
Abstract
The Rab family GTPases regulate many major biological processes during tumor progression such as cell proliferation, cytoskeleton organization, cell movement, and invasion. The present study aims to examine the clinical significance, biological roles, and molecular mechanism of Rab11a in pancreatic cancer progression. We examined expression pattern of Rab11a in 96 cases of pancreatic cancer specimens using immunohistochemistry and found Rab11a overexpression correlated with tumor-node-metastasis (TNM) stage (p = 0.0111). We depleted Rab11a in Bxpc3 cells using small interfering RNA (siRNA) and overexpressed Rab11a in Capan2 cells. Knockdown of Rab11a inhibited cell growth, invasion, and cell cycle progression while its overexpression facilitated cell growth, invasion, and cell cycle progression. In addition, Rab11a overexpression increased gemcitabine resistance and inhibited gemcitabine-induced apoptosis in Capan2 cells while its depletion reduced drug resistance. We investigated the role of Rab11a in the regulation of Wnt/β-catenin signaling and we demonstrated that Rab11a overexpression upregulated GSK3β phosphorylation and nuclear β-catenin accumulation. Rab11a depletion inhibited while its overexpression enhanced β-catenin/T-cell factor (TCF) transcriptional activity with corresponding change of Wnt target genes including cyclin D1, cyclin E, MMP7, and c-myc. Wnt inhibitor (FH535) partly attenuated the effects of Rab11a on cell proliferation and Wnt target genes. In conclusion, the present study demonstrated that Rab11a promotes aggressiveness of pancreatic cancer through GSK3β/Wnt/β-catenin signaling pathway.
Similar content being viewed by others

Introduction
Pancreatic cancer is one of the most common causes of cancer-related death in the world. Despite development of conventional therapies including surgery and chemotherapy, the prognosis of pancreatic cancer remains poor and the 5-year survival rate was below 5 % [1]. Thus, it is necessary to find new oncogenes which could serve as new molecular targets to the development of targeted therapy.
Rab11a belongs to Ras superfamily of small G proteins which regulate membrane vesicle trafficking [2, 3]. Rab11a has been shown to contribute to the recycling of a wide range of receptors to the cell surface and mitotic spindle organization/orientation [4, 5]. Active Rab11a is required for E-cadherin trafficking and lumen formation during epithelial morphogenesis [6]. Rab11a was shown to be overexpressed in breast cancers and esophageal cancer [7, 8]. It is reported that active Rab11a could induce the colorectal cell transformation and migration [9], suggesting its potential role as an oncogene. However, the expression pattern and biological function of Rab11a in human pancreatic carcinoma have not been well explored.
In the present study, we examined clinical significance of Rab11a in pancreatic cancer tissues using immunohistochemistry. We overexpressed and depleted Rab11a in pancreatic cancer cell lines and examined its effects on cell proliferation, invasion, cell cycle progression, and gemcitabine resistance. In addition, we investigated the molecular signaling pathways underlying the biological effects of Rab11a.
Materials and methods
Clinical specimens
The protocol of the present study was approved by the reviewer board of Liaoning Medical University. Clinical tissues were collected from 96 patients who were diagnosed with pancreatic cancer in the First Affiliated Hospital of Liaoning Medical University since 2010 to 2013. The histological diagnosis and tumor grade were evaluated by pathologist according to the WHO classification guidelines. Eight fresh cancer specimens which contain both tumor tissues and corresponding normal tissues were stored at −70 °C after resection for extraction of RNA and protein.
