
Abstract
Background:
Cachexia in both mice and humans is associated with tumour production of a sulphated glycoprotein called proteolysis-inducing factor (PIF). In mice PIF binds with high affinity to a surface receptor in skeletal muscle, but little is known about the human receptor. This study compares the human PIF receptor with the murine.
Methods:
Human PIF was isolated from the G361 melanoma and murine PIF from the MAC16 colon adenocarcinoma. The human PIF receptor was isolated from human skeletal muscle myotubes. Protein synthesis and degradation induced by human and murine PIF was studied in human and murine skeletal muscle myotubes.
Results:
Both the human and murine PIF receptors showed the same immunoreactivity and Mr 40 000. Both murine and human PIF inhibited total protein synthesis and stimulated protein degradation in human and murine myotubes to about the same extent, and this was attenuated by a rabbit polyclonal antibody to the murine PIF receptor, but not by a non-specific rabbit antibody. Both murine and human PIF increased the activity of the ubiquitin–proteasome pathway in both human and murine myotubes, as evidenced by an increased ‘chymotrypsin-like’ enzyme activity, protein expression of the 20S and 19S proteasome subunits, and increased expression of the ubiquitin ligases MuRF1 and MAFbx, and this was also attenuated by the anti-mouse PIF receptor antibody.
Conclusions:
These results suggest that the murine and human PIF receptors are identical.
Similar content being viewed by others


Main
Patients with cancer frequently experience cachexia, a wasting syndrome characterised by massive losses of both muscle and fat. In lung cancer patients muscle mass decreased by 75%, and total body fat by 85% when they had lost 32% of their pre-illness stable weight (Fearon, 1992). Muscle atrophy causes weakness, reducing the ability of the patient to carry out normal activities, while death occurs when the weight loss reaches 25%–30% (Wigmore et al, 1997).
We have shown that both murine and human tumours, which induce muscle atrophy elaborate a catabolic glycoprotein called proteolysis-inducing factor (PIF; Todorov et al, 1996). Proteolysis-inducing factor expression was associated with an increased rate of weight loss in patients with a tumour of the head of the pancreas (Wigmore et al, 2000), and in patients with non-small-cell lung cancer (NSCLC; Wang et al, 2010). In patients with NSCLC PIF expression was negatively related to the survival of the patients (Wang et al, 2010). Proteolysis-inducing factor contains one O-linked and one N-linked sulphated oligosaccharide chain (Todorov et al, 1997), which would facilitate its high affinity binding to a surface receptor of Mr 40 000 in skeletal muscle (Todorov et al, 2007). Antiserum to the N-terminal 19 amino acids of the PIF receptor (PIFR) attenuated protein degradation initiated by PIF in vitro, as well as loss of skeletal muscle and body weight in mice bearing the cachexia-inducing MAC16 tumour (Todorov et al, 2007). The N-terminal peptide also attenuated protein degradation induced by PIF in vitro (Mirza et al, 2011). These results suggest that the receptor is linked to the signalling system that initiates protein degradation in response to PIF.
Since the PIFR may play a central role in the initiation of muscle atrophy in cancer cachexia it could act as a potential target for agents having a therapeutic role in this condition, particularly since antibodies to the PIFR attenuated muscle wasting in a pre-clinical model of cachexia (Todorov et al, 2007). The aim of this study was to determine if the human PIFR was similar to the mouse in terms of immunoreactivity of the N-terminal sequence, to find out if the mouse is a suitable model for pre-clinical testing of PIFR antagonists. The potential application of antibodies to the PIFR for the treatment of cancer cachexia has been previously published (Tisdale et al, 2008).
