
Abstract
DNA breaks can be induced by exogenous stimuli or by endogenous stress, but are also generated during recombination of V, D, and J genes (V(D)J recombination), immunoglobulin class switch recombination (CSR). Among various DNA breaks generated, DNA double strand break (DSB) is the most deleterious one. DNA damage response (DDR) is initiated when DSBs are detected, leading to DNA break repair by non-homologous end joining (NHEJ). The process is critically important for the generation of diversity for foreign antigens; and failure to exert DNA repair leads to immunodeficiency such as severe combined immunodeficiency and hyper-IgM syndrome. In V(D)J recombination, DSBs are induced by RAG1/2; and generated post-cleavage hairpins are resolved by Artemis/DNA-PKcs/KU70/KU80. DDR is initiated by ataxia-telangiectasia mutated as a master regulator together with MRE11/RAD50/NBS1 complex. Finally, DSBs are repaired by NHEJ. The defect of one of the molecules shows various degree of immunodeficiency and radiosensitivity. Upon CSR inducing signal, DSBs induced by activation-induced cytidine deaminase and endonucleases elicit DDR. Broken ends are repaired either by NHEJ or by mismatch repair system. Patients with radiosensitive SCID require hematopoietic cell transplantation as a curative therapy; but the procedures for eradication of recipient hematopoietic cells are often associated with severe toxicity.
Similar content being viewed by others

Introduction
DNA damage response (DDR) is critically important in maintenance of proper cell functions. Among various DNA damage, a double strand DNA break (DSB) is the most deleterious which necessitates proper DNA response, resulting in successful cell survival when appropriately repaired or in cell death when the lesions are misrepaired. Inappropriate DSB repair can result in chromosomal aberration or translocation; the survived cells with the lesions then can develop malignancy when acquire growth advantage.
Cells respond to the DNA breaks with a sophisticated DDR, starting from detection of DNA legions, delivering DNA damage signaling, initiating DNA break repair system, and then deciding the cell fate [1]. The process is executed with participation of multiple molecules in an orchestrated manner. Generated DSBs activate a specific signal cascade that is mainly regulated by the kinase, ataxia-telangiectasia mutated (ATM), and are repaired mainly by non-homologous end joining (NHEJ). Homologous recombination (HR) also functions for the repair of broken ends but does so only in specific occasions when a sister chromatid is present [1,2,3,4,5].
DSB can be induced by exogenous stimuli or by endogenous stress such as replication, but is also generated in cells with a programmed manner. For example, the programmed DSB is observed in lymphocytes development and in meiosis [5, 6].
V(D)J (variable: V, diversity: D, and joining: J) recombination is a DNA rearrangement process that occurs at Immunoglobulin heavy chain (IgH) loci, Immunoglobulin light chain loci (Igκ and Igλ), and at T cell receptor loci (TCR α, β, γ, δ), and thus is critical in development of B and T lymphocytes. Immunoglobulin class switch recombination (CSR) occurs at IgH constant regions, and replaces Cμ to Cγ, Cα, or to Cε, resulting in the production of IgG, IgA, or IgE. RAG1/2 lymphoid specific factors initiate DSB formation in V(D)J recombination; and activation-induced cytidine deaminase (AID) triggers DSB formation in CSR response [5,6,7].
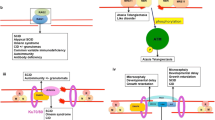
More than twenty molecules are involved in the V(D)J recombination and CSR; and inability to persecute the programmed DDR results in primary immunodeficiency (PID) (Table 1) [3, 6, 8,9,10,11,12,13,14,15]. The patients with defect either in DNA damage signal or in NHEJ process present PID phenotype, and often show one or more of phenotypes that include radiosensitivity, predisposition to malignancy, developmental delay, and neurological deficits (Table 2). These disorders provide us with an insight into physiological importance and function of each molecule in health and disease, specific function of a domain in each molecule, and with how DDR system works as a complex in a regulated manner.
Since detailed machinery of DDR and impact of defective responses to DNA damage on immune cell development and tumorigenesis have been covered by several well-described papers [3, 6, 8, 10, 12,13,14,15], this review will mainly focus on phenotypes of PID due to impaired DDR including newly discovered disorders (Fig. 1).

