
Abstract
Background:
To compare the efficacy and safety of CAPIRI+bevacizumab (Bev) in comparison with FOLFIRI+Bev as first-line treatment for patients with metastatic colorectal cancer (mCRC).
Methods:
Patients were randomised to receive either FOLFIRI plus Bev 5 mg kg−1 every 2 weeks (Arm-A) or CAPIRI plus Bev 7.5 mg kg−1 every 3 weeks (Arm-B).
Results:
Three hundred thirty-three patients (Arm-A=167; Arm-B=166) were enrolled into the study. No difference was observed in median progression-free survival (PFS) (10.0 and 8.9 months; P=0.64), overall survival (25.7 and 27.5 months; P=0.55) or response rates (45.5 and 39.8.7%; P=0.32) for FOLFIRI-Bev and CAPIRI-Bev, respectively. Patients treated with CAPIRI-Bev presented significantly higher incidence of diarrhoea (P=0.005), febrile neutropenia (P=0.003) and hand–foot skin reactions (P=0.02) compared with patients treated with FOLFIRI-Bev. Treatment delays (P=0.05), dose reduction (P<0.001) and treatment discontinuation owing to toxicity (P=0.01) occurred more frequently in the CAPIRI-Bev arm.
Conclusion:
The PFS of FOLFIRI-BEV is not superior to that observed with the CAPIRI-Bev regimen. CAPIRI-Bev has a less favourable toxicity profile, requiring dose reductions, in order to be considered as an option in first-line treatment of patients with mCRC.
Similar content being viewed by others


Main
Despite the improvement in median overall survival (mOS) over the last 10 years, metastatic colorectal cancer (mCRC) remains a major public health problem accounting for 8% of cancer deaths in adults in the western world (Jemal et al, 2011). Combination chemotherapy with infusional 5-fluorouracil (FU) and folinic acid (FA) with either irinotecan (FOLFIRI) or oxaliplatin (FOLFOX) is commonly used in the daily practice. Expansion of mOS and 5-year survival rate have been correlated with the proportion of patients receiving all active chemotherapeutic agents (Grothey et al, 2004) and the increasing use of hepatic or/and pulmonary resection of metastatic lesions (Kopetz et al, 2009).
The addition of bevacizumab (Bev), a monoclonal antibody targeting vascular endothelial growth factor (VEGF), to combination chemotherapy in the first-line setting appears to increase the efficacy of systemic treatment in randomised trial (progression-free survival (PFS) and/or mOS) (Hurwitz et al, 2004; Saltz et al, 2008), but the magnitude of the benefit is debatable. However, one can argue that the benefit from addition of Bev to irinotecan-based chemotherapy (Hurwitz et al, 2004) is generally greater than that observed when it is combined with oxaliplatin-based regimens (Saltz et al, 2008).
Capecitabine, an oral fluoropyrimidine, was designed to mimic continuous infusion 5-FU and to generate 5-FU preferentially in the tumour tissue (Miwa et al, 1998; Schuller et al, 2000). Capecitabine has similar efficacy compared with 5-FU/LV as first-line treatment in mCRC patients (Van et al, 2000); the advantage of capecitabine is its convenient oral administration (Scheithauer et al, 2003). Capecitabine in combination with oxaliplatin (CAPOX) has consistently demonstrated similar efficacy results compared with the FOLFOX regimen (Cassidy et al, 2008). Likewise, the combination of capecitabine with irinotecan (CAPIRI) was proven effective and safe in a large randomised trial (Koopman et al, 2007). Finally, the combination of Bev and capecitabine has a synergistic effect, in an in vivo xenograft model, with a greater duration of tumour growth inhibition than with either agent alone (Kolinsky et al, 2009).
Based on these data, the Hellenic Oncology Research Group (HORG) designed a randomised phase-II trial in order to investigate the efficacy and safety of addition of Bev to FOLFIRI or CAPIRI as front-line treatment of patients with mCRC.