Materials and methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM), McCoys 5a medium and horse serum (HS) were purchased from PAA Laboratories (Somerset, UK), and fetal bovine (FBS) from Biosera (East Sussex, UK). L-[2,3,4,5,6-3H] Phenylalanine (sp.act.4.669 TBq mmol−1) was from Perkin Elmer Ltd (Cambridge, UK). The chymotrypsin substrate (N-Succinyl-Leu-Leu-Val-Tyr-7-amido-4-methylcoumarin) was obtained from Sigma (Dorset, UK). The human melanoma cell line, G361 was from the European Collection of Cell Cultures (Wilts, UK), and the human skeletal muscle myoblasts together with their growth and differentiation medium were from Zen-Bio, Inc. (Durham, NC, USA). Mouse monoclonal antibodies to 20S proteasome α-subunits and p42 were from Affinite Research Products (Exeter, UK), while goat polyclonal antibody to MuRF1 and rabbit polyclonal antibody to MAFbx and β-actin were from Santa Cruz Biotechnology (Heidelberg, Germany). Peroxidase-conjugated donkey antirabbit and sheep anti-mouse antibodies were purchased from GE Healthcare (Bucks, UK) as was the enhanced chemiluminescence (ECL) development hits. Peroxidase-conjugated donkey antigoat antibody was from Abcam (Cambridge, UK), and nitrocellulose membranes were from VWR (Bucks, UK). Polyclonal antisera to the first 19 amino acids of the N-terminal sequence of the murine PIFR (mPIFR; Todorov et al, 2007) was generated in rabbits by Severn Biotech (Worcester, UK).
Cell culture
Human skeletal muscle myoblasts were maintained in growth medium under an atmosphere of 10% CO2 in air at 37°C. When 80% confluency was achieved they were transferred to differentiation medium, with medium changes every 2 days. Differentiation was complete in 6 days. The conditions for C2C12 murine myoblasts were similar except that cells were maintained in DMEM supplemented with 10% FBS and glutamine, under an atmosphere of 10% CO2 in air at 37°C, and differentiation was achieved in DMEM containing 2% HS in 3–5 days.
G361 cells were maintained in McCoys 5a medium supplemented with 10% FBS in a humidified atmosphere of 10% CO2 in air at 37°C. All experiments were performed on cells in the subconfluent state.
Purification of PIF
Murine PIF was isolated from solid MAC16 tumours excised from mice with established weight loss as previously described in detail (Mirza et al, 2011). Since PIF has been shown to be resistant to pronase (Todorov et al, 1996) the bulk protein was reduced by pronase digestion and the PIF was isolated by adhesion to DEAE cellulose and elution with 0.3 M NaCl. Human PIF was isolated from G361 cells by a similar methodology as previously reported (Todorov et al, 1999), except that ammonium sulphate precipitation was replaced by pronase digestion.
Isolation of PIFR
Human and mouse PIFR was isolated from human skeletal muscle myoblasts and C2C12 myoblasts as previously reported (Todorov et al, 2007). The PIFR was purified by wheat germ agglutinin (WGA) agarose affinity chromatography after addition of PIF, based on the observation (Todorov et al, 1996) that this lectin will bind the oligosaccharide chain of PIF.
Measurement of 20S proteasome activity
The functional activity of the 20S proteasome was determined as the ‘chymotrypsin-like’ enzyme activity as previously described (Whitehouse and Tisdale, 2003). Activity was measured by the release of aminomethyl coumarin from a fluorogenic peptide in the absence and presence of the specific proteasome inhibitor lactacystin (10 μ M). Only the activity suppressed by lactacystin was considered to be proteasome specific.
Western blotting
Myotubes were scraped from the substratum and washed with PBS prior to sonication at 4°C in 20 mM Tris-HCl (pH 7.4), 2 mM ATP, 5 mM MgCl2 and 1 mM DTT. Cytosolic protein (5 μg) formed by centrifugation at 18 000 g for 10 min at 4°C was resolved on 12% SDS–PAGE at 180 V for 1 h, followed by transference onto 0.45 μm nitrocellulose membranes at 300 mA for 1 h. The membranes were blocked with 5% Marvel in Tris-buffered saline (pH 7.5) for 1 h at room temperature. The primary antibodies to the 20S proteasome and p42 were used at a dilution of 1 : 1000, while those for MuRF1 and MAFbx were used at a dilution of 1 : 200. The secondary antibodies were used at a dilution of 1 : 5000. Incubation with the primary antibodies was overnight at room temperature, while for the secondary antibodies it was 1 h also at room temperature. Development was by ECL. Blots were scanned by a densitometer to quantify differences.