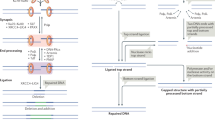
DNA repair proteins involved in V(D)J recombination. RSS recombination signal sequence
Molecular event of V(D)J recombination and CSR
In order for immune cells to respond to various foreign antigens, lymphoid cells generate great diversity of antigen receptors, B-cell receptor (BCR) in B cells and T-cell receptor (TCR) in T cells. The receptors exhibit numerous variety through variable domains generated through the combination of two or three gene regions (VDJ or VJ). V, D, and J domains are consisted as multiple copies, thus enabling the production of diversity by the combination of V, D, J or that of V and J. The coding genes, V, D, and J, are flanked by recombination signal sequences (RSS); and V(D)J recombination is initiated by recognition of the RSS and by cleavage at the site, by sequence-specific recombinase, RAG1 and RAG2 [16]. The programmed DNA break and DNA repair are restricted at the G0/G1 phase; and the generated DSB is repaired by NHEJ. When DSBs are induced, coding ends of BCR or TCR gene loci are sealed by a hairpin. The hairpin is bound by KU70/80 which form DNA dependent protein kinase (DNA-PK) complex together with DNA-PK catalytic subunit (DNA-PKcs coded by PRKDC gene), and the complex recruits Artemis (DCLRE1C) [9]. DNA-PK phosphorylates Artemis; and activated Artemis opens the hairpin via its endo-/exonuclease activity. DNA damage signaling molecules also play a role at this programmed DDR. ATM and the MRN complex (MRE11, RAD50, and NBS1 that are stable only when all the components are present) accumulate at the break site and initiate DDR signal [17]. P53 is one of key substrates of ATM; and thus ATM regulates cell cycle and dictates whether cells survive or die with unrepaired DSBs [18]. 53BP1 and RNF168 also facilitates DDR. RNF168 is shown to ubiquitilate 53BP1 that is thought to have an overlapping role with XLF (DNA repair protein) [19]. Finally, DSBs are repaired by the DNA ligase IV (LIG IV)/XRCC4-XLF complex by NHEJ [20]. Recent research identified a paralog of XRCC4 and XLF (PAXX), and showed a redundant role of PAXX in DNA repair system (the entire process of V(D)J recombination is shown in Fig. 2).

DNA repair proteins involved in Immunoglobulin class switch recombination (CSR). AID activation induced cytidine deaminase, APE apurinic/apyrimidic endonuclease, UNG uracyl N-glycosiase
B cells can undergo CSR, in which a set of IgH region is replaced by another Ig region, allowing B cells to produce different Ig subsets. CSR is initiated by activation of AID that deaminates cytidine to uracil at the transcriptionally active switch region [21]. Introduced uracil is modified by uracil-DNA glycosylase (UNG) and then removed by base excision repair. Abasic site is subsequently cleaved by apurinic/apyrimidinic endonuclease, generating DNA single strand break (SSB). Closely generated DNA SSB at the both strand lead to DSB. Alternatively, mismatch repair create DSBs by the contribution of MMR complex including MSH2, MSH6, MLH1, PMS2, and EXO1. The Sμ/Sx synapse is repaired with assistance of ATM/MRN/53BP1/RNF168/γH2AX, and then repaired with the same machinery used in the final process of V(D)J recombination [11]. Though hairpin is not formed in CSR process, Artemis is supposed to be involved by resolution of complex region generated in CSR [22, 23] (the entire scheme of CSR is shown in Fig. 2).
Disorders mainly present severe combined immunodeficiency (SCID) phenotype
RAG1/2 deficiency
RAG1 and RAG2 are absolutely required for V(D)J recombination; and thus mutations in RAG1 or RAG2 result is defect in B and T cells and show B-T-NK+ severe combined immunodeficiency (SCID) phenotype [6, 10]. Patients with partial loss of the recombination activity exhibits a variety of phenotype. These include Omenn syndrome (OS) that show erythrodermia, hepatosplenomegaly, lymphoadenopathy, alopecia, and eosinophilia with elevated serum IgE. T cells are detected, but are usually oligoclonal. The reason for elevated IgE is largely unknown. Hypomorphic mutation in RAG1 is reported in a patient with common variable immunodeficiency and in IgA deficiency [24, 25]. RAG1/2 deficient SCID is not radiosensitive, does not show any other developmental delay, but requires planning for hematopoietic cell transplantation (HCT) as soon as the diagnosis is made.
