tissue material for additional histological evaluation if necessary and for molecular and immunohistochemical studies. The study was approved by the local Ethics Committee and by the Steering Committee of Tumorothèque des Hospices Civils de Lyon, the local tissue biobank, which provided the tissue samples.
A total of 107 patients, including 53 treated by alkylating agents, were eventually included. For all patients, formalin-fixed, paraffin-embedded (FFPE) tissue samples were available; fresh frozen tissue samples were also available in 47 patients. The following data were collected: age at diagnosis, gender, MEN1 syndrome, primary tumour location (gastrointestinal, pancreas, lung, unknown), functional status, histological grade (Rindi et al, 2006), WHO classification (Rindi et al, 2010), TNM stage (Rindi et al, 2006, 2007; Sobin et al, 2009), and number of metastatic sites.
Clinical and pathological data are summarised in Supplementary Appendix 1. The main primary locations were the pancreas (58%) and the gastrointestinal tract (31%); 74% of tumours were metastatic; histological grade was G1 in 38% of cases and G2 in 53%. Median follow-up from diagnosis was 77 (1–335) months.
Evaluation of MGMT status
MGMT promoter methylation status. MGMT promoter methylation status was studied using two techniques: MS-PCR and pyrosequencing, giving, respectively, qualitative and quantitative information.
The techniques used here were derived from the procedures employed in our institution for gliomas. All samples examined contained more than 80% tumour cells. DNA extraction from FFPE tissue was performed after deparaffinization using a purification kit (MasterPure DNA, Epicentre, Madison, WI, USA). Genomic DNA was modified by bisulfite conversion (EZ DNA Methylation Gold Kit, Zymo, Irvine, CA, USA). For MS-PCR, we used the primers and PCR conditions described by Dong et al (2005). Pyrosequencing was performed using PyroMark Q96 MGMT kit (Qiagen, Courtaboeuf, France) on a PSQTM96 MA system (Biotage, Uppsala, Sweden), as described previously (Karayan-Tapon et al, 2010; Quillien et al, 2012).
For pyrosequencing analysis, a 8% cutoff was used, according to the standards currently used in neuro-oncology (Quillien et al, 2012) and according to our own control data from normal gastroenteropancreatic tissues, in which median promoter methylation value was found to be 7% (data not shown). MGMT promoter was therefore scored ‘methylated’ if more than 8% methylated alleles compared with unmethylated alleles were detected. Results are given as ‘methylated’, ‘not methylated’ or ‘not interpretable’.
Immunohistochemistry. O6-Methylguanine-DNA methyltransferase protein expression was evaluated by an immunohistochemical technique applied to sections of FFPE archival tissue material. An automated immunostaining system was used (Ventana Benchmark, Tucson, AZ, USA). After antigen retrieval by heating, deparaffinized 4-μm-thick tissue sections were incubated with a mouse monoclonal anti-MGMT antibody (clone MT23.2, Invitrogen, Camarillo, CA, USA) diluted 1:200 for 32 min. Revelation was performed using the streptavidin–biotin complex technique. Only cells with unambiguous nuclear staining for MGMT were considered positive. Only cases with positive internal controls (stromal cells, lymphocytes) showing unambiguous nuclear staining were interpreted. The percentage of positive tumour cells was evaluated semi-quantitatively in the areas containing the highest density of positive tumour cells. As previously suggested (Ekeblad et al, 2007), tumours were scored negative if they contained <10% of positive tumour cells in the whole tumour tissue section.
Gene promoter methylation studies
In addition to MGMT gene promoter, the following genes were studied using MS-PCR, as described above, after DNA extraction from fresh frozen tumour tissues obtained in 47 patients of our study group (21 with pancreatic NETs and 26 with ileal NETs): (a) five genes of the so-called classical panel for the definition of CpG island methylated phenotype in colon cancer: p16, hMLH1, MINT1, MINT2, MINT31 (Park et al, 2003; Curtin et al, 2011); (b) six additional genes previously tested in comprehensive surveys of epigenetic alterations in gastroenteropancreatic NETs: HIC, THBS, APC, RASSFA1, TIMP3, CDH1 (encoding E-cadherin; House et al, 2003; Pizzi et al, 2005; Zhang et al, 2006; Arnold et al, 2007; Pizzi et al, 2008). Primers and PCR conditions were designed as previously described (House et al, 2003; Park et al, 2003; Arnold et al, 2007).
Treatment and follow-up
Chemotherapy regimens. Various chemotherapy regimens were classified into three categories:
-
‘Dacarbazine-based’ regimens: all regimens using dacarbazine in association with 5-fluorouracile (5-FU) and/or epirubicin; our usual regimen consisted of the intravenous administration of 500 mg m−2 per day of 5-FU and 250 mg m−2 per day of dacarbazine for 3 days, and 50 mg m−2 of epirubicin on day 1, administered every 21 days (Bajetta et al, 2002; Walter et al, 2010); two patients did not receive epirubicin because of physician choice.
