
Abstract
Semantic dementia (SD) is a unique syndrome in the frontotemporal lobar degeneration spectrum. Typically presenting as a progressive, fluent anomic aphasia, SD is the paradigmatic disorder of semantic memory with a characteristic anatomical profile of asymmetric, selective antero-inferior temporal lobe atrophy. Histopathologically, most cases show a specific pattern of abnormal deposition of protein TDP-43. This relatively close clinical, anatomical and pathological correspondence suggests SD as a promising target for future therapeutic trials. Here, we discuss outstanding nosological and neurobiological challenges posed by the syndrome and propose a pathophysiological model of SD based on sequential, regionally determined disintegration of a vulnerable neural network.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Semantic dementia (SD) is a unique clinicopathological syndrome in the frontotemporal lobar degeneration (FTLD) spectrum. Although the precise nosology continues to be debated, according to an emerging consensus SD subsumes the semantic variant of primary progressive aphasia (Adlam et al. 2006; Grossman 2010; Gorno-Tempini et al. 2011). The SD syndrome is characterised by an insidious deterioration of semantic memory (the memory system mediating knowledge of the meanings of words, objects and concepts) associated with focal asymmetric atrophy chiefly involving the anterior and inferior temporal lobes. Case histories compatible with SD have been described for over a century (Pick 1892; Hodges and Patterson 2007). However, the significance of such cases was not appreciated until the seminal work of Warrington (1975) defining the neuropsychological syndrome and Mesulam (1982) identifying the group of focal ‘language-led dementias’ (the primary progressive aphasias), followed by the coining of the term ‘semantic dementia’ by Snowden et al. (1989). Recent decades have witnessed an intense international research effort devoted to the SD syndrome and its brain correlates using a range of neuropsychological, neuroimaging and neuropathological approaches. The recent identification of TAR-DNA binding protein 43 (TDP-43) as the major component of pathological ubiquitinated inclusions in a substantial proportion of FTLD cases (the ‘TDP-43 proteinopathies’—Neumann 2009) has opened an unprecedented window on the molecular pathophysiology of this group of diseases, including SD. Although it is an uncommon disorder (with estimated prevalence 1–5/100,000 between ages 45 and 64, extrapolating from population-based dementia surveys—Hodges and Patterson 2007), SD is of high clinical and neurobiological importance. Clinically, it is the paradigmatic disorder of human semantic memory and presents a coherent clinical picture with largely predictable evolution and pathology. Neurobiologically, it is a striking illustration of a key contemporary theme in the field of neurodegenerative disease, namely, the selective vulnerability of neuronal populations or networks. Potentially, therefore, SD holds insights both for our understanding of the organisation of human brain knowledge systems and the network pathophysiology of neurodegeneration. However, there are a number of key unresolved issues surrounding the SD syndrome and its brain basis.
Clinical Features
SD usually manifests as an insidious fluent aphasia with reduced vocabulary. The syndrome often presents in late middle life; however, the incidence in the elderly has almost certainly been underestimated. In a recent pathologically ascertained series of 100 cases, the mean age of onset was 60.3 years with a range of 40–79 years (Hodges et al. 2010). It is generally a sporadic disorder (Rohrer et al. 2009; Hodges et al. 2010). The features of the language disturbance are captured in the recent consensus criteria for SD (Gorno-Tempini et al. 2011) and reflect a core deficit of word meaning knowledge. Typically, the patient complains of word-finding difficulty and will ask the meaning of familiar words; on examination, there is profound anomia, impaired generation of words and categories, reduced comprehension of single words and regularisation errors on reading words aloud (for example, sounding the English word ‘yacht’ as ‘yached’), resulting from the inappropriate use of ‘surface’ rules of sound–print correspondence (‘surface’ dyslexia). Speech articulation, phonology, syntax and prosody are intact and repetition of words and phrases and comprehension of grammatical relations (within the limits of single word comprehension) are normal. Initially, speech is generally copious and circumlocutory but the relentless erosion of vocabulary leads (as in other progressive aphasia syndromes) ultimately to mutism.