Immunohistochemistry
Tumor specimens were fixed with 10 % neutral formalin, and 4-μm-thick paraffin sections were made. Immunostaining was performed using the S-P staining kit from MaiXin (Ultrasensitive™, MaiXin, Fuzhou, China). After antigen retrieval in citrate buffer (pH 6.0) for 2 min in an autoclave, 0.3 % hydrogen peroxide was used for 15 min and then the sections were incubated with goat serum. Then sections were incubated with Rab11a rabbit polyclonal antibody at 4 °C overnight (1:300 dilution, Proteintech, USA). Primary antibody incubation was performed at room temperature for 2 h. Biotinylated goat anti-rabbit serum IgG was incubated after washing in phosphate-buffered saline (PBS). Then horseradish peroxidase (HRP)-conjugated streptavidin–biotin was incubated with sections. DAB kit (MaiXin, Fuzhou, China) was used for staining. Counterstaining with hematoxylin was performed, and the sections were dehydrated in ethanol before mounting.
Two independent pathologists examined all slides randomly. Five views were examined per slide. Staining of Rab11a was scored on a semiquantitative scale by evaluating the intensity and percentage of cells showing positive staining. Cytoplasmic staining was considered as positive immunostaining. The intensity of Rab11a cytoplasmic staining was also scored as 0 (none), 1 (weak), and 2 (strong). Percentage scores were assigned as 1, 1–25 %; 2, 26–50 %; 3, 51–75 %; and 4, 76–100 %. The scores were multiplied to give a final score of 0 to 8, and the total expression of Rab11a was determined as either negative/low expression: score <4 or high expression: score ≥4.
Cell culture and transfection
Capan2, Bxpc3, and CFPAC1 cell lines were purchased from ATCC (Manassas, VA, USA), which were cultured in 1640 medium (Invitrogen, USA) with 10 % FBS (Invitrogen). Cells were passaged every 2 days with trypsin.
For small interfering RNA (siRNA) knockdown, DharmaFECT1 was employed (Dharmacon, CO, USA). The ON-TARGETplus siRNA for Rab11a was also obtained from Dharmacon (Dharmacon, CO, USA). Non-Targeting siRNA were used as a negative control.
pCMV6-Rab11a plasmid was obtained from OriGene company (OriGene, Rockville, USA). Attractene Transfection was used for transfection of plasmid (Qiagen, Hilden, Germany). A pCMV6 empty vector was used as a control. FH535 (Santa Cruz, USA) was used as Wnt inhibitor.
Western blot analysis
Total protein was extracted using Pierce Lysis Buffer (Pierce, Rockford, IL). Nuclear protein was extracted using Thermo Scientific NE-PER Nuclear and Cytoplasmic Extraction (Thermo Scientific). Protein quantification was performed using the Bradford method. Fifty micrograms sample protein was added to SDS-PAGE, which was transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, MA, USA). Incubation of primary antibodies was performed overnight at 4 °C. Antibody dilution was listed as follows: Rab11a (1:800, Proteintech, USA), cyclin D1, cyclin E, c-myc, MMP7 (1:1000; Cell signaling, Boston, MA, USA), and mouse monoclonal antibody against GAPDH (1:2000; Santa Cruz). After incubation with HRP-coupled anti-mouse or rabbit IgG antibody (1:1000 dilution, Cell Signaling Technology, USA) at 37 °C for 2 h, target proteins on PVDF membrane were visualized using Pierce ECL kit and captured using a DNR Bio-Imaging System (DNR, Jerusalem, Israel).
Quantitative real-time PCR
Quantitative real-time PCR was performed using SYBR Green master mix kit from Applied Biosystems. PCR was performed using 7500 Real-Time PCR System (Applied Biosystems) β-actin was used as the endogenous control. The relative levels of gene expression were represented as ΔCt = Ct gene–Ct reference, and the fold change of gene expression was calculated by the 2−ΔΔCt Method. The primer sequences are as follows:
-
Rab11a forward, 5′-AAAGCAAGAGCACCATTGGAG-3′, Rab11a reverse, 5′-TGCCCTGCTGTGTCCCAT-3′; cyclin D1 forward, 5′-TGGAGGTCTGCGAGGAACA-3′, cyclin D1 reverse, 5′-TTCATCTTAGAGGCCACGAACAT-3′; cyclin E forward, 5′-AGCCAGCCTTGGGACAATAAT-3′, cyclin E reverse, 5′-GAGCCTCTGGATGGTGCAAT-3′; c-myc forward, 5′-ACGGTGGTGGAGGAGCTCTT-3′, c-myc reverse, 5′-CGGTTGACGCTCTCCACAC-3′; MMP7 forward, 5′-CGGAGGAGATGCTCACTTCG-3′, MMP7 reverse, 5′-GGATCAGAGGAATGTCCCATACC-3′; β-actin forward, 5′-ATAGCACAGCCTGGATAGCAACGTAC-3′, β-actin reverse, 5′-CACCTTCTACAATGAGCTGCGTGTG-3′.