-
‘Temozolomide-based’ regimens: all regimens using temozolomide alone or in association with capecitabine; usually, the protocol consisted of oral temozolomide 200 mg m−2 once daily at bedtime for 5 days (days 10–14) every 28 days, with or without oral capecitabine 750 mg m−2 twice daily for 14 days (days 1–14; Strosberg et al, 2011); in some patients, starting doses were reduced because of baseline renal insufficiency and/or previous haematotoxicity with other chemotherapy regimen.
-
‘Streptozotocin-based’ regimens: all regimens combining streptozotocin with either doxorubicin or 5-FU; the dose of streptozotocin was 500 mg m−2 per day for 5 days, administered every 42 days; 5-FU was given at 400 mg m−2 per day for 5 days, every 42 days; doxorubicin was given at 50 mg m−2 per day on day 1 and day 22, administered every 42 days.
Efficacy assessment. We analysed the response to chemotherapy according to the following criteria: type of regimen; start and end date of chemotherapy; best response obtained in accordance with RECIST criteria (stable disease was defined as disease stable for at least 2 months of treatment); progression-free survival (PFS) and overall survival (OS). Patients were assessed at 2- to 3-month intervals of treatment or earlier if clinically indicated, using clinical examination and spiral computed tomography scan of thorax, abdomen and pelvis and/or magnetic resonance imaging of the liver when appropriate. Imaging results were retrospectively reviewed and RECIST criteria v1.1 were applied (Therasse et al, 2000), even for the earliest period when WHO criteria were still used in clinical practice.
Statistical analysis
Categorical variables were expressed as percentages, and compared by the χ2 test or with Fisher’s exact test when appropriate. Continuous variables were expressed as median with range. The comparison between the techniques used for MGMT status evaluation was calculated with the Cohen’s kappa coefficient (k). Progression-free survival was calculated from initiation of each chemotherapy line to the date of disease progression according to RECIST criteria or death from any cause, whichever occurred first. Overall survival was calculated from initiation of each chemotherapy line to the date of death or last follow-up. Progression-free survival and OS were assessed using Kaplan–Meier analysis and comparisons were performed using the log-rank test. A P value of <0.05 was considered statistically significant. Cox proportional hazard models were developed using relevant clinico-pathologic variables to determine the association of each parameter with PFS or OS. For continuous variables, the cutoff level chosen was their median value. Only variables with a P value of <0.10 at univariate analysis were introduced in the Cox model. Relative risks were expressed as hazard ratios (HRs) with 95% confidence intervals. The cutoff date for the final analysis was 1 July 2013. All statistical analyses were performed using Statistical Package for Social Sciences version 17.0 (Chicago, IL, USA).
Results
MGMT status
O6-Methylguanine-DNA methyltransferase status according to tumour location and histological grade is given in Table 1. There was no significant difference between primary sites or between grades.
MGMT promoter methylation. Molecular studies could not be performed in eight cases, because of insufficient DNA quality after extraction from FFPE tissues. MGMT promoter methylation was detected in 12 cases (12%) by MS-PCR. By pyrosequencing, the rate of promoter methylation ranged from 1 to 46% in the whole series (median: 5%); 24 cases (24%) met our criteria to be declared ‘methylated’ (promoter methylation >8%). Among the 47 cases in which MGMT promoter analysis by MS-PCR could be repeated after DNA extraction from fresh frozen tissue samples, 13 cases (27.6%) were considered methylated, among which 2 were not detected by previous FFPE sample examination.
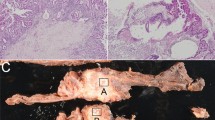
MGMT protein expression. Immunohistochemical evaluation for MGMT protein expression (Figure 1) was not interpretable in 18 cases, mainly because of the lack of positive internal controls (Figure 1C) or because an ambiguous tumour cell labelling, due to a very weak intensity of the nuclear staining. Among the 89 tumours interpretable, the number of positive tumour cells and their apparent staining intensity were variable from one case to another (Figure 1A and D). In addition, the distribution of positive tumour cells was markedly heterogeneous in some cases (Figure 1D). In an attempt to identify only tumours with low MGMT activity, we scored negative only the tumours with less than 10% of positive tumour cells in the whole tumour tissue section. In all, 29 tumours (33%) were scored negative (Figure 1B).

Immunostaining for MGMT protein. Representative examples of strong positive nuclear staining of tumour cells in a pancreatic NET (A) and of unambiguous negative staining of an ileal NET (B) are shown. Note in B the nuclear staining of lymphoid cells (L) and endothelial cells (arrows), which serve as internal positive controls (B). (C) An example of non-interpretable staining in an ileal NET: despite a very faint positivity observed in some tumour cell nuclei (open arrowheads), the absence of any positive internal control, especially in endothelial cells (arrows), prevents the definitive interpretation of the case. In D, an example of highly heterogeneous pancreatic NET is shown, with two distinct tumour cell populations, one with a faint nuclear labelling (arrows) and the other with no detectable labelling. Immunoperoxidase with nuclear counterstaining using Mayer’s haematoxylin. Original magnifications: A, × 120; B, × 350; C, × 380; D, × 240; scale bar=50 μm.