During the course of SD, the meaning of sensory objects is generally also lost with supervening agnosia for visual, auditory (Bozeat et al. 2000; Goll et al. 2010), tactile (Coccia et al. 2004) and chemosensory (Rami et al. 2007; Piwnica-Worms et al. 2010) stimuli: the deficit in these cases particularly affects object recognition, or the association of the sensory percept with meaning (‘associative agnosia’), while perceptual encoding and discrimination are relatively (or entirely) spared. In some patients, nonverbal deficits dominate the clinical presentation: of these nonverbal presentations, the best defined is selective impairment of face recognition (progressive prosopagnosia), and such cases usually develop significant aphasia later in the course. Nonverbal associative agnosias can be demonstrated within the respective sensory modalities (i.e. after removing potentially confounding effects from verbal labelling or other cross-modal recognition procedures—Hodges and Patterson 2007; Goll et al. 2010; Piwnica-Worms et al. 2010). Furthermore, analogous kinds of deficits appear in separate modalities (Hodges and Patterson 2007): thus, for nonverbal objects, as for words, recognition becomes heavily dependent on familiarity and typicality, with better recognition of superordinate categories than specific exemplars and a tendency to make ‘typicalisation’ errors (the patient may identify a peacock only as a bird or later, as an animal, and may also omit to draw its distinctive tail or may represent it with four legs when asked to draw a peacock from memory—the nonverbal visual equivalent of ‘surface dyslexia’). In cognitive neuropsychological terms, this decline into typicality and generality could be characterised as a ‘pruning’ of the semantic space corresponding to stored concepts (Lambon Ralph et al. 2010). From one perspective, the development of deficits extending across modalities in SD implies an underlying essentially ‘pan-modal’ defect of semantic memory. Integral to the SD syndrome is initially intact (or relatively preserved) performance in a range of other cognitive domains, including episodic (autobiographical) memory and perceptual, spatial, praxic and nonverbal executive functions (Warrington 1975; Graham et al. 2000; Hodges and Patterson 2007).
Although (in contrast to the behavioural variant of frontotemporal dementia) behavioural disturbances are not usually prominent at presentation in SD, these generally do become significant as the disease evolves and include obsessionality and mental rigidity, clock-watching, preoccupation with games and puzzles, irritability and disinhibition, alterations of eating behaviour with odd food preferences, and loss of emotional understanding and responsiveness (Edwards-Lee et al. 1997; Hodges and Patterson 2007; Rohrer and Warren 2010). These behaviours overlap closely with the spectrum of frontotemporal dementia and are likely to arise both from impaired understanding of social signals (for example, culturally sanctioned food combinations—Piwnica-Worms et al. 2010) and spread of disease to extratemporal areas, in particular orbitofrontal cortex. Other neurological (extrapyramidal and rarely, amyotrophic) features may emerge later in the course of SD (Hodges and Patterson 2007; Östberg P and Bogdanovic 2010; Kremen et al. 2011) though these are seldom evident at presentation. Patients may remain in a state of dependency for a number of years; the median clinical disease duration to death in the large series reported by Hodges et al. (2010) was 12.8 years.
The differential diagnosis of SD includes other forms of primary progressive aphasia, behavioural variant frontotemporal dementia and clinical Alzheimer’s disease (Kertesz et al. 2010). However, the striking and disproportionate involvement of semantic memory, with loss of word knowledge despite well-preserved language structure and fluency, generally allows SD to be distinguished from these other possibilities, at least in its earlier stages. While semantic impairment does accompany Alzheimer’s disease (Garrard et al. 1998), it is rarely the leading feature, as in SD.
Brain Anatomy: Structure and Function
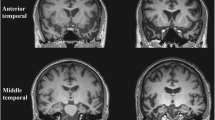
The clinical syndrome of SD is associated with a characteristic neuroanatomical profile, which usually secures the diagnosis (see Fig. 1). Structural volumetric MRI typically reveals selective atrophy of the antero-inferior temporal lobe, usually bilateral but asymmetric, and frequently more severe in the left hemisphere. Atrophy is most marked in temporal polar cortex, fusiform gyrus and mesial temporal structures (Chan et al. 2001) with a clear antero-posterior gradient; knife-blade atrophy at the pole gives way to relatively normal cortex in more posterior temporal and peri-Sylvian regions. This atrophy profile distinguishes SD from both frontotemporal dementia (Rosen et al. 2002) and Alzheimer’s disease (Chan et al. 2001). As might be anticipated in an essentially sporadic disease, little information is available concerning the very earliest anatomical changes in SD; however, longitudinal imaging using MRI volumetry (Czarnecki et al. 2008; Rohrer et al. 2009) and tensor-based morphometry (Brambati et al. 2009) has shown that disease evolution is associated with extension of atrophy to inferior frontal, insular and more posterior temporal lobe cortices and to the contralateral temporal lobe, so that the less affected temporal lobe ‘catches up’ over the course of the disease; post mortem data indicate a largely symmetric distribution of atrophy (Davies et al. 2009). This evidence further strengthens the case for regarding the left and right temporal lobe ‘variants’ of SD as a single coherent clinic–anatomical syndrome (Seeley et al. 2005). Atrophy rates during the phase of early established clinical disease are sufficiently large and uniform that either whole brain or regional lobar atrophy could feasibly serve as a biomarker in future clinical trials: in terms of projected sample sizes required to detect a moderate MRI treatment effect, SD compares favourably with other FTLD subtypes (Rohrer et al. 2009; Knopman et al. 2009; Krueger et al. 2010; Gordon et al. 2010).