CCK-8 cell viability assay
Cell proliferation and viability were analyzed using Cell Counting Kit-8 (CCK-8) kit (Dojindo, Japan) according to the protocol. Briefly, 48 h after transient plasmid or siRNA transfection, cells were seeded in 96-well plates and treated with CCK-8 solution. After 3 h, wells were measured in a microplate reader at 450 nm.
Wnt reporter assay
T-cell factor (TCF)/LEF TOP-FLASH reporter plasmid (0.2 μg) and Renilla luciferase reporter plasmid were transfected into pancreatic cancer cells using Attractene reagent (Qiagen, Germany). About 48 h after transfection, dual luciferase reporter kit (Promega, USA) was used to detect luciferase activity.
Cell cycle analysis
Forty-eight hours after transfection, cells were harvested and fixed using 1 % paraformaldehyde. Then cells were washed with PBS and stained in 5 mg/ml propidium iodide for 30 min at room temperature. Flow cytometry was performed using BD FACSCalibur flow cytometer systems (Becton Dickinson, USA).
Cell apoptosis detection
Cell apoptosis detection was performed with Annexin V/PI double staining. Forty-eight hours after transfection, cells were harvested and washed in chill PBS. Then cells were resuspended in 250 μl of binding buffer. Annexin V/FITC solution and propidium iodide solution were added in cell suspension. After incubation in for 30 min, the cells were analyzed by BD FACSCalibur flow cytometer (Becton Dickinson, USA).
Invasion assay
Invasion assay was performed using a 24-well transwell chamber with 20 μl Matrigel from BD Bioscience (1:5 dilution, BD Bioscience, CA, USA). Briefly, cells were suspended in 100 μl of serum-free medium and added to the upper chambers. Six hundred microliters of medium with 15 % serum was added to the lower chamber. After 16–20 h, the cells on the upper chamber were wiped out with cotton tips. Cells that invaded through the filter to the low chamber were stained with hematoxylin. The invading cell number was counted under the microscope.
Immunofluorescence assay
Cells cultured in the chamber slides were fixed using paraformaldehyde, treated with Triton X-100, and then incubated with primary antibody overnight. After washed with PBS, the slides were incubated with Alexa Fluor 488-conjugated anti-IgG (Molecular Probes). Cell images were captured using an Olympus BX53 fluorescence microscope (Olympus, Tokyo, Japan).
Statistical analysis
SPSS software was used to obtain statistical analyses. χ 2 test was used to examine the clinicopathologic data. t test was used to compare densitometry data. p < 0.05 was considered to indicate statistical significance.
Results
Expression of Rab11a in normal and pancreatic cancer tissues
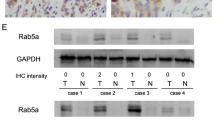
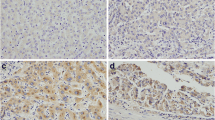
We investigated Rab11a protein expression in a panel of 96 pancreatic cancer specimens by immunohistochemistry. Rab11a showed negative/weak staining in normal pancreatic tissue (Fig. 1a). In cancer tissues, Rab11a protein was significantly upregulated, which was localized in the cytoplasmic compartment of cancer cells. We observed Rab11a overexpression in 32 out of 96 (33.3 %) pancreatic cancer tissues (Fig. 1b–d). We analyzed the relationship between Rab11a expression and clinical factors. No statistical difference was found between Rab11a overexpression and the characteristics of age, gender, T stage, nodal status, and differentiation (Table 1). Rab11a overexpression significantly correlated with tumor-node-metastasis (TNM) stage (p = 0.0111). We also confirmed Rab11a upregulation in eight-paired pancreatic cancer tissues using western blot and real-time reverse transcription PCR (RT-PCR). As shown in Fig. 1e, f, significant Rab11a protein upregulation was observed at both protein (mean gray value: 54.8 ± 14.6 vs 35.2 ± 12.7, p < 0.05) and messenger RNA (mRNA) levels in cancer tissues compared with adjacent normal tissues, especially in case 1, 2, 5, and 6.