Measurement of protein synthesis
Myotubes were formed in six-well multiwell dishes, and were supplemented with DMEM without HS for 18 h before experimentation. Proteolysis-inducing factor-receptor antibody and non-specific rabbit IgG were added to the cultures 2 h before adding PIF. Protein synthesis was carried out after 4 h incubation with PIF. Cultures were supplemented with 2 μl (370 kBq) L-[2,3,4,5,6-3H] phenylalanine in 8 μl sterile PBS, and the plates were incubated for 4 h at 37°C under an atmosphere of 10% CO2 in air. Protein synthesis was linear over a 4-h period. The reaction was arrested by washing three times with 1 ml ice-cold sterile PBS. Following removal of PBS, 1 ml ice-cold 0.2 M perchloric acid was added, and the plates were kept at 4°C for 20 min. The perchloric acid was substituted with 1 ml of 0.3 M NaOH/well, and incubation was continued for 30 min at 4°C, followed by a further incubation at 37°C for 20 min. The NaOH extract was removed and combined with a further 1-ml wash of each well, and 0.5 ml of 0.2 M perchloric acid was added and left on ice for 20 min. The extract was then centrifuged at 700 g for 5 min at 4°C. The protein-containing pellet was dissolved in 1 ml of 0.3 M NaOH, and 0.5 ml of the solution was counted for radioactivity after mixing with 8 ml Ultima Gold XR scintillation fluid.
Measurement of total protein degradation in myotubes
This has previously been described in detail (Whitehouse and Tisdale, 2003). Myotubes were differentiated in six-well multiwell plates and labelled for 24 h with L-[2,3,4, 5,6 −3H] phenylalanine (2 μCi) at 37°C. Then the plates were washed extensively with PBS and incubated for 2 h in fresh medium to allow degradation of short-lived proteins. Protein degradation was determined by incubation for a further 24 h in DMEM free of phenol red, but supplemented with 2% HS, and in the presence of 2 mM phenylalanine to prevent reincorporation of the radioactivity in the absence or presence of PIF (4.2 nM). Some myotubes were also treated with anti-receptor or non-specific antibody (5 μg ml−1) for 2 h prior to PIF addition. The radioactivity released was expressed as a fraction of the total incorporated into the myotubes.
Statistical analysis
Results are expressed as mean values±s.e. for at least three replicate experiments. The significance of differences in mean values between groups was determined by one-way ANOVA followed by Tukey–Kramer multiple comparison test. P values <0.05 were considered significant.
Results
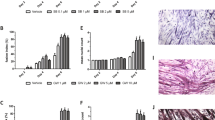
The human PIFR (hPIFR) was isolated from human skeletal muscle myotubes, produced by differentiation of skeletal muscle myoblasts in vitro, using a WGA affinity column, as previously described for the mPIFR; Todorov et al, 2007). Western blotting showed that the hPIFR showed immunoreactivity to the rabbit polyclonal antibody raised against the mPIFR (Figure 1), and that both the hPIFR and the mPIFR had the same Mr 40 000 on SDS–PAGE. We have previously reported that in skeletal muscle PIF inhibits protein synthesis, as well as stimulating protein degradation, both in vitro (Eley and Tisdale, 2007) and in vivo (Lorite et al, 1997). In this study human PIF, isolated from the G361 human melanoma (Todorov et al, 1999) and murine PIF isolated from the MAC16 colon adenocarcinoma (Todorov et al, 1997), when used at the optimum concentration (4.2 nM), previously observed (Eley and Tisdale, 2007; Whitehouse and Tisdale, 2003), both significantly inhibited protein synthesis in murine C2C12 myotubes (Figure 2A and B), and in both cases this was attenuated by a rabbit polyclonal antibody to the mPIFR, but not by a non-specific rabbit polyclonal antibody. A previous study (Todorov et al, 2007) established that a concentration of 5 μg ml−1 of antibody gave optimal results. A similar situation was observed in human myotubes (Figure 2C and D) with both murine and human PIF inhibiting protein synthesis, which was attenuated by the anti-mPIFR antibody, but not by a non-specific polyclonal antibody.