Representative coronal T1-weighted MRI sections of the brain of a patient with semantic dementia (clinical disease duration 3 years). The sections show key structures at the level of the temporal pole (TP), anterior temporal lobe (aTL) and posterior temporal lobe (pTL); the left hemisphere is shown on the left in all sections. The sections show a characteristic profile of asymmetric focal atrophy most markedly affecting the anterior and inferior temporal lobes
More recently, MR volumetry has been complemented by other structural and functional imaging techniques and these provide a convergent picture of regional neural network breakdown in SD. Regional resting metabolic changes overlap with (but may extend beyond) the zone of maximal cortical atrophy (Diehl et al. 2004; Diehl-Schmid et al. 2006; Desgranges et al. 2007; Acosta-Cabronero et al. 2011); however, task-related PET and fMRI has demonstrated more widespread alterations associated with verbal semantic processing (Mummery et al. 1999), reading (Wilson et al. 2009), topographical recognition (Cipolotti and Maguire 2003) and autobiographical memory (Maguire et al. 2010), suggesting distributed functional reorganisation involving networks coupled to the primary semantic network centred on the anterior temporal lobes. Diffusion tensor tractography suggests possible anatomical substrates for these functional changes: SD is associated with signal alterations in major white matter tracts traversing the temporal lobe (the inferior longitudinal, arcuate and uncinate fasciculi) and the genu of the corpus callosum but sparing the fronto-parietal superior longitudinal fasciculus (Whitwell et al. 2010; Agosta et al. 2010; Acosta-Cabronero et al. 2011). Resting-state fMRI has demonstrated that atrophy in SD targets a leftward-asymmetric intrinsic connectivity network linking the temporal pole, amygdala and subgenual cingulate (Seeley et al. 2009): these findings align SD with an emerging paradigm of neurodegenerative disease, according to which large-scale brain networks present in the healthy brain are selectively targeted by particular neurodegenerative pathologies (Seeley 2008; Zhou et al. 2010). To date, these networks are best understood for Alzheimer’s disease and the behavioural variant of frontotemporal dementia, but network disintegration is likely to be of general relevance as a pathophysiological paradigm potentially explaining a range of neurodegenerative diseases.
Particular features of the SD syndrome have been shown to correlate with the regional distribution of atrophy. Verbal deficits are especially associated with left (dominant) temporal lobe involvement while nonverbal deficits (including prosopagnosia, other forms of object agnosia and impaired knowledge of living things) have been linked to right temporal lobe involvement (Joubert et al. 2003; Thompson et al. 2003; Snowden et al. 2004; Mendez et al. 2010; Mion et al. 2010). More specifically, atrophy of the right anterior fusiform gyrus has been linked to deficits of face recognition (Josephs et al. 2008; Omar et al. 2010a). Behavioural disturbances too are often more prominent in association with predominantly right-sided atrophy (Seeley et al. 2005; Hodges and Patterson 2007). Rather than any strict partition of functions between hemispheres or within the temporal lobe, the balance of evidence suggests disruption of an integrated bi-temporal network that mediates semantic processing across sensory modalities, with relative anatomical selectivity for particular aspects of semantic knowledge or particular knowledge modalities within this network (Snowden et al. 2004). The temporal poles (a site of particularly severe damage in SD) may serve a key role, perhaps by instantiating a cross-modal or amodal network ‘hub’ for integration of modality-specific information into coherent concepts (Lambon Ralph et al. 2007, 2010), though other candidate regions for such a hub (in particular, the anterior fusiform gyrus) have been advanced (Mion et al. 2010). Precise one-to-one mappings between regional atrophy and cognitive deficits are often difficult to substantiate: for example, the severity of hippocampal atrophy in SD is comparable to that seen in Alzheimer’s disease (Chan et al. 2001), yet episodic memory deficits in SD (in contrast to Alzheimer’s) are generally not a significant clinical issue. The key to resolving this apparent paradox is likely to lie with the network basis of SD (and other neurodegenerative disorders): thus, hippocampal atrophy in SD is associated with atrophy of an anterior network focussed on surrounding neocortical areas in the temporal and inferior frontal lobes, whereas hippocampal atrophy in Alzheimer’s is associated with breakdown of a posteriorly directed network including the posterior cingulate gyrus (Pereira et al. 2009; Lehmann et al. 2010; Acosta-Cabronero et al. 2011).
