
Abstract
Background:
To evaluate the efficacy and tolerability of the urokinase plasminogen activator (uPA) inhibitor upamostat in combination with gemcitabine in locally advanced pancreatic adenocarcinoma (LAPC).
Methods:
Within a prospective multicenter study, LAPC patients were randomly assigned to receive 1000 mg m−2 of gemcitabine IV weekly either alone (arm A) or in combination with 200 mg (arm B) or 400 mg (arm C) oral upamostat daily. Efficacy endpoints of this proof-of-concept study included response rate, time to first metastasis, progression-free and overall survival (OS).
Results:
Of the 95 enroled patients, 85 were evaluable for response and 93 for safety. Median OS was 12.5 months (95% CI 8.2–18.2) in arm C, 9.7 months (95% CI 8.4–17.1) in arm B and 9.9 months (95% CI 7.4–12.1) in arm A; corresponding 1-year survival rates were 50.6%, 40.7% and 33.9%, respectively. More patients achieved a partial remission (confirmed responses by RECIST) with upamostat combination therapy (arm C: 12.9%; arm B: 7.1%; arm A: 3.8%). Overall, only 12 patients progressed by developing detectable distant metastasis (arm A: 4, arm B: 6, arm C: 2). The most common adverse events considered to be related to upamostat were asthenia, fever and nausea.
Conclusion:
In this proof-of-concept study targeting the uPA system in LAPC, the addition of upamostat to gemcitabine was tolerated well; similar survival results were observed for the three treatment arms.
Similar content being viewed by others
Main
Urokinase plasminogen activator (uPA) is a serine proteinase and has an important role in cancer invasion and metastasis: uPA bound to its receptor (uPAR) mediates mitogenic and migratory processes and the activation of growth factors (Muehlenweg et al, 2001; Ulisse et al, 2009; Mack and Marshall, 2010). Elevated levels of uPA and uPAR correlate with increased malignant behaviour and poor patient outcome (Janicke et al, 2001; Sperl et al, 2001). Preclinical studies conducted in different tumour models showed that uPA-inhibitors could effectively decrease tumour invasiveness as well as the number of metastases (Alonso et al, 1996; Setyono-Han et al, 2005). Upamostat (WX-671) is the oral pro-drug of the active metabolite WX-UK1, a novel uPA inhibitor (Setyono-Han et al, 2005); pharmacokinetic data on WX-UK1 and upamostat were collected in a total of 8 previous studies (WILEX AG, data on file). In head and neck cancer patients receiving upamostat in the 2 weeks before their scheduled surgery, WX-UK1 tissue levels exceeded the inhibition constant of WX-UK1 by 1.5–2-fold and were comparable for daily administration of both 200 and 400 mg upamostat (Meyer et al, 2008). In a trial of 149 patients with advanced pancreatic cancer, pre-treatment serum uPA levels were elevated in about 40% of patients and elevated serum uPA was associated with a shorter survival compared with patients with normal uPA levels (Ali et al, 2004).
This randomised trial is the first to evaluate the efficacy and safety of an uPA inhibitor administered to cancer patients. Locally advanced pancreatic cancer (LAPC) was chosen for this small initial proof-of-concept study as upamostat and its active metabolite WX-UK1 demonstrated both anti-metastatic and anti-tumour activity in a CA20948 pancreatic adenocarcinoma rat model. In addition, an additive effect of a combination therapy of upamostat with gemcitabine was observed in the mammary tumour rat model BN472 (WILEX AG, data on file).
Patients and Methods
Patient population
This study included patients with locally advanced, unresectable, non-metastatic, histologically or cytologically proven pancreatic adenocarcinoma (locoregional lymph nodes were not considered metastases). The assessment of surgical non-resectability was determined locally at each participating centre. Other inclusion criteria were age ⩾18 years, no prior (or concomitant) chemo- or radiotherapy, ECOG performance status 0–1 and adequate bone marrow, liver and renal function. All patients gave written informed consent before any study specific procedure. The study complied with the provisions of the Declaration of Helsinki and Good Clinical Practice Guidelines; it was registered at ClinicalTrials.gov (NCT00499265).