Patients and methods
Eligibility criteria
Untreated patients with mCRC were eligible for the study. Patients who had received prior adjuvant chemotherapy with fluoropyrimidines±oxaliplatin were eligible if they had remained free of disease for at least 6 months after completion of treatment. Other eligibility criteria were as follows: age ⩾18 years; performance status (ECOG) 0–2; at least one measurable lesion according to the Response Evaluation Criteria in Solid Tumors (RECIST) criteria (Therasse et al, 2000); adequate haematologic parameters (absolute neutrophil count ⩾1.5 × 109/l and platelets ⩾100 × 109/l); creatinine and total bilirubin ⩽1.25 times the upper limit of normal; aspartate and alanine aminotransferases ⩽3.0 times the upper limit of normal; absence of active infection or malnutrition; and absence of a second primary tumour except a skin squamous carcinoma or an in situ carcinoma of the uterine cervix. Patients with liver metastases involving more than 50% of the liver parenchyma; chronic diarrhoea; myocardial infarction within 1 year before treatment initiation; stroke; pre-existing bleeding diatheses or coagulopathy, or need for full-dose anticoagulation therapy or history of deep vein thrombosis within 6 months prior to registration; uncontrolled hypertension; pre-treatment proteinuria ⩾ grade-2; and central nervous system metastases were excluded.
The study was approved by the Ethics and Scientific Committees of each participating centre and all patients gave written informed consent prior to study enrolment.
Treatment protocol
Patients were randomised to receive either FOLFIRI-Bev (Arm-A: irinotecan at the dose of 180 mg m−2, iv, on day 1; FA at the dose of 200 mg m−2, iv, on days 1 and 2; and 5-FU at the dose of 400 mg m−2day−1, iv, bolus and 600 mg m−2 day−1, as a 22-h iv continuous infusion, on days 1 and 2, plus 5 mg kg−1 Bev on day 1, every 2 weeks) or CAPIRI-Bev (Arm-B: capecitabine at the dose of 2000 mg m−2, p.o., on days 1–14; irinotecan at the dose of 250 mg m−2, iv, on day 1; and Bev at the dose of 7.5 mg kg−1, iv, every 3 weeks). Stratification factors were age (⩽65 years vs >65 years), extent of metastatic disease (liver limited vs other) and prior adjuvant chemotherapy (yes vs no). Routine antiemetic prophylaxis with a 5-hydroxytryptamine-3-receptor antagonist was used in both arms. Treatment was administered until disease progression or unacceptable toxicity, or consent withdrawal.
Patients were assessed for toxicity before each cycle using the National Cancer Institute Common Toxicity Criteria version 3.0. Chemotherapy was delayed until recovery if neutrophils were less than 1.5 × 109/l or platelets less than 100 × 109/l, or for significant (more than grade-II) persisting non-haematologic toxicity.
Doses of all drugs were reduced by 15% in subsequent cycles in case of grade-4 neutropenia or grade-3–4 thrombocytopenia lasting for more than 3 days, or in case of febrile neutropenia. No prophylactic administration of granulocyte colony-stimulating factor was allowed. Doses of irinotecan and 5-FU or capecitabine were reduced by 15% in subsequent cycles in case of grade-3 or 4 diarrhoea. The 5-FU or capecitabine dose was reduced in case of grade-3–4 stomatitis or dermatitis. Bevacizumab was permanently discontinued in patients developing gastrointestinal perforation, wound dehiscence requiring medical intervention, serious bleeding, nephrotic syndrome or hypertensive crisis. Temporary discontinuation of Bev administration was implemented in patients with evidence of moderate-to-severe proteinuria and in patients with severe hypertension that was not controlled with medical management.
Patient evaluation
Pre-treatment evaluation included medical history and physical examination, complete blood cell count (CBC) with differential and platelet count, blood chemistry, serum levels of carcinoembryonic antigen, and computed tomographic (CT) scans of the chest and imaging of the abdomen (CT or MRI). Pre-treatment evaluation had to be performed within 2 weeks prior to study entry. During treatment, a CBC with was performed weekly. In addition, patients were clinically assessed and blood chemistry was performed before each treatment cycle. Response to treatment was evaluated every 8 weeks according to the RECIST criteria (Therasse et al, 2000) in order to gain comparable efficacy results between the two treatment regimens.
Statistical considerations
The primary endpoint of the study was PFS. Secondary endpoints were mOS, response to treatment and safety profile in terms of adverse events incidence, dose reductions and treatment delays. Based on the results of the BICC trial (Fuchs et al, 2007) the study was designed in order to detect a 3-month difference (8 vs 11 months) in PFS with an 80% power at a significance level of 0.05. In order to achieve the statistical hypothesis, 165 patients (per arm) should be enrolled in 36 months, with an additional follow-up period of 24 months.