Histopathology
Among the syndromes in the FTLD spectrum, the histopathological substrate for SD is one of the best defined: at pathological examination, approximately 75% of cases have features conforming to subtype 1 in the original Sampathu classification of the TDP-43 proteinopathies or type C in the recently proposed consensus classification system (Sampathu et al. 2006; Cairns et al. 2007; Grossman 2010; Mackenzie et al. 2011). This subtype is characterised by the presence of numerous, long tortuous dystrophic neurites staining positive for TDP-43 in superficial cortical laminae (predominantly layers II and III) with sparse neuronal cytoplasmic inclusions and few or no neuronal intranuclear inclusions. An important minority of cases of SD have alternative histopathological and molecular associations, including other TDP-43 proteinopathies (Östberg P and Bogdanovic 2010), mutations in the gene coding microtubule-associate protein tau (Bessi et al. 2010), classical Pick’s disease (Hodges et al. 2010) and Alzheimer’s disease (Grossman 2010). The aphasia associated with mutations in the progranulin gene may resemble SD but can generally be distinguished clinically based on the presence of grammatical and phonological errors and associated parietal signs (Rohrer et al. 2010). However, such cases underline the need for more precise diagnostic criteria, both to identify the ‘core’ syndrome of SD and to distinguish it from overlap and ‘halo’ syndromes. This issue is not merely of academic interest: while the relatively specific mapping between clinical syndrome, anatomical profile and tissue pathology makes SD an attractive target for future trials of disease modifying therapies, the success of such therapies will depend on accurate prediction of the target disease process (Kertesz et al. 2010).
Outstanding Difficulties
Here, we argue that the design and evaluation of rational therapies for SD will depend ultimately on a detailed characterisation of the pathophysiology of the syndrome, and that this in turn will require an understanding of the phenomenology of regional network dysfunction and the mechanisms that translate the molecular lesion of SD to a large-scale ensemble of neurones. At present, these remain elusive goals.
From a phenomenological perspective, the essential characteristics of the SD syndrome are well understood but the cognitive basis for the syndrome has not been fully defined and unresolved problems remain. These chiefly concern the scope of the syndrome, the relations between the component deficits and the boundaries of the core deficit. Thus, SD affects all modalities of object knowledge, yet certain self-contained, specialised symbolic systems (in particular, mathematics and music) may be relatively spared (Hailstone et al. 2009; Omar et al. 2010b; Julien et al. 2008; Weinstein et al. 2011), suggesting that the core deficit lies with world-based (referential) knowledge rather than truly global semantic failure. At least earlier in the course of SD, the erosion of semantic function is not uniform across modalities (for example, verbal semantic deficits generally lead nonverbal semantic deficits: Hodges and Patterson 2007) nor always within modalities (category effects, though uncommon, are documented—Lambon Ralph et al. 2003). It is uncertain to what extent these effects reflect the testing procedures used to document the deficits, the intrinsic cognitive demands of processing in different semantic domains or the underlying disease process. However, this evidence suggests at least some neuropsychological differentiation within the damaged semantic system. Despite the emphasis on semantic impairment, deficits of apperceptive processing (structural representation of objects) have been reported in SD for both the visual and auditory modalities (Joubert et al. 2003; Caine et al. 2009; Goll et al. 2010): it is not clear whether such deficits simply reflect spread of disease to adjacent cortical areas mediating apperceptive processing or point to a more fundamental disruption of object analysis at the interface between perceptual and associative mechanisms. While the balance of evidence supports network dysfunction as a key mechanism in the pathogenesis of SD, the anatomical status of the putative culprit network is less well established than (for example) the default mode network implicated in the pathogenesis of Alzheimer’s disease. Taken together with the neuropsychological data (in particular, apparent disparities between verbal and nonverbal deficits), this has led to the ‘suggestion that two or more separate networks are essentially involved in SD, with implications for the nosology of the syndrome in relation to the progressive aphasias’ (Mesulam et al. 2003). These issues may only be settled by joint neuropsychological and neuroimaging (especially, functional imaging) experiments designed to test specified hypotheses about the network organisation of object knowledge and semantic memory more broadly.
From a molecular perspective, the mechanisms by which abnormal proteins lead to selective neuronal destruction remain largely unknown, though a number of candidates have been identified. Axonal transport deficits might reduce growth factor supply to projection neurones resulting in spreading axonal degeneration (Salehi et al. 2009), or abnormal proteins might themselves spread transynaptically between neurones (as in the case of prion protein—Bartz et al. 2002). Abnormally folded tau has been shown to induce abnormal folding in nearby tau molecules (Frost et al. 2009). Information concerning the in vivo behaviour of TDP-43 remains limited; however, it is likely to play a key role in gene transcription and splicing regulation, in addition to other cellular processes including apoptosis, cell division, mRNA stabilisation, regulation of neuronal plasticity and nuclear ‘scaffolding’ (Neumann 2009). These activities of TDP-43 could potentially have downstream effects via loss of function or toxic gain of function mechanisms. If a cortical convergence zone or ‘hub’ were affected by the molecular process, this strategic localisation would tend to amplify its effects with rapid disintegration of an entire connected neuronal system (Seeley et al. 2009).