DCLRE1C (Artemis) deficiency
Artemis is phosphorylated by DNA-PKcs and by ATM, but is mainly activated by the former phosphatidylinositol (PI)-3-kinase. Defect in DCLRE1C gene is observed in B-T-NK+ radiosensitive SCID [26,27,28,29]. Mutations in DCLRE1C are usually observed in the N-terminal region leading to the loss of nuclease activity. The most frequent mutations (>50%) were large deletions of exons 1–3 or exons 1–4 due to a homologous recombination of the wild-type DCLRE1C with a pseudo-DCLRE1C gene located 5′ to the DCLRE1C start codon. Hypomorphic DCLRE1C mutation exhibits OS phenotype. Most of the patients with leaky SCID were compound heterozygous for one loss-of-function and one hypomorphic allele, with significant residual levels of recombination and DNA repair activity [29]. Patients with defective Artemis show predisposition to lymphoma. Radiosensitivity in Artemis deficiency is not prominent compared to that in LIG IV deficiency or XLF deficiency; and the patients do not show growth retardation or neurological deficits.
DNA ligase IV (LIG IV) deficiency
DNA ligase IV (LIG IV) is a ligase that resolves the DSBs during V(D)J recombination among three DNA ligase family. LIG IV deficiency was originally found in a patient with leukemia who showed severe side effect of chemotherapy. Subsequently, the disorder was reported in patients with microcephaly, growth retardation, pancytopenia, and various degree of immune dysfunction. Commonly associated features include growth failure, severe microcephaly, pancytopenia and a predisposition to lymphoid malignancy [30]. Most cases reported harbor missense LIG IV mutations or nucleotide deletions displaying a variety of presentation. The missense mutations with reduced ligase IV activity (typically 5–10%) present combined immunodeficiency. The patients show a wide range of immunological abnormality with decreased B, T, and NK cells.
XLF (NHEJ1, Cernunnos) deficiency
XLF deficiency was identified in patients with radiosensitive SCID and cytopenia [31]. It was postulated that XLF is critical in V(D)J recombination given the fact that it is a component of LIG IV complex; however, the immune system is relatively mildly affected compared to other SCID. Recent study demonstrates that XLF is dispensable for V(D)J recombination [32]. After cleavage in vitro, the RAG proteins remain associated with the DNA ends in a post-cleavage complex (PCC), which serves a crucial function in joining coding and signal ends. The RAG complex may play a role in assisting repair of RAG-mediated DSBs in the absence of XLF [32]. XLF, instead, stimulate N-nucleotide insertion hereby generating receptor diversity [33]. Defective XLF is associated with impaired survival and premature aging of hematopoietic stem cells, leading to cytopenia [34, 35].
XLF-related molecules and phenotype of the defect (non-SCID)
XRCC4 and XLF are a member of the same protein family and show structural similarity playing a role in NHEJ in concert with LIG IV. It has been postulated that defect in XRCC4 would display a phenotype of SCID as observed in immunodeficiency caused by a defect in XLF or in LIG IV. Recent study has identified a compound heterozygous mutation in XRCC4 in a patient with microcephaly and cerebellar ataxia with normal immune cell composition and response [36]. The cells showed severe defect in DSB repair, but displayed normal V(D)J recombination capacity using an artificial substrate.
PRKDC (DNA-PKcs) deficiency
Gene analysis of B-T-NK + SCID patient without any developmental delay or mutation in RAG1/2 revealed homozygous mutation in PRKDC in 2009 [27]. The mutated DNA-PKcs did not showed normal protein expression and possessed normal kinase activity, but lacked in a capacity to activate Artemis. Another PRKDC deficient patient showed reduced DNA-PKcs protein expression, undetectable kinase activity, and impaired DSB rejoining. The patient also exhibited microcephaly, developmental delay, and severe neurological deficit [37]. These findings indicate DNA-PK is critical in neurological development. Mutated DNA-PKcs failed to induce autoimmune regulator (AIRE)-dependent gene expression in vitro, which correlated with the presence of anti-calcium sensor receptor antibody in the patient. DNA-PKcs and KU70/80 are particularly abundant in human; and may play an additional role in genome stability such as protection of telomere damage. In fact, mice or dogs lacking in DNA-PKcs display SCID phenotype but show almost normal growth and neurological development.