The Kaplan–Meier method was used to estimate PFS and survival curves, and log-rank test was used to compare curves. Cox proportional hazards modelling was used to calculate hazard ratios (HRs) and confidence intervals (CIs). Heterogeneity tests were performed in order to determine whether the effect size for the subgroups varies significantly from the main effect. Forest plots were used in order to investigate the effect of the studied variables apart in accordance to the overall effect for each case. χ2-Tests were used to compare toxicity and confirmed response rates. P-values less than 0.05 were considered statistically significant for all comparisons. Progression-free survival was defined as the interval from the time of enrolment to the date of first documented disease progression or patient's death from any cause. Overall survival is considered the time interval from the date of enrolment until the date of death from any cause. The duration of response was measured from the first documentation of response to disease progression.
Results
Patients’ characteristics
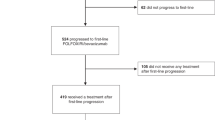
From June 2005 to June 2008, 336 patients with unresectable mCRC were enrolled into the study at 23 institutions throughout Greece. Two patients, in the CAPIRI+Bev Arm-B and one patient in the FOLFIRI+Bev Arm-A received no study treatment because they were found ineligible. The remained 333 were randomly allocated to receive front-line treatment and received at least one chemotherapy cycle (167 in Arm-A and 166 in Arm-B), and were analysed for efficacy and safety (Figure 1). Patients’ characteristics were typical for mCRC in the western world (Table 1). More specifically, about one half of patients in both arms were >65 years old, the vast majority (97% in Arm-A and 98% in Arm-B) had PS of 0–1, one-third had received prior adjuvant chemotherapy, whereas 37% in Arm-A and 38% in Arm-B had metastatic disease limited to the liver. Similarly, in one-third of patients in each arm, metastases were synchronous to diagnosis of the primary tumour. Overall, 25% and 22% of the patients in Arm-A and Arm-B, respectively, were classified as high risk according to the Kohne prognostic index (Kohne et al, 2002).

CONSORT diagram of the study.
Compliance with treatment
In the FOLFIRI-Bev arm, 1494 treatment cycles were administered compared with 871 cycles in the CAPIRI-Bev arm. The median number of cycles was 11 (range 1–20) and 6 (range 1–10) per patient treated with the FOLFIRI-Bev and CAPIRI-Bev regimen, respectively; however, the median treatment period was similar (5.5 months) for both arms.
Treatment delays were observed in 134 (9.0%) chemotherapy courses in the FOLFIRI-Bev arm and 136 (15.6%) in the CAPIRI-Bev arm (P=0.05); the median duration of delay was 4 days (range 1–14) in the FOLFIRI-Bev arm and 7 days (range 1–18) in the CAPIRI-Bev arm (P=0.23). In the FOLFIRI-Bev arm the reasons of delay were haematologic (n=60, 4.0%), non-haematologic (n=34, 2.2%) or combined (n=54, 3.6%) toxicity. In the CAPIRI-Bev arm treatment was delayed due to haematologic (n=44, 5.2%), non-haematologic (n=82, 9.4%) or combined (n=10, 1.1%) toxicity. The incidence of non-haematologic toxicity was significantly higher in the CAPIRI-Bev arm (P=0.031). The median interval between cycles was 14 (range 14–28) and 21 (range 21–39) days in the FOLFIRI-Bev and CAPIRI-Bev arms, respectively. Dose reduction was required in 65 (4.3%) cycles in the FOLFIRI-Bev arm and 95 (10.9%) cycles in the CAPIRI-Bev arm (P<0.001). The main reasons for dose reduction were haematologic (FOLFIRI-Bev (n=21, 1.4%) and CAPIRI-Bev (n=25, 2.8%)), non-haematologic (FOLFIRI-Bev (n=30, 2.0%) and CAPIRI-BEV (n=49, 5.6%)) or both (FOLFIRI-Bev (n=9, 0.6%) and CAPIRI-BEV (n=21, 2.4%)) toxicities. Treatment was discontinued in seven (4.2%) patients enrolled in the FOLFIRI-Bev arm and 17 (10.2%) in the CAPIRI-Bev arm (P=0.04). The delivered relative dose intensity was 90% for irinotecan, 92% for 5-FU/FA and 94% for Bev of the protocol-planned doses in the FOLFIRI-Bev arm, and 79% for irinotecan, 82% for capecitabine and 97% for Bev in the CAPIRI-Bev arm.