A Pathophysiological Model of Semantic Dementia
Taking these caveats and uncertainties into account, we outline a pathophysiological model of SD which makes certain key assumptions. The model is schematised in Fig. 2. First and most fundamentally, we assume that the SD syndrome arises from disintegration of a distributed neural network with specific intrinsic anatomical and functional connectivity. We further assume that semantic information about objects and concepts is distributed within this network. For present purposes, we do not assume a particular form of semantic network organisation amongst the several which have been proposed (for a review see Lambon Ralph et al. 2007, 2010). However, the model should generate two key features of the SD syndrome: the progressive neuropsychological erosion of fine-grained object concepts with ‘pruning’ of concept boundaries (Lambon Ralph et al. 2010) and anatomical asymmetry within and between the temporal lobes. Each of these features could arise if a gradient of disease-related damage were established following an initial stochastic insult (or insults) within the network. Here, we make a key assumption that the pathological process acts trans-synaptically and spreads in an anatomically restricted manner via projection neurones linking regions within each temporal lobe as well as homologous regions in both temporal lobes. In this scenario, the most heavily connected network elements (e.g. convergence zones in the anterior temporal lobe) that are most likely to integrate detailed (feature-rich) semantic representations would be relatively more vulnerable to the effects of the instigating insult. In contrast, more remote network elements (e.g. those in the posterior ipsilateral and contralateral temporal lobes) would be relatively less vulnerable. This feature of the model is in line with recent empirical data in SD (Acosta-Cabronero et al. 2011).

A pathophysiological model of semantic dementia. The schematic represents a distributed neural network in both temporal lobes, linked via commissural fibres (C); the temporal lobes are outlined in coronal section (compare Fig. 1) and stylised ‘neurons’ represent different elements of the network. In this representation, the ‘left’ temporal lobe is the more affected (atrophic) but the model does not assume a particular lateralisation. An initial (stochastic) neurodegenerative event (here indicated as cross-hatching at location I) propagates trans-synaptically through the network. The schematic shows a single instigating neurodegenerative focus; however, this could be multifocal. The neurodegenerative process is assumed to be ‘diluted’ as it traverses successive synaptic relays (here coded as grey within the initially maximally affected temporal lobe, and black in the contralateral temporal lobe): though the instigating molecular lesion has not been characterised precisely, such a dilution could in principle result from spread of a toxic molecule or loss of function effects (e.g. synaptic dysregulation or loss of trophic support). Trans-synaptic processes are assumed to act bidirectionally (i.e. potentially both anterograde and retrograde); however, in general there will be a net ‘direction’ of effects according to whether the triggering event is local or remote from the involved network element. The overall result of the process is a gradient of neural damage across the network. At a given disease stage, areas with high synaptic convergence (locations II and III) are more severely affected by the neurodegenerative process (represented as unfilled circles), establishing a gradient of atrophy within the temporal lobe. In addition, there is an inter-hemispheric gradient of damage manifesting as asymmetric temporal lobe atrophy. A precise mapping of commissural pathways between homologous areas in opposite temporal lobes and mirrored network connectivity patterns in each temporal lobe are together assumed to recapitulate the sequence of regional atrophy evolution in the initially less affected temporal lobe
A critical (and still undetermined) feature of the model is the nature of the underlying molecular lesion that allows the putative tissue gradients to become established. This lesion could in principle act via a toxic gain of function or loss-of-function mechanism: axonal transfer of a toxic molecule, synaptic dysregulation or loss of trophic support could allow a gradual trans-synaptic spread of disease restricted to target projection elements within the network. In this regard, we note that cortical laminae II and III (a major focus of pathology in the most common histopathological substrate of SD) are key targets for inter-hemispheric cortico-cortical afferent fibres, and lamina III is the principal source of cortico-cortical efferent fibres (Miller 1996). We suggest that a disease process with these characteristics could produce a graded and anatomically selective atrophy profile like that found in SD. One strong (and empirically testable) prediction of this model is a precise sequential spread of atrophy ‘mirrored’ between homologous subregions in each temporal lobe: empirical data, though limited, are consistent with such a longitudinal atrophy profile (Czarnecki et al. 2008). This mirroring process would be facilitated by inter-hemispheric disease spread (i.e. via commissural pathways); however, involvement of commissural pathways need not be the sole mechanism. If network connectivity is itself mirrored in each temporal lobe, this would ensure that the sequence of disease spread is recapitulated in the initially less affected temporal lobe without necessarily invoking inter-hemispheric effects. This principle could also account for a similar pattern of atrophy evolution arising from different kinds of molecular lesions (i.e. different histopathlogical substrates), in line with the pathological heterogeneity of the SD syndrome. It follows that network morphology, rather than the precise histopathological insult, would be the key determinant of the macroscopic and clinical manifestations of SD. More speculatively, intrinsic asymmetries of network organisation in the left and right temporal lobes might account for the over-representation of cases with leftward asymmetric temporal lobe atrophy (though this is potentially attributable to various factors, including ascertainment bias).