Disorder mainly present CSR deficiency
AID deficiency and UNG deficiency
The most common identified cause of autosomal recessive hyper-IgM syndrome results from defects in the AICDA gene encoding for AID, an enzyme required for CSR and somatic hypermutation (SHM) in B cells [7, 38, 39]. Expression of AID is upregulated in response to signal through CD40 in B cells and it initiates CSR by deaminating deoxycytosine in the switch regions of the IgH genes generating deoxyuracils in both DNA strands. UNG then removes deoxyuracils from DNA and initiates the DNA repair pathway. Defects in UNG also result in hyper-IgM syndrome [7, 38]. AID deficiency and UNG deficiency have lymph node hyperplasia caused by the presence of giant germinal centers. Six of 22 patients with AID defects had autoimmunity or inflammation: diabetes mellitus, polyarthritis, autoimmune hepatitis, hemolytic anemia, immune thrombocytopenia, Crohn’s disease and chronic uveitis.
INO80 deficiency
Defect in INO80 was recently reported in two patients with normal IgM, decreased IgG and IgA [11]. INO80 is a component of chromatin remodeling complex and is involved in DNA repair. The presence of both INO80 and cohesion subunit SMC were detected on Sα and Eμ regions in model B cells, suggesting the role of INO80 in S-region synapsis during CSR. INO80 may modulate cohesin activity in B cells.
Mismatch repair deficiency syndrome
Constitutional mismatch repair deficiency (CMMRD) syndrome is a disorder associated with a high risk of malignancy [40]. Causative mutations are detected in DNA MMR genes, PMS2, MSH6, MSH2 or MLH1. The mutations are reported in patients with hereditary non-polyposis colorectal cancer (HNPCC) or Lynch syndrome. CMMRD is characterized by childhood brain tumors, hematological malignancies, and gastrointestinal cancer later in life. Modest decrease of IgG, particularly IgG2 subclass, but not IgA was reported in MSH6 deficiency [41]. The defect leads to a partial Ig-CSR defect and an abnormal SHM pattern. MSH6 seems to be not only able to convert staggered DSBs to blunt DSBs but is also involved in S junction DSB repair.
RIDDLE syndrome (RNF168)
RIDDLE (radiosensitivity, immunodeficiency, dysmorphic features, and learning difficulties) syndrome is a novel immunodeficiency disorder associated with defective DSB repair [42, 43]. Immunologically the patients showed decreased IgG and IgA. RIDDLE syndrome shares overlapping clinical features with ataxia telangiectasia (A-T). The cells from a RIDDLE patient exhibit impaired re-localization of 53BP1 and BRCA1 to DSBs, while MDC1 and NBS1 remain unaffected. The responsible gene for the syndrome is RNF168 that mediates ubiquitylation of 53BP1 [19]. The modification is required for the recruitment of 53BP1 at the sites of DSBs and for its function in DNA damage repair, checkpoint regulation and genomic integrity.
Disorders involved in DDR and NHEJ signaling
Ataxia telangiectasia
Ataxia telangiectasia (A-T) is an autosomal recessive neurodegenerative disorder characterized by early onset ataxia, telangiectasia at around 6–7 years old, and an increased incidence of malignancy, and the disorder is caused by mutation in ATM [44, 45]. About 30% of A-T patients exhibit immunodeficiency, showing decreased T cell number due to impaired efficiency in V(D)J rearrangement and decreased T cell neogenesis. Decreased serum IgG2 and IgA are also noted, which is caused by impaired CSR. Increase of serum alpha-fetoprotein (AFP) and chromosomal translocations involving chromosomes 7 and 14 are frequently noted in A-T patients. The most prominent feature of A-T is radiosensitivity. ATM kinase plays a central role in DDR by phosphorylation of various DDR molecules, and in involved not only in DSB repair, cell cycle regulation, cell fate decision, but also in transcriptional regulation, telomere maintenance, mitochondria maintenance, nonsense mediated decay, and autophagy [2, 46]. Defect in ATM leads to predisposition to malignancy, especially to leukemia and lymphoma.
M/R/N complex deficiency
Deficiency in NBN(NBS1) is known as Nijmegen Breakage syndrome (NBS), a chromosomal instability syndrome characterized by microcephaly, ‘‘bird-like’’ face, growth retardation, mild mental retardation, immunodeficiency, and a predisposition to malignancy [47]. Immunological studies reveal mild or severe leukopenia in nearly half of the patients with evident reduction in lymphocytes. Absolute number of T cells decrease in majority of patients, accompanied by a reduced CD4+ T cells. The number of B cells is reduced in three quarter of NBS patients. The humoral immunodeficiency in NBS patients ranges from agammaglobulinemia to a moderate reduction in the immune response. IgG2 subclass deficiency is also reported in NBS, indicating involvement of NBN in CSR.