Efficacy
After a median follow-up period of 32 months (range 1–64 months), 143 (86%) patients in FOLFIRI-Bev and 138 (83%) in CAPIRI-Bev experienced disease progression, whereas 90 (54%) and 87 (52%) patients, respectively died. There was no statistical difference in terms of median PFS between the two arms: 10.0 months (95% CI: 8.9–11.1 months) for patients treated with FOLFIRI-Bev compared with 8.9 months (95% CI: 7.3–10.2 months) for those treated with CAPIRI-Bev (HR=0.99; 95% CI: 0.90–1.09; P=0.85; Figure 2A). Similarly, there was no statistical difference in terms of mOS between the two regimens (Figure 2B). Patients treated with the FOLFIRI-Bev regimen presented an mOS of 25.7 months (95% CI: 23.0–28.4 months) whereas those treated with CAPIRI-Bev showed an mOS of 27.5 months (95% CI: 22.6–32.3 months) (HR=1.08; 95% CI: 0.94–1.24; P=0.30). In addition, no difference in PFS (Figure 2C) or mOS (Figure 2D) has been observed in subgroup analysis.

(A) Progression-free survival of patients treated with FOLFIRI+Bev or CAPIRI+Bev. (B) Overall survival of patients treated with FOLFIRI+Bev or CAPIRI+Bev. (C) Forest plots of PFS of patients treated with FOLFIRI+Bev or CAPIRI+Bev. (D) Forest plots of OS of patients treated with FOLFIRI+Bev or CAPIRI+Bev.
In the ITT population, the overall response rate (ORR) was 45.5% (95% CI: 38.0–53.1%) in the FOLFIRI-Bev arm and 39.8% (95% CI: 32.3–47.2%) in the CAPIRI-Bev arm (P=0.32). More specifically, complete responses (CRs) were recorded in 11 (6.6%) and partial responses (PRs) in 65 (38.9%) patients treated with FOLFIRI-Bev, whereas 9 (6.5%) and 53 (31.9%) patients experienced CRs and PRs, respectively, in the CAPIRI-Bev arm. The median time of response duration was 8.2 (95% CI: 7.6–8.9) and 8.0 months (95% CI: 6.6–9.5) in the FOLFIRI-Bev and CAPIRI-Bev arm, respectively (P=0.58). Fifty (29.9%) patients treated with FOLFIRI-Bev and 52 (31.3%) patients treated with CAPIRI-Bev experienced stabilisation of disease, whereas 41 (24.6%) and 48 (28.9%) patients, respectively, had progression of their disease at the first efficacy evaluation. Secondary R0 metastasectomy was performed in six (3.6%) patients treated with FOLFIRI-Bev and three (1.8%) patients treated with CAPIRI-Bev (P=0.38). In patients with liver-limited disease, R0 resections were obtained in 5 (8%) and 3 (5%) patients in the FOLFIRI-Bev and the CAPIRI-Bev arm, respectively (P=0.88).
Toxicity
Patients treated with CAPIRI-Bev had a significantly higher incidence of grade-3/4 febrile neutropenia (P<0.001), diarrhoea (P=0.003) and hand–foot skin reaction (P=0.03) compared with patients treated with FOLFIRI-Bev (Table 2). All other adverse events were equally distributed between the two treatment arms. Bevacizumab-related serious adverse events were rare in both arms. Grade-3/4 hypertension was observed in 3.8% and 4.2% of the patients in the FOLFIRI-Bev and CAPIRI-Bev arm, respectively. One patient in each arm presented with a large bowel perforation, which was lethal in one of them (in FOLFIRI-Bev arm). Three additional deaths, all due to febrile neutropenia combined with diarrhoea, occurred in the CAPIRI-Bev arm during treatment. The death rates within the first 60 days of treatment were 2.4% (95% CI: 1.0–4.1%) for patients treated with the FOLFIRI-Bev regimen and 4.1% (95% CI: 2.3–5.9%) for those treated with the CAPIRI-Bev regimen (P=0.42).