Future Directions
Aside from being a promising candidate target for future clinical trials and a compelling experiment of nature, SD is a key model system for understanding how neurodegeneration translates to regional brain dysfunction. The essential linkage in this pathophysiological cascade appears to be neural network disintegration. If indeed SD is a specific ‘network-opathy’, future progress in understanding and ultimately treating this focal dementia will require a detailed analysis of the nature of the network dysfunction and the mechanisms by which such specific dysfunction could arise within a neuronal ensemble. This enterprise will require a multidisciplinary approach drawing on neuropsychology, structural and functional brain imaging and molecular physiology. We anticipate that the fusion of these traditionally disparate levels of analysis will herald a paradigm shift in neurodegenerative disease research and a new era of rational therapies.
References
Acosta-Cabronero J., Patterson K., Fryer T.D., Hodges J.R., Pengas G., Williams G..B and Nestor P.J. (2011) Atrophy, hypometabolism and white matter abnormalities in semantic dementia tell a coherent story. Brain [Epub ahead of print].
Adlam AL, Patterson K, Rogers TT et al (2006) Semantic dementia and fluent primary progressive aphasia: two sides of the same coin? Brain 129:3066–3080
Agosta F, Henry RG, Migliaccio R et al (2010) Language networks in semantic dementia. Brain 133:286–299
Bartz JC, Kincaid AE, Bessen RA (2002) Retrograde transport of transmissible mink encephalopathy within descending motor tracts. J Virol 76:5759–5768
Bessi V, Bagnoli S, Nacmias B, Tedde A, Sorbi S, Bracco L (2010) Semantic dementia associated with mutation V363I in the tau gene. J Neurol Sci 296:112–114
Bozeat S, Lambon Ralph MA, Patterson K, Garrard P, Hodges JR (2000) Nonverbal semantic impairment in semantic dementia. Neuropsychologia 38:1207–1215
Brambati SM, Rankin KP, Narvid J et al (2009) Atrophy progression in semantic dementia with asymmetric temporal involvement: a tensor-based morphometry study. Neurobiol Aging 30:103–111
Caine D, Breen N, Patterson K (2009) Emergence and progression of ‘non-semantic’ deficits in semantic dementia. Cortex 45:483–494
Cairns NJ, Bigio EH, Mackenzie IRA et al (2007) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 114:5–22
Chan D, Fox NC, Scahill RI et al (2001) Patterns of temporal lobe atrophy in semantic dementia and Alzheimer's disease. Ann Neurol 49:433–442
Cipolotti L, Maguire EA (2003) A combined neuropsychological and neuroimaging study of topographical and non-verbal memory in semantic dementia. Neuropsychologia 41:1148–1159
Coccia M, Bartolini M, Luzzi S, Provinciali L, Lambon Ralph MA (2004) Semantic memory is an amodal, dynamic system: evidence from the interaction of naming and object use in semantic dementia. Cogn Neuropsychol 21:513–527
Czarnecki K, Duffy JR, Nehl CR et al (2008) Very early semantic dementia with progressive temporal lobe atrophy: an 8-year longitudinal study. Arch Neurol 65:1659–1663
Davies RR, Halliday GM, Xuereb JH, Kril JJ, Hodges JR (2009) The neural basis of semantic memory: evidence from semantic dementia. Neurobiol Aging 30:2043–2052
Desgranges B, Matuszewski V, Piolino P et al (2007) Anatomical and functional alterations in semantic dementia: a voxel-based MRI and PET study. Neurobiol Aging 28:1904–1913
Diehl J, Grimmer T, Drzezga A, Riemenschneider M, Förstl H, Kurz A (2004) Cerebral metabolic patterns at early stages of frontotemporal dementia and semantic dementia. A PET study. Neurobiol Aging 25:1051–1056
Diehl-Schmid J, Grimmer T, Drzezga A et al (2006) Longitudinal changes of cerebral glucose metabolism in semantic dementia. Dement Geriatr Cogn Disord 22:346–351
Edwards-Lee T, Miller BL, Benson DF et al (1997) The temporal variant of frontotemporal dementia. Brain 120:1027–1040
Frost B, Ollesch J, Wille H, Diamond MI (2009) Conformational diversity of wild-type Tau fibrils specified by templated conformation change. J Biol Chem 284:3546–3551
Garrard P, Patterson K, Watson PC, Hodges JR (1998) Category specific semantic loss in dementia of Alzheimer’s type. Functional–anatomical correlations from cross-sectional analyses. Brain 121:633–646