Comparison between the techniques used for MGMT status evaluation. We first examined (Table 2) the concordance between MS-PCR and pyrosequencing for evaluation of MGMT promoter methylation in FFPE tissues. Results were concordant in 87 of 99 cases examined (88%; κ of 0.602). All cases in which MGMT promoter was considered non-methylated by pyrosequencing were also considered non-methylated by MS-PCR.
We next examined the concordance between molecular studies and immunohistochemistry. Concordance rate was slightly higher between pyrosequencing and immunohistochemistry (89%, 72 out of 81 tumours with both results available, κ of 0.731, and 72 out of 107 (67%) of all specimens) than between MS-PCR and immunochemistry (84%, 68 of 81 tumours with both results available, κ of 0.561, and 68 out of 107 (64%) of all specimens). However, whereas all tumours in which MGMT promoter was considered methylated by MS-PCR scored negative by immunohistochemistry, three tumours in which MGMT promoter was considered methylated by pyrosequencing (with CpG island methylation rates of respectively, 10%, 11% and 13%) scored positive by immunohistochemistry.
Evaluation of MGMT status as a prognostic marker
The 107 patients were included in this part of the study. The following data were analysed: age at diagnosis, gender, MEN1 syndrome, primary tumour location (gastrointestinal, pancreas, lung, unknown), functional status, histological grade, WHO classification, MGMT status, metastatic status and number of metastatic sites. In univariate analysis, MGMT promoter methylation determined by either MS-PCR or pyrosequencing was found not to be a prognostic factor of OS from diagnosis (respectively, P=0.38 and P=0.49; n=99). In the same way, loss of MGMT expression by immunochemistry had no prognostic value in univariate analysis (P=0.15; n=89). In contrast, three parameters were of prognostic significance in both univariate and multivariate analysis: primary tumour resection (HR=6.9 (2.3–20.8), P<0.001), low tumour grade (HR=0.14 (0.04–0.54) for high grade, P=0.004) and localised disease (HR=0.15 (0.02–0.97) for metastatic disease, P=0.047). The number of metastatic sites was the fourth factor associated with poorer prognosis in univariate analysis but it did not remain significant in multivariate analysis.
Evaluation of MGMT status as a predictive marker of response to alkylating agents
Fifty-three patients were included in this part of the study. All received at least one chemotherapy regimen including an alkykating agent; 16 received two successive regimens. In total, there were 69 chemotherapy regimens evaluable (34 dacarbazine-based, 20 streptozotocin-based and 15 temozolomide-based treatments). The description and efficacy by regimens are shown in Table 3. Streptozotocin-based chemotherapy was given as first-line treatment in 50% of cases, whereas temozolomide-based treatment was given as a third-line or even more advanced line of treatment in 60% of cases. The median number of cycles administered to each patient was 6 (range: 1–37). Objective response and stable disease were obtained in 20% (14/69) and 64% (44/69) of patients, respectively, after alkylant use. The median PFS and OS were, respectively, 14 (9–18) and 45 (29–62) months.
We first verified whether MGMT status was associated with tumour response. Patients with methylated MGMT promoter in tumour tissue assessed by pyrosequencing had a 50% (8 out of 16) rate of objective response, in contrast to 11% (5 out of 47) for the other patients (P=0.003; Table 4). OR was achieved in 62% of patients with tumours scored as MGMT negative by immunohistochemistry as compared with 7% patients with MGMT-positive tumours (P<0.001). The nine patients with progressive disease had MGMT-positive tumours. Three patients with MGMT-positive tumours responded to treatment but they received the alkylating agent in combination with another drug. Despite the fact that the number of methylated cases and the sensitivity to alkylating agents were higher in pancreatic NETs than in gastrointestinal NETs, the association between MGMT status and response to alkylating agents was verified in patients with either gastrointestinal or pancreatic NETs (Table 5).
We then evaluated the association of MGMT status with PFS and OS from first alkylant use in multivariate analysis. Median PFS was found to be significantly longer for patients with MGMT promoter methylation in tumour tissue assessed by pyrosequencing (26.4 vs 10.8 months, HR=0.29 (0.13–0.64), P=0.002; Figure 2A). In multivariate analysis, no other factor tested was significantly associated with PFS. The difference in PFS according to MGMT status was verified for streptozotocin-based treatments (P=0.04) as well as for dacarbazine-based treatments (P=0.004; Table 4); it was not significant (P=0.23) for temozolomide-based treatments considered alone, but statistical significance could be reached by grouping temozolomide-based and dacarbazine-based treatments (P=0.003), which is justified as the two drugs act through the same metabolite. Median PFS was also significantly longer for patients with MGMT-negative tumours by immunohistochemistry (20.2 vs 7.6 months, HR=0.19 (0.08–0.48), P<0.001; Figure 2B). Finally, OS from first alkylant use was significantly longer in patients with MGMT promoter methylation assessed by pyrosequencing (77 vs 43 months, P=0.026) and in patients with MGMT-negative tumours by immunohistochemistry (105 vs 34 months, P=0.006; Figure 2C and D). No significant correlation was found between PFS or OS and MGMT promoter methylation assessed by MS-PCR (data not shown). We also did the analysis, with similar results (Table 4), at the patient-level in keeping only the first treatment of patients who received two treatments.