Study design and treatment plan
This three-arm trial was an open, randomised, multicenter, proof-of-concept phase II study. Locally advanced pancreatic cancer patients were centrally randomised in a 1 : 1 : 1 ratio to receive either gemcitabine (1000 mg m−2 IV over 30 min weekly for 7 weeks in the first 8 weeks, followed by weekly gemcitabine for 3 weeks of a 4-week cycle; arm A), or combination therapy with the same dose and schedule of gemcitabine and a daily oral dose of either 200 mg (arm B) or 400 mg (arm C) upamostat. Dose selection for this study was based on previous preclinical data, on phase I data of WX-UK1 and upamostat in healthy volunteers and on clinical data obtained from two studies in head and neck patients receiving upamostat before surgery (Wilex AG, data on file; Meyer et al, 2008). In order to assess a potential dose-response relationship two dose levels of 200 and 400 mg upamostat daily were selected. Treatment duration was until disease progression or unacceptable toxicity. Pre-defined efficacy endpoints for this proof-of-concept study included objective response rate, time to first metastasis, progression-free survival (PFS) and overall survival (OS). We assumed that the s.e. of response rates in excess of 10% was in the order of 5–10% for a sample size of 30. Hence, a sample size of 90 patients, 30 patients per group, was chosen for this study.
Dose adjustments
Dose modifications were performed according to the recommendations of the summary of product characteristics of gemcitabine. No dose adjustments were permitted for upamostat. The patient was to be discontinued if toxicity was observed and reduction of gemcitabine did not lead to improvement.
Evaluation of efficacy and safety
Computed tomography (CT) scans of chest and abdomen were performed at baseline and every 8 weeks. Computed tomography data were analysed centrally according to RECIST (version 1.0) by an experienced reader. Progression-free survival was measured from the time of randomisation until progression of disease as determined by a centrally evaluated CT scan, clinical evaluation or death. The population contributing to this endpoint therefore consisted of patients who had at least one CT staging following baseline. Overall survival was the interval between randomisation and death from any cause. The endpoint time to first metastasis was measured as the time from randomisation until development of metastasis (new lesions only) as determined by a centrally evaluated CT scan. Toxicities were graded according to the NCI Common Toxicity Criteria (NCI-CTCAE v3.0).
Statistical analysis
All statistical tests were exploratory: categorical data were described using absolute and relative frequencies, continuous data were presented using descriptive statistics and time to event data were estimated by the Kaplan–Meier method.
Results
Patient characteristics
A total of 95 patients from 29 centres were randomised between June 2007 and August 2008; 93 patients received study medication and were included in the safety population (see CONSORT diagram in Supplementary Figure 1S). Demographic variables according to treatment arm are summarised in Table 1.
Treatment duration and dose intensity
Data capture ended in December 2009 according to a protocol amendment. The main reasons for discontinuation of the protocol treatment were disease progression (n=45; determined either by CT imaging or by clinical progression as assessed by the local investigator), patient refusal (n=19), unrelated medical illness or complications (n=13) and unacceptable toxicity (n=4); no treatment-related death occurred. The median duration of gemcitabine treatment was 22 weeks for patients who received gemcitabine only, 16.6 weeks for patients who received gemcitabine +200 mg upamostat and 16.1 weeks for patients who received gemcitabine +400 mg upamostat. The gemcitabine exposure in the three treatment arms was comparable (Supplementary Table 1S).
Response and survival
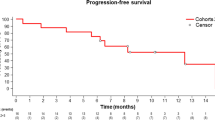
Detailed data on objective response, PFS and OS are summarised within Table 2. More patients achieved a partial remission as their best response with upamostat combination therapy (arm C: 35.5%; arm B: 21.4%; arm A: 15.4%; these responses were not confirmed by a follow-up CT scan, as requested by RECIST). As pre-defined by the study protocol, OS was monitored until 75% of patients had died; the OS analysis was performed for the intention-to-treat population. At the time of final analysis, 75 of 95 patients (79%) had died. Median OS was 9.9 months in arm A, 9.7 months in arm B and 12.5 months in arm C (Table 2, Supplementary Figure 2S). During the course of protocol treatment and follow-up, the majority of patients progressed without having developed detectable distant metastasis. Events (occurrence of confirmed distant metastases) were observed in 4 (arm A), 6 (arm B) and 2 (arm C) patients only. Owing to the small size of evaluable cohorts, a calculation of the pre-defined endpoint time to first metastasis was not performed.