Goll JC, Crutch SJ, Loo JHY et al (2010) Non-verbal sound processing in the primary progressive aphasias. Brain 133:272–285
Gordon E, Rohrer JD, Kim LG et al (2010) Measuring disease progression in frontotemporal lobar degeneration: a clinical and MRI study. Neurology 74:666–673
Gorno-Tempini ML, Hillis AE, Weintraub S et al (2011) Classification of primary progressive aphasia and its variants. Neurology 76:1006–1014
Graham KS, Simons JS, Pratt KH, Patterson K, Hodges JR (2000) Insights from semantic dementia on the relationship between episodic and semantic memory. Neuropsychologia 38:313–324
Grossman M (2010) Primary progressive aphasia: clinicopathological correlations. Nat Rev Neurol 6:88–97
Hailstone JC, Omar R, Warren JD (2009) Relatively preserved knowledge of music in semantic dementia. J Neurol Neurosurg Psychiatry 80:808–809
Hodges JR, Patterson K (2007) Semantic dementia: a unique clinicopathological syndrome. Lancet Neurol 6:1004–1014
Hodges JR, Mitchell J, Dawson K et al (2010) Semantic dementia: demography, familial factors and survival in a consecutive series of 100 cases. Brain 133:300–306
Josephs KA, Whitwell JL, Vemuri P et al (2008) The anatomic correlate of prosopagnosia in semantic dementia. Neurology 71:1628–1633
Joubert S, Felician O, Barbeau E, Sontheimer A, Barton JJ, Ceccaldi M, Poncet M (2003) Impaired configurational processing in a case of progressive prosopagnosia associated with predominant right temporal lobe atrophy. Brain 126:2537–2550
Julien CL, Thompson JC, Neary D, Snowden JS (2008) Arithmetic knowledge in semantic dementia: is it invariably preserved? Neuropsychologia 46:2732–2744
Kertesz A, Jesso S, Harciarek M, Blair M, McMonagle P (2010) What is semantic dementia? A cohort study of diagnostic features and clinical boundaries. Arch Neurol 67:483–489
Knopman DS, Jack CR, Kramer JH et al (2009) Brain and ventricular volumetric changes in frontotemporal lobar degeneration over 1 year. Neurology 72:1843–1849
Kremen SA, Mendez MF, Tsai PH, Teng E (2011) Extrapyramidal signs in the primary progressive aphasias. Am J Alzheimers Dis Other Demen 26:72–77
Krueger CE, Dean DL, Rosen HJ et al (2010) Longitudinal rates of lobar atrophy in frontotemporal dementia, semantic dementia, and Alzheimer's disease. Alzheimer Dis Assoc Disord 24:43–48
Lambon Ralph MA, Patterson K, Garrard P, Hodges JR (2003) Semantic dementia with category specificity: a comparative case-series study. Cogn Neuropsychol 20:307–326
Lambon Ralph MA, Lowe C, Rogers TT (2007) Neural basis of category-specific semantic deficits for living things: evidence from semantic dementia, HSVE and a neural network model. Brain 130:1127–1137
Lambon Ralph MA, Sage K, Jones RW, Mayberry EJ (2010) Coherent concepts are computed in the anterior temporal lobes. Proc Natl Acad Sci USA 107:2717–2722
Lehmann M, Rohrer JD, Clarkson MJ et al (2010) Reduced cortical thickness in the posterior cingulate gyrus is characteristic of both typical and atypical Alzheimer's disease. J Alzheimers Dis 20:587–598
Mackenzie I.R., Neumann M., Baborie A. et al. (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol [Epub ahead of print]
Maguire EA, Kumaran D, Hassabis D, Kopelman MD (2010) Autobiographical memory in semantic dementia: a longitudinal fMRI study. Neuropsychologia 48:123–136
Mendez MF, Kremen SA, Tsai P-H, Shapira JS (2010) Interhemispheric differences in knowledge of animals among patients with semantic dementia. Cogn Behav Neurol 23:240–246
Mesulam MM (1982) Slowly progressive aphasia without generalized dementia. Ann Neurol 11:592–598
Mesulam MM, Grossman M, Hillis A, Kertesz A, Weintraub S (2003) The core and halo of primary progressive aphasia and semantic dementia. Ann Neurol 54(Suppl 5):S11–S14
Miller R (1996) Neural assemblies and laminar interactions in the cerebral cortex. Biol Cybern 75:253–261
Mion M, Patterson K, Acosta-Cabronero J et al (2010) What the left and right anterior fusiform gyri tell us about semantic memory. Brain 133:3256–3268
Mummery CJ, Patterson K, Wise RJ, Vandenberghe R, Price CJ, Hodges JR (1999) Disrupted temporal lobe connections in semantic dementia. Brain 122:61–73