Defect in RAD50 resembles phenotype of NGS. The patients presented microcephaly, mental retardation, ‘bird-like’ face, and short stature, but had normal lymphocyte counts and immunoglobulin level [48].
Patients with MRE11 deficiency display Ataxia telangiectasia like disorder (ATLD) with ataxia, cerebellar atrophy, ocular apraxia, but without telangiectasia [49, 50]. No immunological abnormality has been reported so far; but the disorder show predisposition to malignancy.
MRE11, RAD50, and NBN function as a M/R/N complex at DDR signal; and each component is indispensable for the stable complex formation. However, contribution of each component to immune response, especially in V(D)J recombination and CSR could differ.
Other genomic instability disorders that can exhibit immunodeficiency
Fanconi anemia (FA) is a heterogeneous disorder that produces a range of phenotypes that are highly variable. These include bone marrow failure (pancytopenia), developmental delay, malformation, and high prevalence of hematological malignancies [51]. At least 19 genes are found to cause FA so far [52]. The protein encoded by the genes function together in a common pathway called as the FA pathway. The FA pathway uses components of other known DNA repair processes to achieve proper repair of ICLs [53]. In addition, Fanconi anaemia proteins have functions in genome maintenance including the stabilization of replication forks and the regulation of cytokinesis. Recent study (Sekinaka Y, et al. J Clin Immunol, in press) has demonstrated that FANC mutations are involved in impaired lymphogenesis probably by the accumulation of DNA replication stress, leading to phenotype of common variable immunodeficiency. Some of the patients show undetectable T-cell receptor excision circles and Kappa deleting recombination excision circles.
Therapeutic measures for immunodeficiency due to defective DDR
Patients with B-T-radiosensitive SCID phenotype regardless of presence or absence of NK cells suffer from recurrent or severe infection and thus require reconstitution of their immune system as a curative measure. These disorders include deficiency in DCLRE1C (Artemis), DNA ligase IV (LIG IV), XLF (Cernunnos). Results of hematopoietic cell transplantation for radiation-sensitive SCID suggest that minimizing exposure to alkylating agents and ionizing radiation is important for optimizing survival and minimizing late effects [54]. LIG IV deficiency and XLF deficiency hardly survive myeloablative conditioning regimen, thus needs reduced intensity conditioning regimen. On the other hand, those with residual NK activity can reject donor hematopoietic cells. Artemis deficiency does not have hematopoietic stem cell defect, necessitating agents to remove host hematopoietic cells and to open marrow niches before the transplantation. One of the conditioning regimen currently used is a combination of Flurarabine, low dose cyclophosphamide, and serotherapy to remove T cells. Development of non-genotoxic conditioning regimen to suppress residual NK cell activity and generate marrow niches is awaited.
References
Sirbu BM, Cortez D. DNA damage response: three levels of DNA repair regulation. Cold Spring Harbor Perspect Biol. 2013;5(8):a012724.
Guleria A, Chandna S. ATM kinase: much more than a DNA damage responsive protein. DNA Repair. 2016;39:1–20.
O’Driscoll M. Diseases associated with defective responses to DNA damage. Cold Spring Harb Perspect Biol. 2012;4:a012773.
Hanawalt PC. Historical perspective on the DNA damage response. DNA Repair. 2015;36:2–7.
Bednarski JJ, Sleckman BP. Lymphocyte development: integration of DNA damage response signaling. Adv Immunol. 2012;116:175–204.
de Villartay JP. Congenital defects in V(D)J recombination. Br Med Bull. 2015;114(1):157–67.
Durandy A, Taubenheim N, Peron S, Fischer A. Pathophysiology of B-cell intrinsic immunoglobulin class switch recombination deficiencies. Adv Immunol. 2007;94:275–306.
de Miranda NF, Bjorkman A, Pan-Hammarstrom Q. DNA repair: the link between primary immunodeficiency and cancer. Ann N Y Acad Sci. 2011;1246:50–63.
de Villartay JP, Fischer A, Durandy A. The mechanisms of immune diversification and their disorders. Nat Rev Immunol. 2003;3(12):962–72.
de Villartay JP, Poinsignon C, de Chasseval R, Buck D, Le Guyader G, Villey I. Human and animal models of V(D)J recombination deficiency. Curr Opin Immunol. 2003;15(5):592–8.