Progression-free survival (A and B) and overall survival (C and D) from treatment initiation in patients treated by alkylating agents. In A and C, patients are compared with MGMT promoter methylation in tumour tissue, assessed by pyrosequencing (group 1: patients with methylated MGMT; group 2: patients with unmethylated MGMT). In B and D, patients are compared according to MGMT protein expression, assessed by immunohistochemistry (group 1: patients with MGMT-negative tumours; group 2: patients with MGMT-positive tumours).
Association between MGMT status and promoter methylation in a gene panel
The occurrence of promoter methylation for each of the 12 genes studied by MS-PCR after DNA extraction from fresh frozen tissues in 47 patients of the study group is given in Supplementary Appendix 2. As expected, HIC, RASSFA1 and APC were the most frequently methylated genes, in 98%, 74.5% and 36% of tumours, respectively. MGMT promoter hypermethylation was detected in 13 cases (27.5%).
The distribution of the 47 tumours tested according to the number of methylated genes is given in Figure 3A. Median OS was significantly shorter in patients with tumours harbouring >3 genes methylated (log-rank test, P=0.017; Figure 3B).

Hypermethylated phenotype in NETs. In A, the distribution of tumours according to the number of methylated genes detected in tumour tissue is given. Open bars represent cases with methylated MGMT; solid bars represent cases with non-methylated MGMT. In B, overall survival curves from diagnosis are compared between patients with tumours containing ⩽3 methylated genes (group 1) and those with tumours containing >3 methylated genes (group 2). The difference is statistically significant (log-rank test, P=0.017). In C, overall survival curves from diagnosis are compared for the subset of patients with tumours containing >3 methylated genes according to their MGMT status (group 1: methylated MGMT, group 2: non-methylated MGMT). The difference is not statistically significant.
MGMT promoter methylation was present in 10 out of 17 tumours with >3 genes methylated, as compared with only 3 out of 30 tumours with ⩽3 genes methylated; the difference was statistically significant (χ2 test, P=0.001; Figure 3A). Among the 17 patients with tumours harbouring >3 genes methylated, median OS was not different according to MGMT status (log-rank test, P=0.925; Figure 3C).
Discussion
In this study, we confirm that MGMT alterations, including MGMT promoter methylation and/or loss of MGMT protein expression, could be detected in a significant proportion of NETs, including not only pancreatic NETs but also NETs of various other origins. These alterations were associated with tumour response to alkylating agents, used alone or in combination, and including not only temozomolide but also dacarbazine (a precursor of the same active metabolite than temozolomide) and streptozotocin. However, they were not a prognostic factor of OS from diagnosis. Finally, we show that MGMT alterations are associated with a high number of methylated genes in tumour tissue.
For MGMT status analysis, we combined two molecular techniques for evaluation of MGMT promoter methylation, MS-PCR and pyrosequencing, along with immunohistochemistry for assessment of MGMT protein. Such a combined strategy is used in gliomas (Karayan-Tapon et al, 2010; Quillien et al, 2012), but so far, only one report has appeared in NETs (Schmitt et al, 2014). As expected, the various techniques employed had different sensitivities and provided different types of information (Wick et al, 2014).
With MS-PCR, we detected MGMT promoter methylation in 12% of tumours, irrespective of their origin. This figure is within the range of 6–16% reported for all NETs in previous studies using the same technique (Liu et al, 2005; Arnold et al, 2007). The range of variation is higher in some NET subsets: for instance, the rate of detection varied from 0 (Chan et al, 2003) to 40% (House et al, 2003) in pancreatic NETs and from 0 (Liu et al, 2005) to 12.5% (Toyooka et al, 2001) in lung NETs. In literature, MGMT promoter methylation was rarely detected in ileal NETs, but could be found in up to 25% of cases from the other digestive segments (Chan et al, 2003; Liu et al, 2005).
We then compared conventional MS-PCR with the more recently developed pyrosequencing technique (Wick et al, 2014). We verified that, as observed in other tumours (Karayan-Tapon et al, 2010, Quillien et al, 2012), pyrosequencing is more sensitive than MS-PCR to detect MGMT promoter methylation in FFPE tissue samples: in our study, the rate of detection was 24% using pyrosequencing as compared with 12% using MS-PCR. Pyrosequencing results have to be interpreted by reference to the upper limit of ‘normal’ methylation accepted for the gene considered: in our study, we retain the same standard (8%) as for central nervous tumours (Karayan-Tapon et al, 2010; Quillien et al, 2012), as it was corroborated by our tests in normal, age-matched gastrointestinal tissues.