Second-line treatment
Although that second-line treatments were not specified by the protocol, the regimens administered after disease progression were recorded. An oxaliplatin-based second-line treatment was administered in 76% and 72% of patients after progression to FOLFIRI-BEV or CAPIRI-BEV, respectively (Table 3). Cetuximab either as monotherapy or in combination with irinotecan was administered in 38% of the patients after progression to FOLFIRI-BEV and 30% of those with progression after CAPIRI-Bev. Bevacizumab administration was continued in the second-line setting in 23% of the patients in each arm (Table 4).
Discussion
Addition of monoclonal antibodies targeting either VEGF or EGFR to irinotecan–5-FU/FA combination chemotherapy in some studies has demonstrated an increase in RR, PFS and mOS compared with chemotherapy alone (Hurwitz et al, 2004; Van et al, 2009). To the best of our knowledge the current study is the first randomised trial comparing the combination of Bev with the standard FOLFIRI regimen with its combination with the outpatient CAPIRI regimen. The primary endpoint, a 3-months increase in PFS, was not met as no statistically significant difference has been observed between the two treatment arms (HR=0.99; 95% CI: 0.90–1.09; P=0.85). Similarly, no differences have been observed in terms of mOS (HR=1.08; 95% CI: 0.94–1.24; P=0.30) and of ORR (P=0.32).
The efficacy parameters of FOLFIRI-Bev are in the same range with that reported in previous phase-II (Kopetz et al, 2010) and III trials (Fuchs et al, 2008), and compare favourably with those reported for FOLFIRI alone (Tournigand et al, 2004; Souglakos et al, 2006; Van et al, 2009). In addition, in the present study, the PFS and mOS for CAPIRI-Bev compare favourably with those reposted for CAPIRI alone (Bajetta et al, 2004; Borner et al, 2005; Koopman et al, 2007), and are in the same range with those recently reported in a phase-II study (Garcia-Alfonso et al, 2010). The CAPIRI regimen has proved its efficacy in several phase-II studies (Bajetta et al, 2004; Borner et al, 2005; Rea et al, 2005), and in a large randomised trial (Koopman et al, 2007) with PFS and mOS of 8.0 and 17.5 months, respectively. Despite that, major concerns regarding the efficacy of the regimen have been raised from the BICC-C trial (Fuchs et al, 2007). This trial reported that administration of CAPIRI led to significantly lower PFS (5.8 months) in comparison with FOLFIRI, whereas the mOS was comparable between the two arms. Overall, the results of the current investigation support that FOLFIRI-Bev and CAPIRI-Bev are equally effective in terms of ORR, PFS and mOS.
Overall survival was not the primary endpoint of the study as it is quite difficult to drawn conclusions from a randomised phase-II study. Taking into account these limitations, it is noteworthy that the mOS observed in the current study in both arms is one of the highest reported in randomised trials. Despite the fact that patients with massive liver infiltration from the tumour (>50% of the total parenchyma) or with central nervous system metastasis were excluded from the study, and that this may be considered as a selection bias, the long mOS observed in the current study could not be explained from the characteristics of the patients enrolled into the study. A significant proportion of patients had received prior adjuvant treatment and the percentage of patients with favourable characteristics, such as disease limited to the liver, low risk according to the Kohne index and metachronous metastatic disease, were in the same range with those recorded in other trials (Hurwitz et al, 2004; Van et al, 2009; Moosmann et al, 2011). The percentage of patients who underwent secondary resection was low (3.5% and 1.8% for FOLFIRI-Bev and CAPIRI-Bev, respectively) but in the same range with that observed in studies with combination of irinotecan plus fluoropyrimidines and Bev (Hurwitz et al, 2004). Thus, it seems difficult to explain of the high mOS observed in the current study. A significant proportion of patients received effective second-line treatment. Approximately 60% of patients were treated with oxaliplatin-based regimens, whereas monoclonal antibodies against EGFR were administered in >30% of the cases and Bev beyond progression in approximately one-quarter of the cases. It is generally accepted that the mOS is significantly correlated with the proportion of patients receiving all active chemotherapeutic agents over the disease course (Grothey et al, 2004), and that salvage treatment with anti-EGFR monoclonal antibodies could increase PFS and OS in KRAS wt patients (Amado et al, 2008; Karapetis et al, 2008). In addition, data from observational cohort studies support that use of Bev beyond progression could be associated with improved mOS (Grothey et al, 2008), although this point has not yet been investigated in prospective randomised trials.