Western blot of murine (lane 1) and human (lane 2) PIF receptors on 12% SDS–PAGE using rabbit polyclonal antiserum to the murine PIF receptor. The PIF receptors were purified as detailed in methods.

Effect of anti-PIF-receptor antibody on the PIF-induced depression of protein synthesis in murine myotubes ( A ) and ( B ) and human myotubes ( C ) and ( D). Protein synthesis in C2C12 myotubes after 4 h incubation with either murine (A) or human (B) PIF (4.2 nM) and protein synthesis in human myotubes after murine (C) or human (D) PIF (4.2 nM) in the absence or presence of rabbit anti-mPIFR antibody (rAb; 5 μg ml−1) or non-specific rabbit antibody (nsAb; 5 μg ml−1). Both antibodies were added 2 h prior to PIF. The experiment was repeated three times. Differences from control are indicated as a, P<0.05, while differences in the presence of antibody are shown as d, P<0.05.
In murine myotubes both human and murine PIF also significantly stimulated total protein degradation, as determined by the release of [3H] phenylalanine from pre-labelled cells (Figure 3A and B), and in both cases this was attenuated by a polyclonal antibody to the mPIFR, but not by a non-specific polyclonal antibody. The extent of protein degradation by human and murine PIF was approximately the same. In human myotubes both murine and human PIF also initiated an increase in total protein degradation (Figure 3C and D), to about the same extent. Again the protein degradation induced by both human and murine PIF was completely attenuated by the rabbit polyclonal antibody to the mPIFR, but not by a non-specific rabbit polyclonal antibody (Figure 3C and D).

Effect of anti-PIF-receptor antibody on PIF-induced protein degradation in murine myotubes ( A ) and ( B ) and human myotubes ( C ) and ( D). Total protein degradation in C2C12 myotubes after 24 h incubation with either murine (A) or human (B) PIF (4.2 nM) and in human myotubes incubated with either murine (C) or human (D) PIF (4.2 nM) in the absence or presence of rabbit anti-murine PIFR antibody (rAb; 5 μg ml−1), or non-specific rabbit antibody (nsAb; 5 μg ml−1). Both antibodies were added 2 h prior to PIF. The experiment was repeated three times. Differences from control are indicated as b, P<0.01 or c, P<0.001, while differences in the presence of antibody are shown as e, P<0.01 or f, P<0.001.
The predominant catabolic pathway for protein degradation in skeletal muscle is the ubiquitin–proteasome pathway (Lecker et al, 1999). The functional activity of the proteasome can be measured as the ‘chymotrypsin-like’ enzyme activity, which is located on the β-5 subunit of the 20S proteasome. As with total protein degradation the proteasome ‘chymotrypsin-like’ enzyme activity was increased by both murine and human PIF in murine myotubes, and attenuated by the polyclonal antisera to the mPIFR (Figure 4A and B). Both murine and human PIF also increased the ‘chymotrypsin-like’ enzyme activity of human muscle to about the same extent (Figure 4C and D), and in both cases this was attenuated by the rabbit anti-mPIFR antibody.

Proteasome ‘chymotrypsin-like’ enzyme activity in C2C12 myotubes after treatment with either murine ( A ) or human ( B ) PIF (4.2 n M ) and in human myotubes in the presence of murine ( C ) and human ( D ) PIF (4.2 n M ) for 24 h in the absence, or presence of the anti-mPIFR antibody (rAb; 5 μ g ml−1) added 2 h prior to the PIF. The experiment was repeated three times. Differences from control are indicated as b, P<0.01 or c, P<0.001, while differences in the presence of the anti-mPIFR antibody are shown as d, P<0.05, e, P<0.01 or f, P<0.001.