Finally, we evaluated MGMT protein expression. Only three previous studies have addressed this issue, usually in selected (mainly pancreatic) NET subsets, and with different interpretation criteria (Ekeblad et al, 2007; Kulke et al, 2009; Schmitt et al, 2014). In our series, MGMT protein was undetectable in 33% of tumours, and the proportion was comparable for all NET subsets, including pancreatic (36%), lung (33%) and gastrointestinal (24%) tumours. Therefore, we could not verify the findings of the previous study of Kulke et al (2009), in which the absence of detectable MGMT protein was restricted to pancreatic NETs: this difference may be related to sample bias in a relatively short series and/or to differences in the immunohistochemical technique used to evaluate MGMT protein expression.
The use of immunohistochemistry for evaluating MGMT status in NETs has been advocated because it is easily performed in FFPE tissue sections. However, numerous issues have to be addressed before MGMT immunostaining might be proposed as a routine test and a reliable alternative to molecular techniques. A first issue is the only partial concordance between MGMT promoter methylation and loss of MGMT protein expression. This point has been verified in our study as in all previous ones in many tumour types (Hegi et al, 2005; Weller et al, 2010; Quillien et al, 2012), including in NETs (Schmitt et al, 2014). This is likely to be due to the existence of various regulatory mechanisms of MGMT expression, in addition to promoter methylation (Wick et al, 2014). A second issue is the presence of several pitfalls in the interpretation and standardisation of MGMT immunohistochemistry. In a significant proportion of our cases, it was not possible to obtain unambiguous internal positive controls, especially in archival tissues stored for long periods of time. In other cases, tumour cell nuclear staining was faint and disputable. Even in interpretable cases, as previously underlined (Ekeblad et al, 2007), the distribution of positive tumour cells may be heterogeneous. In the absence of recommendations for the interpretation of MGMT immunostaining, we decided to score negative only tumours with a very low percentage (<10%) of positive cells over the whole tumour tissue section, in order to be the most discriminant possible.
We then aimed at evaluating the prognostic and predictive relevance of MGMT alterations in NETs. In our retrospective series, we could not demonstrate any relation between MGMT status and OS from diagnosis; this is in contrast with results reported in other tumours, such as gliomas (Gorlia et al, 2008, Weller et al, 2009), and in one previous study of NETs, but based on a very limited sample (Kulke et al, 2009). In a recent study (Schmitt et al, 2014), loss of MGMT protein was correlated with poor outcome but was not independent from other known histoprognostic factors, such as grade and stage in multivariate analysis (Schmitt et al, 2014).
In several tumour types, it has been suggested that, despite its lack of overall prognostic relevance, MGMT status might be predictive of the response to the alkylating agent temozomolide. Previous studies in NETs reported contrasting results. In one study (Ekeblad et al, 2007), no relation was found between MGMT status and response to temozolomide. In two other ones (Kulke et al, 2009; Schmitt et al, 2014), a predictive value was found, but only for pancreatic NETs. In our series, both MGMT promoter methylation, assessed by pyrosequencing, and MGMT protein status were found to be associated with response to alkylating agents, as shown by significant differences in PFS and OS after first alkylant use according to MGMT status. Two of the main interests of our study are: (a) to strongly suggest that MGMT status may be predictive of response not only to temozolomide, but also to other alkylating agents used in the treatment of NETs, including dacarbazine, which has the same active metabolite than temozolomide, and streptozotocin, which remains one the main first-line treatments for advanced pancreatic NETs; (b) to suggest that this predictive value is not restricted to pancreatic NETs but might be verified in all NET subsets. However, our results must be interpreted with some caution because of the retrospective design of our study and the fact that alkylating agents have been used either alone or in combination, making it difficult to properly assess their contribution to treatment efficacy. Further prospective studies are required to confirm our findings. One important further question would be to evaluate to which extent MGMT status is correlated between the primary tumour and its metastases, which are the actual target of systemic chemotherapy. In one previous study, based on a small number of cases, MGMT methylation was found to be comparable in primary NETs and their secondary locations (House et al, 2003), whereas in another one (Christmann et al, 2011), MGMT activity has been shown to be higher in metastases than in primaries.
In many tumours, MGMT promoter methylation is known to be associated with a ‘hypermethylator’ phenotype. In our series, MGMT methylation was found to be much more frequent in NETs harbouring a high number of methylated genes (>3). In keeping with all previous studies (Toyooka et al, 2001; Chan et al, 2003; House et al, 2003; Liu et al, 2005; Arnold et al, 2007), we found that the presence of a high number of methylated genes (suggestive of a so-called ‘hypermethylator’ phenotype, even if no consensus definition of this term is available for NETs) is associated with more progressive disease and shorter survival. Our findings therefore suggest that MGMT methylation is part of a ‘hypermethylator’ phenotype, which in turn, behaves as an adverse prognostic factor. This may explain the apparent discrepancy between the absence of prognostic relevance of MGMT status and its predictive value.