The main difference recorded in the present study concerns the toxicity profile of the two regimens. Indeed, patients treated with CAPIRI-Bev had a significantly higher incidence of diarrhoea (P=0.005), febrile neutropenia (P=0.003) and hand–foot skin reactions (P=0.02) compared with patients treated with FOLFIRI-Bev. The timing of safety assessments was different between the two treatments arm (every 2 weeks in the FOLFIRI+Bev arm and every 3 weeks in the CAPIRI+Bev arm). Despite that, the differences in safety profile could not be explained from the dissimilarity in the timing of safety assessment as the higher toxicity grade was recorded in each assessment and the worst toxicity per patient is reported. Moreover, despite the fact that the delivered relative dose intensity was comparable between the two arms, treatment delays (P=0.05), dose reductions (P<0.001) and treatment discontinuation owing to toxicity (P=0.01) occurred more frequently in the CAPIRI-Bev arm. The incidence of grade-3/4 toxicities in the FOLFIRI-Bev was in the same range with those reported for FOLFIRI alone (Tournigand et al, 2004; Souglakos et al, 2006). The incidence of severe toxicities with CAPIRI-Bev was, also, comparable with those observed in the BICC-C and CAIRO studies (Fuchs et al, 2007; Koopman et al, 2007) for CAPIRI alone. The additional gastro-intestinal toxicity of the CAPIRI-Bev regimen observed in the current study should not be considered as specific to the combination of capecitabine with irinotecan. In fact, the incidence of severe diarrhoea is higher with the XELOX regimen in comparison with FOLFOX4 (Cassidy et al, 2008).
Overall, addition of Bev in either arm does not seem to increase the incidence of adverse events. Recently, data from studies investigating lower doses of CAPIRI in combination with either Bev (Garcia-Alfonso et al, 2010) or cetuximab (Moosmann et al, 2011) reported a more favourable toxicity profile with lower incidence of diarrhoea and neutropenia, as well as lower rates of dose reductions and treatments delays.
Overall, the results of the current study show that the CAPIRI-Bev regimen at the doses used in this study demonstrated comparable efficacy with FOLFIRI-Bev but with increased incidence of diarrhoea, neutropenia and hand–foot skin reactions. Owing to the increase toxicity and frequent dose modification, lower doses of cytotoxics would be considered in future trials using the CAPIRI-Bev regimen as front-line treatment for patients with mCRC.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Amado RG, Wolf M, Peeters M, Van CE, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD (2008) Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 26 (10): 1626–1634
Bajetta E, Di BM, Mariani L, Cassata A, Artale S, Frustaci S, Pinotti G, Bonetti A, Carreca I, Biasco G, Bonaglia L, Marini G, Iannelli A, Cortinovis D, Ferrario E, Beretta E, Lambiase A, Buzzoni R (2004) Randomized multicenter phase II trial of two different schedules of irinotecan combined with capecitabine as first-line treatment in metastatic colorectal carcinoma. Cancer 100 (2): 279–287
Borner MM, Bernhard J, Dietrich D, Popescu R, Wernli M, Saletti P, Rauch D, Herrmann R, Koeberle D, Honegger H, Brauchli P, Lanz D, Roth AD (2005) A randomized phase II trial of capecitabine and two different schedules of irinotecan in first-line treatment of metastatic colorectal cancer: efficacy, quality-of-life and toxicity. Ann Oncol 16 (2): 282–288
Cassidy J, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A, Wong R, Koski S, Lichinitser M, Yang TS, Rivera F, Couture F, Sirzen F, Saltz L (2008) Randomized phase III study of capecitabine plus oxaliplatin compared with fluorouracil/folinic acid plus oxaliplatin as first-line therapy for metastatic colorectal cancer. J Clin Oncol 26 (12): 2006–2012
Fuchs CS, Marshall J, Barrueco J (2008) Randomized, controlled trial of irinotecan plus infusional, bolus, or oral fluoropyrimidines in first-line treatment of metastatic colorectal cancer: updated results from the BICC-C study. J Clin Oncol 26 (4): 689–690