Kracker S, Di Virgilio M, Schwartzentruber J, Cuenin C, Forveille M, Deau MC, et al. An inherited immunoglobulin class-switch recombination deficiency associated with a defect in the INO80 chromatin remodeling complex. J Allergy Clin Immunol. 2015;135(4):998 e6–1007 e6.
Mizutani S, Takagi M. XCIND as a genetic disease of X-irradiation hypersensitivity and cancer susceptibility. Int J Hematol. 2013;97(1):37–42.
Nakada S. Abnormalities in DNA double-strand break response beyond primary immunodeficiency. Int J Hematol. 2011;93(4):425–33.
Prochazkova J, Loizou JI. Programmed DNA breaks in lymphoid cells: repair mechanisms and consequences in human disease. Immunology. 2016;147(1):11–20.
Woodbine L, Gennery AR, Jeggo PA. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair (Amst). 2014;16:84–96.
Schatz DG, Ji Y. Recombination centres and the orchestration of V(D)J recombination. Nature Rev Immunol. 2011;11(4):251–63.
Paull TT. Mechanisms of ATM activation. Annu Rev Biochem. 2015;84:711–38.
Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14(4):197–210.
Bohgaki M, Bohgaki T, El Ghamrasni S, Srikumar T, Maire G, Panier S, et al. RNF168 ubiquitylates 53BP1 and controls its response to DNA double-strand breaks. Proc Natl Acad Sci USA. 2013;110(52):20982–7.
Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211.
Keim C, Kazadi D, Rothschild G, Basu U. Regulation of AID, the B-cell genome mutator. Genes Dev. 2013;27(1):1–17.
Du L, van der Burg M, Popov SW, Kotnis A, van Dongen JJ, Gennery AR, et al. Involvement of Artemis in nonhomologous end-joining during immunoglobulin class switch recombination. J Exp Med. 2008;205(13):3031–40.
Rivera-Munoz P, Soulas-Sprauel P, Le Guyader G, Abramowski V, Bruneau S, Fischer A, et al. Reduced immunoglobulin class switch recombination in the absence of Artemis. Blood. 2009;114(17):3601–9.
Abolhassani H, Wang N, Aghamohammadi A, Rezaei N, Lee YN, Frugoni F, et al. A hypomorphic recombination-activating gene 1 (RAG1) mutation resulting in a phenotype resembling common variable immunodeficiency. J Allergy Clin Immunol. 2014;134(6):1375–80.
Kato T, Crestani E, Kamae C, Honma K, Yokosuka T, Ikegawa T, et al. RAG1 deficiency may present clinically as selective IgA deficiency. J Clin Immunol. 2015;35(3):280–8.
Le Deist F, Poinsignon C, Moshous D, Fischer A, de Villartay JP. Artemis sheds new light on V(D)J recombination. Immunol Rev. 2004;200:142–55.
van der Burg M, Ijspeert H, Verkaik NS, Turul T, Wiegant WW, Morotomi-Yano K, et al. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J Clin Investig. 2009;119(1):91–8.
Schlissel MS. Does artemis end the hunt for the hairpin-opening activity in V(D)J recombination? Cell. 2002;109(1):1–4.
Felgentreff K, Lee YN, Frugoni F, Du L, van der Burg M, Giliani S, et al. Functional analysis of naturally occurring DCLRE1C mutations and correlation with the clinical phenotype of ARTEMIS deficiency. J Allergy Clin Immunol. 2015;136(1):140 e7–140e7.
Altmann T, Gennery AR. DNA ligase IV syndrome; a review. Orphanet J Rare Dis. 2016;11(1):137.
Revy P, Malivert L, de Villartay JP. Cernunnos-XLF, a recently identified non-homologous end-joining factor required for the development of the immune system. Curr Opin Allergy Clin Immunol. 2006;6(6):416–20.
Lescale C, Abramowski V, Bedora-Faure M, Murigneux V, Vera G, Roth DB, et al. RAG2 and XLF/Cernunnos interplay reveals a novel role for the RAG complex in DNA repair. Nat Commun. 2016;7:10529.
Ij H, Rozmus J, Schwarz K, Warren RL, van Zessen D, Holt RA, et al. XLF deficiency results in reduced N-nucleotide addition during V(D)J recombination. Blood. 2016;128(5):650–9.