Expression pattern of Rab11a in pancreatic cancer tissues. a Negative Rab11a staining in normal pancreatic tissue. b Negative Rab11a staining in pancreatic ductal carcinoma. c Moderate cytoplasmic staining of Rab11a in pancreatic cancer. d Strong cytoplasmic staining of Rab11a in pancreatic cancer (magnification 400×). e Protein expression of Rab11a in eight cases of paired pancreatic cancer tissues and corresponding normal tissues. Western blot showed that Rab11a protein was higher in tumor tissues in most cases. f Real-time RT-PCR showed that mRNA expression was upregulated in pancreatic cancer tissues
Rab11a regulates pancreatic cell proliferation and cell cycle
We examined Rab11a protein expression in three pancreatic cancer cell lines by western blot and immunofluorescence. High Rab11a expression was found in Bxpc3 cell line, and low Rab11a level was found in Capan2 cell line (Fig. 2a, b). Immunofluorescence showed that Rab11a was mainly located in the cytoplasm of cancer cells (Fig. 2b). To investigate biological function of Rab11a, specific siRNAs were transfected in Bxpc3 cell line and Rab11a plasmid was introduced into Capan2 cell line. Knockdown and transfection efficiency was confirmed by western blot and real-time RT-PCR (Fig. 3c, d). The role of Rab11a on cancer proliferation rate was checked using the CCK-8 assay and colony formation assay. As shown in Fig. 3a, Rab11a depletion decreased cell growth rate in Bxpc3 cells (absorbance at day 5; control siRNA vs Rab11a siRNA: 1.908 ± 0.026 vs 1.407 ± 0.016, p < 0.05). Rab11a overexpression promoted cell proliferation rate in Capan2 cells (absorbance at day 5; pCMV6 vs Rab11a plasmid: 1.027 ± 0.038 vs 1.361 ± 0.027, p < 0.05). In addition, colony formation assay showed that Rab11a depletion decreased the number of Bxpc3 colonies after 2 weeks incubation. Rab11a overexpression increased the colony numbers in Capan2 cells (Fig. 3b). Furthermore, we checked cell cycle distribution after treatment with siRNA or plasmid. As shown in Fig. 3c, we found that the percentage of G1 phase in Rab11a overexpressed Capan2 cells was decreased compared with control and the percentage of S phase was increased. In Rab11a depleted Bxpc3 cells, the percentage of G1 phase was increased and the percentage of S phase was decreased. These results indicated that Rab11a could induce cell proliferation by facilitating cell cycle transition in pancreatic cancer.