In conclusion, the evaluation of MGMT status in NETs opens new perspectives for a better use of alkylating agents. However, much remains to be done for the standardisation of the techniques and of their interpretation: this is necessary for the prospective, multicentric studies required to verify the encouraging, but preliminary results reported here.
Change history
03 February 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Arnold CN, Sosnowski A, Schmitt-Graff A, Arnold R, Blum HE (2007) Analysis of molecular pathways in sporadic neuroendocrine tumors of the gastro-entero-pancreatic system. Int J Cancer 120: 2157–2164.
Auernhammer CJ, Goke B (2011) Therapeutic strategies for advanced neuroendocrine carcinomas of jejunum/ileum and pancreatic origin. Gut 60: 1009–1021.
Bajetta E, Ferrari L, Procopio G, Catena L, Ferrario E, Martinetti A, Di Bartolomeo M, Buzzoni R, Celio L, Vitali M, Beretta E, Seregni E, Bombardieri E (2002) Efficacy of a chemotherapy combination for the treatment of metastatic neuroendocrine tumours. Ann Oncol 13: 614–621.
Chan AO, Kim SG, Bedeir A, Issa JP, Hamilton SR, Rashid A (2003) CpG island methylation in carcinoid and pancreatic endocrine tumors. Oncogene 22: 924–934.
Chan JA, Blaszkowsky L, Stuart K, Zhu AX, Allen J, Wadlow R, Ryan DP, Meyerhardt J, Gonzalez M, Regan E, Zheng H, Kulke MH (2013) A prospective, phase 1/2 study of everolimus and temozolomide in patients with advanced pancreatic neuroendocrine tumor. Cancer 119: 3212–3218.
Chan JA, Stuart K, Earle CC, Clark JW, Bhargava P, Miksad R, Blaszkowsky L, Enzinger PC, Meyerhardt JA, Zheng H, Fuchs CS, Kulke MH (2012) Prospective study of bevacizumab plus temozolomide in patients with advanced neuroendocrine tumors. J Clin Oncol 30: 2963–2968.
Christmann M, Verbeek B, Roos WP, Kaina B (2011) O(6)-Methylguanine-DNA methyltransferase (MGMT) in normal tissues and tumors: enzyme activity, promoter methylation and immunohistochemistry. Biochim Biophys Acta 1816: 179–190.
Curtin K, Slattery ML, Samowitz WS (2011) CpG island methylation in colorectal cancer: past, present and future. Patholog Res Int 2011: 902674.
Delaunoit T, Ducreux M, Boige V, Dromain C, Sabourin JC, Duvillard P, Schlumberger M, de Baere T, Rougier P, Ruffie P, Elias D, Lasser P, Baudin E (2004) The doxorubicin-streptozotocin combination for the treatment of advanced well-differentiated pancreatic endocrine carcinoma; a judicious option? Eur J Cancer 40: 515–520.
Dong SM, Lee EJ, Jeon ES, Park CK, Kim KM (2005) Progressive methylation during the serrated neoplasia pathway of the colorectum. Mod Pathol 18: 170–178.
Ekeblad S, Sundin A, Janson ET, Welin S, Granberg D, Kindmark H, Dunder K, Kozlovacki G, Orlefors H, Sigurd M, Oberg K, Eriksson B, Skogseid B (2007) Temozolomide as monotherapy is effective in treatment of advanced malignant neuroendocrine tumors. Clin Cancer Res 13: 2986–2991.
Gorlia T, van den Bent MJ, Hegi ME, Mirimanoff RO, Weller M, Cairncross JG, Eisenhauer E, Belanger K, Brandes AA, Allgeier A, Lacombe D, Stupp R (2008) Nomograms for predicting survival of patients with newly diagnosed glioblastoma: prognostic factor analysis of EORTC and NCIC trial 26981-22981/CE.3. Lancet Oncol 9: 29–38.
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R (2005) MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 352: 997–1003.
House MG, Herman JG, Guo MZ, Hooker CM, Schulick RD, Lillemoe KD, Cameron JL, Hruban RH, Maitra A, Yeo CJ (2003) Aberrant hypermethylation of tumor suppressor genes in pancreatic endocrine neoplasms. Ann Surg 238: 423–431.
Karayan-Tapon L, Quillien V, Guilhot J, Wager M, Fromont G, Saikali S, Etcheverry A, Hamlat A, Loussouarn D, Campion L, Campone M, Vallette FM, Gratas-Rabbia-Ré C (2010) Prognostic value of O6-methylguanine-DNA methyltransferase status in glioblastoma patients, assessed by five different methods. J Neurooncol 97: 311–322.
Koumarianou A, Antoniou S, Kanakis G, Economopoulos N, Rontogianni D, Ntavatzikos A, Tsavaris N, Pectasides D, Dimitriadis G, Kaltsas G (2012) Combination treatment with metronomic temozolomide, bevacizumab and long-acting octreotide for malignant neuroendocrine tumours. Endocr Relat Cancer 19: L1–L4.