Fuchs CS, Marshall J, Mitchell E, Wierzbicki R, Ganju V, Jeffery M, Schulz J, Richards D, Soufi-Mahjoubi R, Wang B, Barrueco J (2007) Randomized, controlled trial of irinotecan plus infusional, bolus, or oral fluoropyrimidines in first-line treatment of metastatic colorectal cancer: results from the BICC-C study. J Clin Oncol 25 (30): 4779–4786
Garcia-Alfonso P, Munoz-Martin AJ, varez-Suarez S, Jerez-Gilarranz Y, Riesco-Martinez M, Khosravi P, Martin M (2010) Bevacizumab in combination with biweekly capecitabine and irinotecan, as first-line treatment for patients with metastatic colorectal cancer. Br J Cancer 103 (10): 1524–1528
Grothey A, Sargent D, Goldberg RM, Schmoll HJ (2004) Survival of patients with advanced colorectal cancer improves with the availability of fluorouracil-leucovorin, irinotecan, and oxaliplatin in the course of treatment. J Clin Oncol 22 (7): 1209–1214
Grothey A, Sugrue MM, Purdie DM, Dong W, Sargent D, Hedrick E, Kozloff M (2008) Bevacizumab beyond first progression is associated with prolonged overall survival in metastatic colorectal cancer: results from a large observational cohort study (BRiTE). J Clin Oncol 26 (33): 5326–5334
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350 (23): 2335–2342
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61 (2): 69–90
Karapetis CS, Khambata-Ford S, Jonker DJ, O′Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, Langer C, Moore MJ, Zalcberg JR (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 359 (17): 1757–1765
Kohne CH, Cunningham D, Di CF, Glimelius B, Blijham G, Aranda E, Scheithauer W, Rougier P, Palmer M, Wils J, Baron B, Pignatti F, Schoffski P, Micheel S, Hecker H (2002) Clinical determinants of survival in patients with 5-fluorouracil-based treatment for metastatic colorectal cancer: results of a multivariate analysis of 3825 patients. Ann Oncol 13 (2): 308–317
Kolinsky K, Shen BQ, Zhang YE, Kohles J, Dugan U, Zioncheck TF, Heimbrook D, Packman K, Higgins B (2009) In vivo activity of novel capecitabine regimens alone and with bevacizumab and oxaliplatin in colorectal cancer xenograft models. Mol Cancer Ther 8 (1): 75–82
Koopman M, Antonini NF, Douma J, Wals J, Honkoop AH, Erdkamp FL, de Jong RS, Rodenburg CJ, Vreugdenhil G, Loosveld OJ, van BA, Sinnige HA, Creemers GJ, Tesselaar ME, Slee PH, Werter MJ, Mol L, Dalesio O, Punt CJ (2007) Sequential versus combination chemotherapy with capecitabine, irinotecan, and oxaliplatin in advanced colorectal cancer (CAIRO): a phase III randomised controlled trial. Lancet 370 (9582): 135–142
Kopetz S, Chang GJ, Overman MJ, Eng C, Sargent DJ, Larson DW, Grothey A, Vauthey JN, Nagorney DM, McWilliams RR (2009) Improved survival in metastatic colorectal cancer is associated with adoption of hepatic resection and improved chemotherapy. J Clin Oncol 27 (22): 3677–3683
Kopetz S, Hoff PM, Morris JS, Wolff RA, Eng C, Glover KY, Adinin R, Overman MJ, Valero V, Wen S, Lieu C, Yan S, Tran HT, Ellis LM, Abbruzzese JL, Heymach JV (2010) Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J Clin Oncol 28 (3): 453–459
Miwa M, Ura M, Nishida M, Sawada N, Ishikawa T, Mori K, Shimma N, Umeda I, Ishitsuka H (1998) Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur J Cancer 34 (8): 1274–1281
Moosmann N, von Weikersthal LF, Vehling-Kaiser U, Stauch M, Hass HG, Dietzfelbinger H, Oruzio D, Klein S, Zellmann K, Decker T, Schulze M, Abenhardt W, Puchtler G, Kappauf H, Mittermuller J, Haberl C, Schalhorn A, Jung A, Stintzing S, Heinemann V (2011) Cetuximab plus capecitabine and irinotecan compared with cetuximab plus capecitabine and oxaliplatin as first-line treatment for patients with metastatic colorectal cancer: AIO KRK-0104 – a randomized trial of the German AIO CRC Study Group. J Clin Oncol 29 (8): 1050–1058
Rea DW, Nortier JW, Ten Bokkel Huinink WW, Falk S, Richel DJ, Maughan T, Groenewegen G, Smit JM, Steven N, Bakker JM, Semiond D, Kerr DJ, Punt CJ (2005) A phase I/II and pharmacokinetic study of irinotecan in combination with capecitabine as first-line therapy for advanced colorectal cancer. Ann Oncol 16 (7): 1123–1132