Tilgner K, Neganova I, Singhapol C, Saretzki G, Al-Aama JY, Evans J, et al. Brief report: a human induced pluripotent stem cell model of cernunnos deficiency reveals an important role for XLF in the survival of the primitive hematopoietic progenitors. Stem Cells. 2013;31(9):2015–23.
Avagyan S, Churchill M, Yamamoto K, Crowe JL, Li C, Lee BJ, et al. Hematopoietic stem cell dysfunction underlies the progressive lymphocytopenia in XLF/Cernunnos deficiency. Blood. 2014;124(10):1622–5.
Guo C, Nakazawa Y, Woodbine L, Bjorkman A, Shimada M, Fawcett H, et al. XRCC4 deficiency in human subjects causes a marked neurological phenotype but no overt immunodeficiency. J Allergy Clin Immunol. 2015;136(4):1007–17.
Mathieu AL, Verronese E, Rice GI, Fouyssac F, Bertrand Y, Picard C, et al. PRKDC mutations associated with immunodeficiency, granuloma, and autoimmune regulator-dependent autoimmunity. J Allergy Clin Immunol. 2015;135(6):1578.e5–1588.e5.
Qamar N, Fuleihan RL. The hyper IgM syndromes. Clin Rev Allergy Immunol. 2014;46(2):120–30.
Casellas R, Basu U, Yewdell WT, Chaudhuri J, Robbiani DF, Di Noia JM. Mutations, kataegis and translocations in B cells: understanding AID promiscuous activity. Nat Rev Immunol. 2016;16(3):164–76.
Wimmer K, Kratz CP, Vasen HF, Caron O, Colas C, Entz-Werle N, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium ‘care for CMMRD’ (C4CMMRD). J Med Genet. 2014;51(6):355–65.
Gardes P, Forveille M, Alyanakian MA, Aucouturier P, Ilencikova D, Leroux D, et al. Human MSH6 deficiency is associated with impaired antibody maturation. J Immunol. 2012;188(4):2023–9.
Blundred RM, Stewart GS. DNA double-strand break repair, immunodeficiency and the RIDDLE syndrome. Expert Rev. 2011;7(2):169–85.
Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136(3):420–34.
Perlman SL, Boder Deceased E, Sedgewick RP, Gatti RA. Ataxia-telangiectasia. Handbook. 2012;103:307–32.
Ambrose M, Gatti RA. Pathogenesis of ataxia-telangiectasia: the next generation of ATM functions. Blood. 2013;121(20):4036–45.
Shiloh Y. ATM: expanding roles as a chief guardian of genome stability. Exp Cell Res. 2014;329(1):154–61.
Chrzanowska KH, Gregorek H, Dembowska-Baginska B, Kalina MA, Digweed M. Nijmegen breakage syndrome (NBS). Orphanet J Rare Dis. 2012;7:13.
Waltes R, Kalb R, Gatei M, Kijas AW, Stumm M, Sobeck A, et al. Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder. Am J Hum Genet. 2009;84(5):605–16.
Taylor AM, Groom A, Byrd PJ. Ataxia-telangiectasia-like disorder (ATLD)-its clinical presentation and molecular basis. DNA Repair. 2004;3(8–9):1219–25.
Uchisaka N, Takahashi N, Sato M, Kikuchi A, Mochizuki S, Imai K, et al. Two brothers with ataxia-telangiectasia-like disorder with lung adenocarcinoma. J Pediatr. 2009;155(3):435–8.
Schneider M, Chandler K, Tischkowitz M, Meyer S. Fanconi anaemia: genetics, molecular biology, and cancer—implications for clinical management in children and adults. Clin Genet. 2015;88(1):13–24.
Dong H, Nebert DW, Bruford EA, Thompson DC, Joenje H, Vasiliou V. Update of the human and mouse Fanconi anemia genes. Hum Genomics. 2015;9:32.
Ceccaldi R, Sarangi P, D’Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol. 2016;17(6):337–49.
Cowan MJ, Gennery AR. Radiation-sensitive severe combined immunodeficiency: the arguments for and against conditioning before hematopoietic cell transplantation—what to do? J Allergy Clin Immunol. 2015;136(5):1178–85.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Morio, T. Recent advances in the study of immunodeficiency and DNA damage response. Int J Hematol 106, 357–365 (2017). https://doi.org/10.1007/s12185-017-2263-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-017-2263-8