Knockdown and overexpression of Rab11a in pancreatic cancer cell lines. a Protein expression of Rab11a in pancreatic cancer cell lines. Bxpc3 cell line has relatively high expression, and Capan2 cell line has relatively low expression. b Immunofluorescence showed strong Rab11a protein expression in Bxpc3 cell line. c Western blot showed that siRNA treatment markedly decreases Rab11a levels in Bxpc3 cells and Rab11a transfection significantly increased its expression in Capan2 cells. d Real-time RT-PCR analysis showed that siRNA treatment markedly decreases Rab11a mRNA in Bxpc3 cells and Rab11a transfection significantly increased its mRNA in Capan2 cells. * p < 0.05

Rab11a knockdown inhibits and Rab11a overexpression promotes cell proliferation and cell cycle progression in pancreatic cancer cell lines. a CCK-8 assay showed that Rab11a knockdown inhibited proliferation in Bxpc3 cell line. Rab11a overexpression promoted proliferation in Capan2 cell line. b Colony formation assay showed that Rab11a knockdown decreased colony number in Bxpc3 cell line. Rab11a overexpression increased colony number in Capan2 cell line. c Rab11a knockdown increased G1 phase percentage and decreased S phase percentage in Bxpc3 cells. Rab11a overexpression increased S phase percentage and decreased G1 phase cell percentage in Capan2 cells. * p < 0.05
Rab11a induces gemcitabine resistance and promotes invasion
To characterize the impact of Rab11a on gemcitabine resistance, CCK-8 and flow cytometry were carried out in Bxpc3 and Capan2 cells with gemcitabine treatment (1 μm). As shown in Fig. 4a, Rab11a significantly downregulated cell viability in Bxpc3 cells at 48 and 72 h of gemcitabine treatment. On the contrary, Rab11a overexpression increased gemcitabine resistance in Capan2 cell line. Annexin V/PI staining was used to detect the degree of apoptosis (Fig. 4b). An increased percentage of gemcitabine-induced apoptosis (1 μm, 48 h) was observed in Bxpc3 cells transfected with Rab11a siRNA. Rab11a overexpression in Capan2 cells inhibited gemcitabine-induced apoptosis, demonstrating that Rab11a confers gemcitabine resistance in pancreatic cancer cells. Then we checked change of invading ability. As shown in Fig. 4c, Rab11a siRNA decreased invading ability of Bxpc3 cells (control vs Rab11a siRNA: 325 ± 19 vs 187 ± 11, p < 0.05) while Rab11a overexpression increased Capan2 invasion (pCMV vs Rab11a plasmid: 152 ± 9 vs 235 ± 16, p < 0.05).

Rab11a increases gemcitabine resistance and invasion. a Depletion of Rab11a expression decreases sensitivity to chemotherapeutic drug gemcitabine. Rab11a overexpression increased Capan2 cell survival treated with gemcitabine. The absorbance rate of cancer cells treated with gemcitabine was measured by the CCK-8 assay. Cell survival rate was calculated by comparing absorbance of the treated groups to that of the untreated group. b AnnexinV/PI analysis showed that Rab11a knockdown increased gemcitabine-induced apoptosis and its overexpression inhibited gemcitabine-induced apoptosis. c Rab11a depletion downregulated and its overexpression upregulated invading ability of pancreatic cancer cell lines. * p < 0.05
Rab11a regulates malignant behavior through Wnt signaling pathway
To check the potential mechanism of Rab11a induced cell proliferation, invasion, and chemoresistance, we examined several related proteins. Western blot and real-time RT-PCR analysis showed that Rab11a depletion downregulated protein and mRNA expression of cyclin D1, cyclin E, MMP7, and c-myc (Fig. 5a) while Rab11a overexpression upregulated protein and mRNA levels of cyclin D1, cyclin E, MMP7, and c-myc. Since these proteins are Wnt downstream targets, we checked if Rab11a could regulate Wnt activity using the luciferase reporter assay. As shown in Fig. 5b, Rab11a siRNA downregulated TOP-Flash luciferase activity while Rab11a plasmid upregulated luciferase activity, suggesting Rab11a regulates malignant behavior through Wnt signaling pathway. In addition, western blot and immunofluorescence showed that protein expression of nuclear β-catenin was upregulated after Rab11a overexpression and downregulated after Rab11a depletion (Fig. 5c, d). To validate the involvement of Wnt signaling in Rab11a induced biological effects, we employed Wnt inhibitor FH535 in Rab11a transfected Capan2 cells. As shown in Fig. 6a, Wnt inhibitor blocked the Rab11a-induced cyclin D1, MMP7, and c-myc. FH535 treatment also blocked the promoting effect of Rab11a on cell proliferation (Fig. 6b). In addition, we found that Rab11a upregulated phosphorylation of GSK3β while Rab11a depletion downregulated GSK3β phosphorylation (Fig. 5c), which is an upstream regulator of nuclear β-catenin.