Saltz LB, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A, Wong R, Koski S, Lichinitser M, Yang TS, Rivera F, Couture F, Sirzen F, Cassidy J (2008) Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 26 (12): 2013–2019
Scheithauer W, McKendrick J, Begbie S, Borner M, Burns WI, Burris HA, Cassidy J, Jodrell D, Koralewski P, Levine EL, Marschner N, Maroun J, Garcia-Alfonso P, Tujakowski J, Van HG, Wong A, Zaluski J, Twelves C (2003) Oral capecitabine as an alternative to i.v. 5-fluorouracil-based adjuvant therapy for colon cancer: safety results of a randomized, phase III trial. Ann Oncol 14 (12): 1735–1743
Schuller J, Cassidy J, Dumont E, Roos B, Durston S, Banken L, Utoh M, Mori K, Weidekamm E, Reigner B (2000) Preferential activation of capecitabine in tumor following oral administration to colorectal cancer patients. Cancer Chemother Pharmacol 45 (4): 291–297
Souglakos J, Androulakis N, Syrigos K, Polyzos A, Ziras N, Athanasiadis A, Kakolyris S, Tsousis S, Kouroussis C, Vamvakas L, Kalykaki A, Samonis G, Mavroudis D, Georgoulias V (2006) FOLFOXIRI (folinic acid, 5-fluorouracil, oxaliplatin and irinotecan) vs FOLFIRI (folinic acid, 5-fluorouracil and irinotecan) as first-line treatment in metastatic colorectal cancer (MCC): a multicentre randomised phase III trial from the Hellenic Oncology Research Group (HORG). Br J Cancer 94 (6): 798–805
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van GM, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92 (3): 205–216
Tournigand C, Andre T, Achille E, Lledo G, Flesh M, Mery-Mignard D, Quinaux E, Couteau C, Buyse M, Ganem G, Landi B, Colin P, Louvet C, de GA (2004) FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol 22 (2): 229–237
Van CE, Findlay M, Osterwalder B, Kocha W, Dalley D, Pazdur R, Cassidy J, Dirix L, Twelves C, Allman D, Seitz JF, Scholmerich J, Burger HU, Verweij J (2000) Capecitabine, an oral fluoropyrimidine carbamate with substantial activity in advanced colorectal cancer: results of a randomized phase II study. J Clin Oncol 18 (6): 1337–1345
Van CE, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D′Haens G, Pinter T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M, Nippgen J, Rougier P (2009) Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 360 (14): 1408–1417
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Registration number: clinicaltrials.gov. NCT00469443
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Souglakos, J., Ziras, N., Kakolyris, S. et al. Randomised phase-II trial of CAPIRI (capecitabine, irinotecan) plus bevacizumab vs FOLFIRI (folinic acid, 5-fluorouracil, irinotecan) plus bevacizumab as first-line treatment of patients with unresectable/metastatic colorectal cancer (mCRC). Br J Cancer 106, 453–459 (2012). https://doi.org/10.1038/bjc.2011.594
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2011.594
Keywords
This article is cited by
-
Different doses of bevacizumab in combination with chemotherapy for advanced colorectal cancer: a meta-analysis and Bayesian analysis
International Journal of Colorectal Disease (2023)
-
Magnitude of advantage in tumor response contributes to a better correlation between treatment effects on overall survival and progression-free survival: a literature-based meta-analysis of clinical trials in patients with metastatic colorectal cancer
International Journal of Clinical Oncology (2020)
-
MINOAS: A Single-arm Translational Phase II Trial of FOLFIRI Plus Aflibercept as First-line Therapy in Unresectable, Metastatic Colorectal Cancer
Targeted Oncology (2019)
-
Efficacy and safety of bevacizumab plus chemotherapy compared to chemotherapy alone in previously untreated advanced or metastatic colorectal cancer: a systematic review and meta-analysis
BMC Cancer (2016)
-
Study protocol of the Asian XELIRI ProjecT (AXEPT): a multinational, randomized, non-inferiority, phase III trial of second-line chemotherapy for metastatic colorectal cancer, comparing the efficacy and safety of XELIRI with or without bevacizumab versus FOLFIRI with or without bevacizumab
Chinese Journal of Cancer (2016)