Kulke MH, Anthony LB, Bushnell DL, de Herder WW, Goldsmith SJ, Klimstra DS, Marx SJ, Pasieka JL, Pommier RF, Yao JC, Jensen RT North American Neuroendocrine Tumor Society (NANETS) (2010) NANETS treatment guidelines: well-differentiated neuroendocrine tumors of the stomach and pancreas. Pancreas 39: 735–752.
Kulke MH, Hornick JL, Frauenhoffer C, Hooshmand S, Ryan DP, Enzinger PC, Meyerhardt JA, Clark JW, Stuart K, Fuchs CS, Redston MS (2009) O6-Methylguanine DNA methyltransferase deficiency and response to temozolomide-based therapy in patients with neuroendocrine tumors. Clin Cancer Res 15: 338–345.
Kulke MH, Stuart K, Enzinger PC, Ryan DP, Clark JW, Muzikansky A, Vincitore M, Michelini A, Fuchs CS (2006) Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J Clin Oncol 24: 401–406.
Liu L, Broaddus RR, Yao JC, Xie S, White JA, Wu TT, Hamilton SR, Rashid A (2005) Epigenetic alterations in neuroendocrine tumors: methylation of RAS-association domain family 1, isoform A and p16 genes are associated with metastasis. Mod Pathol 18: 1632–1640.
Modlin IM, Oberg K, Chung DC, Jensen RT, de Herder WW, Thakker RV, Caplin M, Delle Fave G, Kaltsas GA, Krenning EP, Moss SF, Nilsson O, Rindi G, Salazar R, Ruszniewski P, Sundin A (2008) Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol 9: 61–72.
Naumann SC, Roos WP, Jost E, Belohlavek C, Lennerz V, Schmidt CW, Christmann M, Kaina B (2009) Temozolomide- and fotemustine-induced apoptosis in human malignant melanoma cells: response related to MGMT, MMR, DSBs, and p53. Br J Cancer 100: 322–333.
Park SJ, Rashid A, Lee JH, Kim SG, Hamilton SR, Wu TT (2003) Frequent CpG island methylation in serrated adenomas of the colorectum. Am J Pathol 162: 815–822.
Pizzi S, Azzoni C, Bottarelli L, Campanini N, D’Adda T, Pasquali C, Rossi G, Rindi G, Bordi C (2005) RASSF1A promoter methylation and 3p21.3 loss of heterozygosity are features of foregut, but not midgut and hindgut, malignant endocrine tumours. J Pathol 206: 409–416.
Pizzi S, Azzoni C, Tamburini E, Bottarelli L, Campanini N, D’Adda T, Fellegara G, Luong TV, Pasquali C, Rossi G, Delle Fave G, Camisa R, Bordi C, Rindi G (2008) Adenomatous polyposis coli alteration in digestive endocrine tumours: correlation with nuclear translocation of beta-catenin and chromosomal instability. Endocr Relat Cancer 15: 1013–1024.
Quillien V, Lavenu A, Karayan-Tapon L, Carpentier C, Labussiere M, Lesimple T, Chinot O, Wager M, Honnorat J, Saikali S, Fina F, Sanson M, Figarella-Branger D (2012) Comparative assessment of 5 methods (methylation-specific polymerase chain reaction, MethyLight, pyrosequencing, methylation-sensitive high-resolution melting, and immunohistochemistry) to analyze O6-methylguanine-DNA-methyltranferase in a series of 100 glioblastoma patients. Cancer 118: 4201–4211.
Rindi G, Klimstra DS, Arnold R, Kloppel G, Bosman FT, Komminoth P, Capella C, Solcia E (2010) Nomenclature and classification of neuroendocrine neoplasms of the digestive system. In WHO Classification of Tumours of the Digestive System, FT Bosman, F Carneiro, RH Hruban, ND Theise, (eds), pp 13–14. IARC: Lyon.
Rindi G, Kloppel G, Alhman H, Caplin M, Couvelard A, de Herder WW, Erikssson B, Falchetti A, Falconi M, Komminoth P, Körner M, Lopes JM, McNicol AM, Nilsson O, Perren A, Scarpa A, Scoazec JY, Wiedenmann B (2006) TNM staging of foregut (neuro)endocrine tumors: a consensus proposal including a grading system. Virchows Arch 449: 395–401.
Rindi G, Kloppel G, Couvelard A, Komminoth P, Korner M, Lopes JM, McNicol AM, Nilsson O, Perren A, Scarpa A, Scoazec JY, Wiedenmann B (2007) TNM staging of midgut and hindgut (neuro) endocrine tumors: a consensus proposal including a grading system. Virchows Arch 451: 757–762.
Schmitt AM, Pavel M, Rudolph T, Dawson H, Blank A, Komminoth P, Vassella E, Perren A (2014) Prognostic and predictive roles of MGMT protein expression and promoter methylation in sporadic neuroendocrine neoplasms. Neuroendocrinology 100: 35–44.