Western blotting of components of the ubiquitin–proteasome pathway showed human PIF to increase expression of the 20S and 19S proteasome subunits in murine myotubes (Figure 5A and B), as well as the ubiquitin ligases MuRF1 (Figure 5C) and MAFbx (Figure 5D) and this was attenuated by the anti-murine PIF antibody. Similarly in human myotubes murine PIF increased expression of the 20S and 19S proteasome subunits (Figure 6A and B), MuRF1 (Figure 6C) and MAFbx (Figure 6D), and this was also attenuated by the anti-murine PIF antibody. These results suggest that the murine and human PIFR are essentially identical.

Western blots showing expression of the proteasome 20S α -subunit ( A ), 19S subunit ( B ), MuRF1 ( C ) and MAFbx ( D ) in C2C12 myotubes after treatment with human PIF (4.2 n M ) for 24 h in the absence or presence of anti-mPIFR antibody (5 μ g ml−1) added 2 h prior to PIF. Actin was used as a loading control. The densitometric analysis is based on three separate western blots. Differences from control are indicated as a, P<0.05 or b, P<0.01, while differences in the presence of the anti-mPIFR antibody are indicated as d, P<0.05.

Western blots showing expression of the proteasome 20S α -subunit ( A ), 19S subunit ( B ), MuRF1 ( C ) and MAFbx ( D ) in human myotubes after treatment with murine PIF (4.2 n M ) for 24 h in the absence or presence of the anti-mPIFR antibody (5 μ g ml−1) added 2 h prior to the PIF. Actin was used as loading control. The densitometric analysis is based on three separate western blots. Differences from control are indicated as a, P<0.05 or b, P<0.01, while differences in the presence of the anti-mPIFR antibody are shown as d, P<0.05.
Discussion
Both human and murine PIF are identical with regard to the central polypeptide chain (dermcidin; Lowrie et al, 2006), and their oligosaccharide chains and sulphate modifications (Todorov et al, 1997; Todorov et al, 1999). This study shows that the receptors for PIF on human and murine skeletal muscles are also identical in their molecular weights, immunoreactivity, responsiveness to depression in protein synthesis and increase in protein degradation by human and murine PIF, and in their ability to increase the expression of the major components of the ubiquitin–proteasome pathway. The full sequence of neither the human or murine PIFR is known, nor the PIF-binding site, although it is likely to be a region of basic amino acids, since PIF is highly negatively charged (Todorov et al, 1997). The PIFR belongs to a class of structurally conserved protein called DING proteins, which are spread through the animal and plant kingdom, and are highly conserved, particularly in the N-terminal 25–30 amino acids (Berna et al, 2002). Most eukaryotic DING proteins have been identified in extracellular, or cell-surface localisations, and show some structural homology with phosphate-binding proteins (Berna et al, 2002), which might explain the ability to bind PIF.
Binding of PIF to its receptor releases Ca2+ from the endoplasmic reticulum in a Zn2+-dependent process, and N-terminal peptides of the PIFR attenuated the rapid rise in intracellular Ca2+ in skeletal muscle in response to PIF (Mirza and Tisdale, 2012). The rise in Ca2+ initiates a signalling cascade leading to a depression in protein synthesis and an increase in protein degradation. This involves activation of both caspases-3 and -8, which leads to activation of the dsRNA-dependent protein kinase (PKR; Eley et al, 2010). Activation of PKR leads to a depression of global protein synthesis through increased phosphorylation of eukaryotic initiation factor 2 on the α-subunit (eIF2α), and increased protein degradation through the ubiquitin–proteasome proteolytic pathway through activation of the transcription factor nuclear factor-κB (NF-κB; Eley and Tisdale, 2007).