Toxicity
Toxicity data are summarised within Table 3. The overall incidence of adverse events that were reported as possibly, probably, or definitely related to study treatment (i.e., related to either upamostat or gemcitabine) was higher among patients who received gemcitabine plus upamostat (83.3% in arm B, 84.8% in arm C) compared with patients who received gemcitabine (56.7% in arm A). The incidence of adverse events considered possibly, probably, or definitely related to upamostat was lower (43.3% for arm B, 51.5% for arm C) than the incidence of adverse events considered to be related to gemcitabine (83.3% for arm B, 84.8% for arm C). A grade 3/4 toxicity considered related to upamostat was reported for 7 patients in arm B and 5 patients in arm C.
Discussion
The clinical evaluation of upamostat was confined to patients with LAPC in this proof-of-concept study as there is increasing evidence that LAPC and metastatic pancreatic cancer may represent different disease entities (Tempero et al, 2011). In past studies, however, addition of various chemotherapeutic agents to standard gemcitabine has not significantly improved OS. The OS in the gemcitabine reference arm of this study (about 10 months) is consistent with the OS results reported from other LAPC trials (Heinemann et al, 2012). The combination of oral upamostat with gemcitabine showed a higher (although statistically non-significant) response rate and possibly also had an impact on the proportion of patients surviving at 1 year in the current study. Despite some early efficacy signs for upamostat regarding response rate and OS, upamostat had no effect on PFS. Of note, such an inconsistent observation for differences in PFS and OS in LAPC was also reported from the ECOG E4201 study: within this randomised trial, 74 LAPC patients were treated with either gemcitabine or a gemcitabine-based chemoradiotherapy (Loehrer et al, 2011). Although patients treated with chemoradiotherapy had an improved OS (11.1 vs 9.2 months), no difference in PFS was detected (6.0 vs 6.7 months).
Interestingly, data from the present study indicate that the majority of patients progressed without evidence of distant metastasis: in only 4, 6 and 2 patients (arms A, B and C, respectively) distant metastasis was detected as the sign of disease progression. Thus, it cannot be ruled out that the pattern of metastatic spread is different in LAPC compared with initially metastatic disease or to a relapse after surgery. This observation is supported by data from an autopsy series where 30% of patients died with locally destructive pancreatic cancer without metastatic spread. The authors hypothesised that this phenomenon in LAPC may be based on a genetic basis of the Dpc4 status (Iacobuzio-Donahue et al, 2009): it currently is suggested that a loss of the tumour suppressor Dpc4 expression in increases the likelihood of developing widespread metastatic disease (Crane et al, 2011). Regarding toxicities in the current study, the combination of gemcitabine and upamostat produced a higher rate of grade 3/4 adverse events compared with gemcitabine alone (Table 3). However, none of the reported reactions could be attributed exclusively to the use of upamostat and most events were attributable either to the underlying disease or the use of gemcitabine.
In summary, the combination of the novel uPA inhibitor upamostat with gemcitabine was safe and tolerated well. Although the differences in efficacy endpoints between the three treatment arms were not statistically significant, the data warrant a larger powered study to define the role of uPA inhibition in LAPC.