Rab11a regulates Wnt signaling and target genes. a Western blotting and real-time RT-PCR analysis revealed that knockdown of Rab11a decreased the mRNA and protein levels of cyclin D1, cyclin E, c-myc, and MMP7. b Wnt TOP-Flash luciferase reporter assay showed that Rab11a depletion downregulated Wnt activity in Bxpc3 cell line. Rab11a overexpression increased Wnt luciferase activity in Capan2 cells. c Western blotting revealed that knockdown of Rab11a decreased protein levels of nuclear β-catenin and p-GSK3β expression. Rab11a overexpression showed the opposite effects. d Immunofluorescence showed that Rab11a knockdown decreased while its overexpression increased nuclear β-catenin. * p < 0.05

Rab11a regulates proliferation through GSK3β/Wnt signaling. a Western blotting analysis revealed that Wnt inhibitor FH535 blocked the role of Rab11a on cyclin D1, MMP7, and c-myc upregulation in Capan2 cells. b CCK-8 showed that Wnt inhibitor FH535 blocked the promoting effect of Rab11a on Capan2 cell proliferation. * p < 0.05
Discussion
Rab11a has been reported to be involved in intracellular vesicle trafficking protein, control of activity of small GTPases, and orientation of mitotic spindle [2, 3, 4]. Active Rab11a is required for E-cadherin trafficking and lumen formation during epithelial morphogenesis [6]. Rab11a has been indicated in human cancer progression. Rab11a regulates E-cadherin turnover and improves cytoskeleton reorganization for cell migration in colorectal cancer cells. Rab11a and E-cadherin together serve as potential markers for colorectal cancer progression [9]. These studies suggest Rab11a as a potential oncogene in human cancer development. However, its expression pattern, biological function, and molecular mechanism in pancreatic cancer were not clear. In the present study, we found Rab11a expression was upregulated in pancreatic cancer tissues using western blot, real-time RT-PCR, and immunohistochemistry. Statistical analysis showed there was a close correlation between high Rab11a expression and TNM stage. Expression rate of Rab11a was higher in high stage tumors than low stage tumors, suggesting a potential role of Rab11a during pancreatic cancer progression.
Then we employed siRNAs to deplete endogenous Rab11a in the Bxpc3 cells which have high endogenous Rab11a expression and overexpressed Rab11a in Capan2 cells. The CCK-8 assay and colony formation assay showed that Rab11a depletion significantly suppressed proliferation while Rab11a overexpression promoted proliferation. Cell cycle analysis showed that in Bxpc3 cell line, Rab11a depletion inhibited cell cycle transition at G1-S point and Rab11a transfection in Capan2 cells facilitated cell cycle. We also examined change of proliferation-related proteins and found Rab11a could induce cyclin D1, cyclin E and c-myc protein and mRNA. Cyclin D1 and cyclin E participate in G1-S transition. Their levels correlate with aggressiveness of pancreatic cancer cells and decreased postoperative survival [10, 11]. c-myc protein is a transcription factor, which is activated upon many signals such as Wnt and Shh. c-myc activates cell proliferation by upregulating cyclins and downregulating p21 [12]. c-myc also participates in pancreatic cancer cell growth [13, 14] Our results strongly suggested that Rab11a promotes pancreatic cancer proliferation and cell cycle transition by upregulation of cyclin and c-myc protein. In this study, we also found that Rab11a overexpression could depletion sensitized pancreatic cancer cell to gemcitabine treatment and inhibit gemcitabine-induced apoptosis. c-myc functions as an antiapoptotic protein in many cancers and also mediates drug resistance in pancreatic cancers [15–17]. Thus, Rab11a induced gemcitabine resistance of pancreatic cancer possibly through c-myc upregulation.