Since both the N-terminal peptide of the PIFR and anti-PIFR antibody attenuated protein degradation in vitro and loss of skeletal muscle and body weight in vivo in a murine model of cachexia the PIFR may be an appropriate target for the development of agents to counteract muscle wasting in cancer, and that the mouse may be an appropriate model for identifying agents likely to be effective in humans.
Change history
26 August 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Berna A, Bernier F, Scott K, Stuhlmüller B (2002) Ring up the curtain on DING proteins. FEBS Lett 524: 6–10.
Eley HL, Tisdale MJ (2007) Skeletal muscle atrophy, a link between depression of protein synthesis and increase in degradation. J Biol Chem 282: 7087–7097.
Eley HL, Russell ST, Tisdale MJ (2010) Mechanism of activation of dsRNA-dependent protein kinase (PKR) in muscle atrophy. Cell Sig 22: 783–790.
Fearon KC (1992) The mechanisms and treatment of weight loss in cancer. Proc Nutr Soc 51: 251–265.
Lecker SH, Solomon V, Mitch WE, Goldberg AL (1999) Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states. J Nutr 129: 227S–237S.
Lorite MJ, Cariuk P, Tisdale MJ (1997) Induction of muscle protein degradation by a tumour factor. Br J Cancer 76: 1035–1040.
Lowrie AG, Wigmore SJ, Wright DJ, Waddell ID, Ross JA (2006) Dermcidin expression in hepatic cells improves survival without N-glycosylation, but requires asparagine residues. Br J Cancer 94: 1663–1671.
Mirza KA, Wyke SM, Tisdale MJ (2011) Attenuation of muscle atrophy by an N-terminal peptide to the receptor for proteolysis-inducing factor (PIF). Br J Cancer 105: 83–88.
Mirza KA, Tisdale MJ (2012) Role of Ca2+ in proteolysis-inducing factor (PIF)-induced atrophy of skeletal muscle. Cell Sig 24: 2118–2122.
Tisdale MJ, Todorov P, Wyke SM (2008) Antibodies to proteolysis inducing factor (PIF) receptor and methods of use thereof. PCT W02008/071934.
Todorov PT, Cariuk P, McDevitt T, Coles B, Fearon K, Tisdale M (1996) Characterisation of a cancer cachectic factor. Nature 379: 739–742.
Todorov PT, Deacon M, Tisdale MJ (1997) Structural analysis of a tumor-produced sulphated glycoprotein capable of initiating muscle protein degradation. J Biol Chem 272: 12279–12288.
Todorov PT, Field WN, Tisdale MJ (1999) Role of a proteolysis-inducing factor (PIF) in cachexia induced by a human melanoma (G361). Br J Cancer 80: 1734–1737.
Todorov PT, Wyke SM, Tisdale MJ (2007) Identification and characterization of a membrane receptor for proteolysis-inducing factor on skeletal muscle. Cancer Res 67: 11419–11427.
Wang Q, Lu J-B, Wu B, Hao L-Y (2010) Expression and clinicopathologic significance of proteolysis-inducing factor in non-small-cell lung cancer: an immunohistochemical analysis. Clin Lung Cancer 11: 346–351.
Whitehouse AS, Tisdale MJ (2003) Increased expression of the ubiquitin-proteasome pathway in murine myotubes by proteolysis-inducing factor (PIF) is associated with activation of the transcription factor NF-κB. Br J Cancer 89: 1116–1122.
Wigmore SJ, Plester CE, Richardson RA, Fearon KC (1997) Changes in nutritional status associated with unresectable pancreatic cancer. Br J Cancer 75: 106–109.
Wigmore SJ, Todorov PT, Barber MD, Ross JA, Tisdale MJ, Fearon KC (2000) Characteristics of patients with pancreatic cancer expressing a novel cancer cachectic factor. Br J Surg 87: 53–58.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Mirza, K., Tisdale, M. Functional identity of receptors for proteolysis-inducing factor on human and murine skeletal muscle. Br J Cancer 111, 903–908 (2014). https://doi.org/10.1038/bjc.2014.379
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.379