Change history
05 March 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ali SM, Marx JH, Leitzel K, Carney W, Hamer P, Linnartz R, Mietlowski W, Demers L, Harvey HA, Lipton A (2004) Pretreatment serum uPA and survival in patients with advanced pancreatic cancer. J Clin Oncol 22 (14S): ): abstract no. 4247
Alonso DF, Farias EF, Ladeda V, Davel L, Puricelli L, Bal de Kier Joffé E (1996) Effects of synthetic urokinase inhibitors on local invasion and metastasis in a murine mammary tumor model. Breast Cancer Res Treat 40 (3): 209–223
Crane CH, Varadhachary GR, Yordy JS, Staerkel GA, Javle MM, Safran H, Haque W, Hobbs BD, Krishnan S, Fleming JB, Das P, Lee JE, Abbruzzese JL, Wolff RA (2011) Phase II trial of cetuximab, gemcitabine, and oxaliplatin followed by chemoradiation with cetuximab for locally advanced (T4) pancreatic adenocarcinoma: correlation of Smad4(Dpc4) immunostaining with pattern of disease progression. J Clin Oncol 29 (22): 3037–3043
Heinemann V, Haas M, Boeck S (2012) Systemic treatment of advanced pancreatic cancer. Cancer Treat Rev 38 (7): 843–853
Iacobuzio-Donahue CA, Fu B, Yachida S, Luo M, Abe H, Henderson CM, Vilardell F, Wang Z, Keller JW, Banerjee P, Herman JM, Cameron JL, Yeo CJ, Halushka MK, Eshleman JR, Raben M, Klein AP, Hruban RH, Hidalgo M, Laheru D (2009) DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol 27 (11): 1806–1813
Janicke F, Prechtl A, Thomssen C, Harbeck N, Meisner C, Untch M, Sweep CG, Selbmann HK, Graeff H, Schmitt M German N0 Study Group (2001) Randomized adjuvant chemotherapy trial in high-risk, lymph node-negative breast cancer patients identified by urokinase-type plasminogen activator and plasminogen activator inhibitor type 1. J Natl Cancer Inst 93 (12): 913–920
Loehrer PJ, Feng Y, Cardenes H, Wagner L, Brell JM, Cella D, Flynn P, Ramanathan RK, Crane CH, Alberts SR, Benson AB (2011) Gemcitabine alone versus gemcitabine plus radiotherapy in patients with locally advanced pancreatic cancer: an Eastern Cooperative Oncology Group trial. J Clin Oncol 29 (31): 4105–4112
Mack GS, Marshall A (2010) Lost in migration. Nat Biotechnol 28 (3): 214–229
Meyer JE, Brocks C, Graefe H, Mala C, Thäns N, Bürgle M, Rempel A, Rotter N, Wollenberg B, Lang S (2008) The oral serine protease inhibitor WX-671 – first experience in patients with advanced head and neck carcinoma. Breast Care 3 (Suppl 2): 20–24
Muehlenweg B, Sperl S, Magdolen V, Schmitt M, Harbeck N (2001) Interference with the urokinase plasminogen activator system: a promising therapy concept for solid tumours. Expert Opin Biol Ther 1 (4): 683–691
Setyono-Han B, Stürzebecher J, Schmalix WA, Muehlenweg B, Sieuwerts AM, Timmermans M, Magdolen V, Schmitt M, Klijn JG, Foekens JA (2005) Suppression of rat breast cancer metastasis and reduction of primary tumour growth by the small synthetic urokinase inhibitor WX-UK1. Thromb Haemost 93 (4): 779–786
Sperl S, Mueller MM, Wilhelm OG, Schmitt M, Magdolen V, Moroder L (2001) The uPA/uPA receptor system as a target for tumor therapy. Drug News Perspect 14 (7): 401–411
Tempero MA, Berlin J, Ducreux M, Haller D, Harper P, Khayat D, Schmoll HJ, Sobrero A, Van Cutsem E (2011) Pancreatic cancer treatment and research: an international expert panel discussion. Ann Oncol 22 (7): 1500–1506
Ulisse S, Baldini E, Sorrenti S, D’Armiento M (2009) The urokinase plasminogen activator system: a target for anti-cancer therapy. Current Cancer Drug Targets 9 (1): 32–71
Acknowledgements
The authors wish to thank all patients and their families, nurses, study coordinators and investigators for their active participation in this multicenter study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Previous presentation: 46th ASCO Annual Meeting, 4–8 June 2010, Chicago, IL, USA and 35th ESMO Congress, 8–12 October 2010, Milan, Italy.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Heinemann, V., Ebert, M., Laubender, R. et al. Phase II randomised proof-of-concept study of the urokinase inhibitor upamostat (WX-671) in combination with gemcitabine compared with gemcitabine alone in patients with non-resectable, locally advanced pancreatic cancer. Br J Cancer 108, 766–770 (2013). https://doi.org/10.1038/bjc.2013.62
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.62
Keywords
This article is cited by
-
The force awakens: metastatic dormant cancer cells
Experimental & Molecular Medicine (2020)
-
Therapeutics targeting the fibrinolytic system
Experimental & Molecular Medicine (2020)
-
Urokinase-type plasminogen activator receptor (uPAR) expression enhances invasion and metastasis in RAS mutated tumors
Scientific Reports (2017)
-
Deregulated hepsin protease activity confers oncogenicity by concomitantly augmenting HGF/MET signalling and disrupting epithelial cohesion
Oncogene (2016)
-
Influence of Immune Myeloid Cells on the Extracellular Matrix During Cancer Metastasis
Cancer Microenvironment (2016)