The involvement of Wnt signaling pathway has been reported in various human cancers. Canonical Wnt signaling leads to activation of TCF transcription factors, which induces upregulation of target genes including cyclin D1, c-myc, and MMP7. The involvement of Wnt activation has been demonstrated in human pancreatic cancer invasion and proliferation [18–20]. In this study, we found that Rab11a activated Wnt signaling and induced Wnt target genes. Rab11a induced nuclear localization of β-catenin. Blockage of Wnt signals abolished the role of Rab11a on proliferation and related proteins. Furthermore, Wnt signaling has been reported to play a part in drug resistance of pancreatic cancers [21, 22]. Our results suggest that Rab11a-induced activation of Wnt signaling accounts for its roles on cell proliferation and invasion. GSK3β is a kinase which plays a vital role in β-catenin degradation. Phosphorylation of GSK3β serine-9 inhibits its proteasomal degradation of β-catenin [23–25]. Our results revealed that GSK3β Ser-9 phosphorylation was involved in the Rab11a-mediated β-catenin stabilization and nuclear localization. These results together indicated that Rab11a promotes cell proliferation through GSK3β/Wnt/β-catenin signaling activation.
In conclusion, we showed that Rab11a was overexpressed in pancreatic cancers and promoted pancreatic cancer cell growth, invasion, and chemoresistance by regulation of cyclin proteins, MMP7, and c-myc, possibly through Wnt signaling pathway. These data implicated Rab11a as a potential diagnosis and therapeutic target in pancreatic cancer.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29.
Kessler D, Gruen GC, Heider D, Morgner J, Reis H, Schmid KW, et al. The action of small GTPases Rab11 and Rab25 in vesicle trafficking during cell migration. Cell Physiol Biochem. 2012;29(5–6):647–56.
Ramel D, Wang X, Laflamme C, Montell DJ, Emery G. Rab11 regulates cell-cell communication during collective cell movements. Nat Cell Biol. 2013;15(3):317–24.
Hehnly H, Doxsey S. Rab11 endosomes contribute to mitotic spindle organization and orientation. Dev Cell. 2014;28(5):497–507.
Kelly EE, Horgan CP, McCaffrey MW. Rab11 proteins in health and disease. Biochem Soc Trans. 2012;40(6):1360–7.
Desclozeaux M, Venturato J, Wylie FG, Kay JG, Joseph SR, Le HT, et al. Active Rab11 and functional recycling endosome are required for E-cadherin trafficking and lumen formation during epithelial morphogenesis. Am J Phys Cell Physiol. 2008;295(2):C545–56.
Palmieri D, Bouadis A, Ronchetti R, Merino MJ, Steeg PS. Rab11a differentially modulates epidermal growth factor-induced proliferation and motility in immortal breast cells. Breast Cancer Res Treat. 2006;100(2):127–37.
Kazemi-Noureini S, Colonna-Romano S, Ziaee AA, Malboobi MA, Yazdanbod M, Setayeshgar P, et al. Differential gene expression between squamous cell carcinoma of esophageus and its normal epithelium; altered pattern of mal, akr1c2, and rab11a expression. World J Gastroenterol. 2004;10(12):1716–21.
Chung YC, Wei WC, Huang SH, Shih CM, Hsu CP, Chang KJ, et al. Rab11 regulates E-cadherin expression and induces cell transformation in colorectal carcinoma. BMC Cancer. 2014;14:587.
Kornmann M, Ishiwata T, Itakura J, Tangvoranuntakul P, Beger HG, Korc M. Increased cyclin D1 in human pancreatic cancer is associated with decreased postoperative survival. Oncology. 1998;55(4):363–9.
Kornmann M, Arber N, Korc M. Inhibition of basal and mitogen-stimulated pancreatic cancer cell growth by cyclin D1 antisense is associated with loss of tumorigenicity and potentiation of cytotoxicity to cisplatinum. J Clin Invest. 1998;101(2):344–52.