Schraml P, von Teichman A, Mihic-Probst D, Simcock M, Ochsenbein A, Dummer R, Michielin O, Seifert B, Schlappi M, Moch H, von Moos R (2012) Predictive value of the MGMT promoter methylation status in metastatic melanoma patients receiving first-line temozolomide plus bevacizumab in the trial SAKK 50/07. Oncol Rep 28: 654–658.
Sobin LH, Gospodarowicz MK, Wittekind C (2009) TNM Classification of Malignant Tumours 7th edn. Wiley: New York.
Strosberg JR, Fine RL, Choi J, Nasir A, Coppola D, Chen DT, Helm J, Kvols L (2011) First-line chemotherapy with capecitabine and temozolomide in patients with metastatic pancreatic endocrine carcinomas. Cancer 117: 268–275.
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92: 205–216.
Toyooka S, Toyooka KO, Maruyama R, Virmani AK, Girard L, Miyajima K, Harada K, Ariyoshi Y, Takahashi T, Sugio K, Brambilla E, Gilcrease M, Minna JD, Gazdar AF (2001) DNA methylation profiles of lung tumors. Mol Cancer Ther 1: 61–67.
Walter T, Brixi-Benmansour H, Lombard-Bohas C, Cadiot G (2012) New treatment strategies in advanced neuroendocrine tumours. Dig Liver Dis 44: 95–105.
Walter T, Bruneton D, Cassier PA, Hervieu V, Pilleul F, Scoazec JY, Chayvialle JA, Lombard-Bohas C (2010) Evaluation of the combination 5-fluorouracil, dacarbazine, and epirubicin in patients with advanced well-differentiated neuroendocrine tumors. Clin Colorectal Cancer 9: 248–254.
Weatherstone K, Meyer T (2012) Streptozocin-based chemotherapy is not history in neuroendocrine tumours. Target Oncol 7: 161–168.
Welin S, Sorbye H, Sebjornsen S, Knappskog S, Busch C, Oberg K (2011) Clinical effect of temozolomide-based chemotherapy in poorly differentiated endocrine carcinoma after progression on first-line chemotherapy. Cancer 117: 4617–4622.
Weller M, Felsberg J, Hartmann C, Berger H, Steinbach JP, Schramm J, Westphal M, Schackert G, Simon M, Tonn JC, Heese O, Krex D, Nikkhah G, Pietsch T, Wiestler O, Reifenberger G, von Deimling A, Loeffler M (2009) Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J Clin Oncol 27: 5743–5750.
Weller M, Stupp R, Reifenberger G, Brandes AA, van den Bent MJ, Wick W, Hegi ME (2010) MGMT promoter methylation in malignant gliomas: ready for personalized medicine? Nat Rev Neurol 6: 39–51.
Wick W, Weller M, van den Bent M, Sanson M, Weiler M, von Deimling A, Plass C, Hegi M, Platten M, Reifenberger G (2014) MGMT testing—the challenges for biomarker-based glioma treatment. Nat Rev Neurol 10: 372–385.
Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, Abdalla EK, Fleming JB, Vauthey JN, Rashid A, Evans DB (2008) One hundred years after ‘carcinoid’: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 26: 3063–3072.
Zhang HY, Rumilla KM, Jin L, Nakamura N, Stilling GA, Ruebel KH, Hobday TJ, Erlichman C, Erickson LA, Lloyd RV (2006) Association of DNA methylation and epigenetic inactivation of RASSF1A and beta-catenin with metastasis in small bowel carcinoid tumors. Endocrine 30: 299–306.
Acknowledgements
C Vercherat is recipient of a post-doctoral grant from LYRIC (Site de Recherche Intégrée sur le Cancer, Lyon, grant INCa-DGOS 4664).
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Walter, T., van Brakel, B., Vercherat, C. et al. O6-Methylguanine-DNA methyltransferase status in neuroendocrine tumours: prognostic relevance and association with response to alkylating agents. Br J Cancer 112, 523–531 (2015). https://doi.org/10.1038/bjc.2014.660
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.660
Keywords
This article is cited by
-
MGMT is frequently inactivated in pancreatic NET-G2 and is associated with the therapeutic activity of STZ-based regimens
Scientific Reports (2023)
-
CT-based nomogram development and validation to predict SSTR2, VEGFR2 and MGMT expression for pancreatic neuroendocrine neoplasms
Chinese Journal of Academic Radiology (2023)
-
Systemische Therapien neuroendokriner Tumoren und Karzinome
Im Fokus Onkologie (2021)
-
Clinical and in vitro studies of the correlation between MGMT and the effect of streptozocin in pancreatic NET
Cancer Chemotherapy and Pharmacology (2019)
-
The Role of Cytotoxic Chemotherapy in Well-Differentiated Gastroenteropancreatic and Lung Neuroendocrine Tumors
Current Treatment Options in Oncology (2019)