Neumann M (2009) Molecular neuropathology of TDP-43 proteinopathies. Int J Mol Sci 10:232–246
Omar R, Rohrer JD, Hailstone JC, Warren JD (2010a) Structural neuroanatomy of face processing in frontotemporal lobar degeneration. J Neurol Neurosurg Psychiat. doi:10.1136/jnnp.2010.227983
Omar R, Hailstone JC, Warren JE, Crutch SJ, Warren JD (2010b) The cognitive organization of music knowledge: a clinical analysis. Brain 133:1200–1213
Östberg P, Bogdanovic N (2010) Semantic dementia with lower motor neuron disease showing FTLD-TDP type 3 pathology (sensu Mackenzie). Neuropathology 31:271–279
Pereira JM, Williams GB, Acosta-Cabronero J et al (2009) Atrophy patterns in histologic vs clinical groupings of frontotemporal lobar degeneration. Neurology 72:1653–1660
Pick A (1892) Uber die Beziehungen der senilen Hirnatrophie zur Aphasie. Prager Med Wochenschrift 17:165–167
Piwnica-Worms KE, Omar R, Hailstone JC, Warren JD (2010) Flavour processing in semantic dementia. Cortex 46:761–768
Rami L, Loy CT, Hailstone J, Warren JD (2007) Odour identification in frontotemporal lobar degeneration. J Neurol 254:431–435
Rohrer JD, Warren JD (2010) Phenomenology and anatomy of abnormal behaviours in primary progressive aphasia. J Neurol Sci 293:35–38
Rohrer JD, Guerreiro R, Vandrovcova J et al (2009) The heritability and genetics of frontotemporal lobar degeneration. Neurology 73:1451–1456
Rohrer JD, Rossor MN, Warren JD (2010) Syndromes of nonfluent primary progressive aphasia: a clinical and neurolinguistic analysis. Neurology 75:603–610
Rosen HJ, Gorno-Tempini ML, Goldman WP et al (2002) Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology 58:198–208
Salehi A, Wu C, Zhan K, Mobley WC (2009) Axonal transport of neurotrophic signals: an Achilles' heel for neurodegeneration? In: St. George-Hyslop PH, Mobley WC (eds) Intracellular traffic and neurodegenerative disorders. Springer, Berlin, pp 87–101
Sampathu DM, Neumann M, Kwong LK et al (2006) Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol 169:1343–1352
Seeley WW (2008) Selective functional, regional, and neuronal vulnerability in frontotemporal dementia. Curr Opin Neurol 21:701–707
Seeley WW, Bauer AM, Miller BL et al (2005) The natural history of temporal variant frontotemporal dementia. Neurology 64:1384–1390
Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD (2009) Neurodegenerative diseases target large-scale human brain networks. Neuron 62:42–52
Snowden JS, Goulding PJ, Neary D (1989) Semantic dementia: a form of circumscribed cerebral atrophy. Behav Neurol 2:167–182
Snowden JS, Thompson JC, Neary D (2004) Knowledge of famous faces and names in semantic dementia. Brain 127:860–872
Thompson SA, Patterson K, Hodges JR (2003) Left/right asymmetry of atrophy in semantic dementia: behavioural cognitive implications. Neurology 61:1196–1203
Warrington EK (1975) The selective impairment of semantic memory. Q J Exp Psychol 27:635–657
Weinstein J, Koenig P, Gunawardena D, McMillan C, Bonner M, Grossman M (2011) Preserved musical semantic memory in semantic dementia. Arch Neurol 68:248–250
Whitwell JL, Avula R, Senjem ML et al (2010) Gray and white matter water diffusion in the syndromic variants of frontotemporal dementia. Neurology 74:1279–1287
Wilson SM, Brambati SM, Henry RG et al (2009) The neural basis of surface dyslexia in semantic dementia. Brain 132:71–86
Zhou J, Greicius MD, Gennatas ED et al (2010) Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer's disease. Brain 133:1352–1367
Acknowledgements
This work was undertaken at UCLH/UCL who received a proportion of funding from the Department of Health’s NIHR Biomedical Research Centres funding scheme. The Dementia Research Centre is an Alzheimer’s Research UK Co-ordinating Centre. This work was funded by the Wellcome Trust and by the UK Medical Research Council. JDW is supported by a Wellcome Trust Senior Clinical Fellowship.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Fletcher, P.D., Warren, J.D. Semantic Dementia: a specific network-opathy. J Mol Neurosci 45, 629–636 (2011). https://doi.org/10.1007/s12031-011-9586-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-011-9586-3