Lin CJ, Malina A, Pelletier J. c-Myc and eIF4F constitute a feedforward loop that regulates cell growth: implications for anticancer therapy. Cancer Res. 2009;69(19):7491–4.
Shukla SK, Gunda V, Abrego J, Haridas D, Mishra A, Souchek J, et al. MUC16-mediated activation of mTOR and c-Myc reprograms pancreatic cancer metabolism. Oncotarget. 2015;6(22):19118–31.
Koenig A, Linhart T, Schlengemann K, Reutlinger K, Wegele J, Adler G, et al. NFAT-induced histone acetylation relay switch promotes c-Myc-dependent growth in pancreatic cancer cells. Gastroenterology. 2010;138(3):1189–99 .e1-2
Kumar K, Raza SS, Knab LM, Chow CR, Kwok B, Bentrem DJ, et al. GLI2-dependent c-MYC upregulation mediates resistance of pancreatic cancer cells to the BET bromodomain inhibitor JQ1. Sci Rep. 2015;5:9489.
Kim DY, Kim MJ, Kim HB, Lee JW, Bae JH, Kim DW, et al. Suppression of multidrug resistance by treatment with TRAIL in human ovarian and breast cancer cells with high level of c-Myc. Biochim Biophys Acta. 2011;1812(7):796–805.
McNeil CM, Sergio CM, Anderson LR, Inman CK, Eggleton SA, Murphy NC, et al. c-Myc overexpression and endocrine resistance in breast cancer. J Steroid Biochem Mol Biol. 2006;102(1–5):147–55.
Wang L, Heidt DG, Lee CJ, Yang H, Logsdon CD, Zhang L, et al. Oncogenic function of ATDC in pancreatic cancer through Wnt pathway activation and beta-catenin stabilization. Cancer Cell. 2009;15(3):207–19.
Pilarsky C, Ammerpohl O, Sipos B, Dahl E, Hartmann A, Wellmann A, et al. Activation of Wnt signalling in stroma from pancreatic cancer identified by gene expression profiling. J Cell Mol Med. 2008;12(6B):2823–35.
Zhou W, Li Y, Gou S, Xiong J, Wu H, Wang C, et al. MiR-744 increases tumorigenicity of pancreatic cancer by activating Wnt/beta-catenin pathway. Oncotarget. 2015;6(35):37557–69.
Wang B, Zou Q, Sun M, Chen J, Wang T, Bai Y, et al. Reversion of trichostatin A resistance via inhibition of the Wnt signaling pathway in human pancreatic cancer cells. Oncol Rep. 2014;32(5):2015–22.
Cui J, Jiang W, Wang S, Wang L, Xie K. Role of Wnt/beta-catenin signaling in drug resistance of pancreatic cancer. Curr Pharm Des. 2012;18(17):2464–71.
Benelli R, Monteghirfo S, Vene R, Tosetti F, Ferrari N. The chemopreventive retinoid 4HPR impairs prostate cancer cell migration and invasion by interfering with FAK/AKT/GSK3beta pathway and beta-catenin stability. Mol Cancer. 2010;9:142.
Luo J. Glycogen synthase kinase 3beta (GSK3beta) in tumorigenesis and cancer chemotherapy. Cancer Lett. 2009;273(2):194–200.
Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, et al. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8(12):1398–406.
Acknowledgments
This study was supported by China NSFC project (no. 31371177) and Health and Family Planning Commission of Liaoning project Province (LNCCC-D31-2015).We thank Dr. Yang Wang and Dr. Yang Liu for IHC evaluation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
None
Rights and permissions
About this article

Cite this article
Yu, L., Li, X., Li, H. et al. Rab11a sustains GSK3β/Wnt/β-catenin signaling to enhance cancer progression in pancreatic cancer. Tumor Biol. 37, 13821–13829 (2016). https://doi.org/10.1007/s13277-016-5172-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-016-5172-1