
Abstract
Atopic dermatitis (AD) is a clinically defined, highly pruritic, chronic inflammatory skin disease. In AD patients, the combination of a genetic predisposition for skin barrier dysfunction and dysfunctional innate and adaptive immune responses leads to a higher frequency of bacterial and viral skin infections. The innate immune system quickly mobilizes an unspecific, standardized first-line defense against different pathogens. Defects in this system lead to barrier dysfunction which results in increased protein allergen penetration through the epidermis and predisposes to secondary skin infections. Two loss-of-function mutations in the epidermal filaggrin gene are associated with AD. Also, inducible endogenous antibiotics such as the antimicrobial peptides cathelicidin and the beta-defensins may show defective function in lesional AD skin. Eczema herpeticum is a disseminated viral infection almost exclusively diagnosed in AD patients, which is based on unmasking of the viral entry receptor nectin-1, lack of cathelicidin production by keratinocytes, and depletion of Type I IFN-producing plasmacytoid dendritic cells from AD skin. Future therapeutic approaches to AD may include enhancement of impaired innate in addition to downregulation of dysfunctional adaptive immunity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
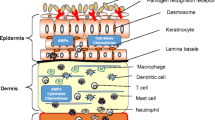
Atopic dermatitis (AD) is a clinically defined chronic inflammatory skin disease with a high incidence in industrialized countries. AD is characterized by a complex genetic background, a disturbed immune system, environmental trigger factors, and typical skin manifestations (Fig. 1) [1, 2]. AD patients show malfunctioning innate and adaptive immune responses and have a strong tendency to mount Th2 responses with the secretion of IL-4 and IL-13 upon exposure to environmental allergens as well as pathogens. The mechanisms of dysfunctional immune regulation in AD have been addressed in numerous clinical and experimental studies. From a functional view, the human immune system may be divided into an adaptive and an innate immune system. Whereas the latter is defined by and has the advantage of a learning capacity, the former does not learn but is quicker in its reaction (Fig. 2). It makes more sense to apply this delineation to immune mechanisms than to entire cells because most cell types play a role in both innate and adaptive immunities.

Immunopathogenesis of atopic dermatitis

Key features of innate versus adaptive immunity
Although many clinical features of AD are direct consequences of the AD patient’s tendency to mount Th2-skewed immune responses using their acquired immune system, innate immunity is also disturbed in AD patients [3]. Secondary cutaneous infections of lesional skin with viruses, bacteria, and fungi play a major role in the clinical course of AD and cause the most severe complications of this disease [4, 5]. In this article, we will review selected aspects and players of the cutaneous innate immune system, present clinical aspects of dysfunctional innate immunity in AD, and discuss current knowledge on the pathophysiology of infectious complications in this disease.
Delineation and Basic Aspects of Innate and Adaptive Immunity
The innate immune system comprises all inborn defense mechanisms which protect an individual by inducing immediate responses against potentially harmful microorganisms such as bacteria, fungi, and viruses. These “no-need-to-learn” mechanisms start with an intact epidermal barrier, but also include the expression and secretion of antimicrobial peptides (AMP) upon recognition of pathogens by toll-like receptors (TLR). Innate immunity includes resident cells such as keratinocytes as well as dendritic cell (DC) and lymphocyte subsets. The risk of autoimmunity in innate immune mechanisms is low but not zero [6], and goes along with the disadvantage of non-improving responses with repeated exposure. Some species such as frogs rely on innate immunity as their one and only defensive system.
In contrast, adaptive immunity summarizes all of the acquired but time-delayed defense mechanisms against pathogens. This part of the immune system is able to “learn”, i.e., it is able to improve its reactions with repeated exposure. In contrast to innate immune mechanisms, it requires somatic recombination which leads to different cell clones and thus is capable of specific protein recognition instead of simple pattern recognition. These immune responses may be built against virtually any structural protein. In contrast to innate immunity, adaptive immunity mechanisms possess a much higher risk of autoimmunity (Fig. 2).
It is important to understand that innate and adaptive immune mechanisms do not simply coexist but are linked to one another in a complex network of immunobiological mechanisms. Though the molecular basis of some relevant mechanisms is known, clinical knowledge suggests that many more pathways are still to be discovered.
Impaired Epidermal Barrier Function
An intact epidermal barrier may be regarded as the first and most essential component of the innate immune system. In lesional skin in AD, numerous histologically and immunobiologically well-characterized changes can be found in contrast to normal human skin, including a thick but dysfunctional epidermal layer, a pronounced lymphohistiocytic infiltrate, and a lack of inducible endogenous antibiotics [1, 7, 8]. Not only lesional but also non-lesional AD skin differs from normal human skin [9]. Non-lesional AD skin shows high transepidermal water loss, low sebum secretion, and minimal signs of an inflammatory infiltrate [10, 11]. This results in increased penetration of protein allergens or other bioactive substances such as pollen-associated lipid mediators through the impaired epidermal barrier [12]. As a consequence, allergic sensitization to a multitude of possible allergens is frequent among AD patients. This can be tested by the atopy patch test technique, which is standardized by the European Task Force on Atopic Dermatitis, and a highly specific procedure. This test relies on intact protein allergen penetration through clinically uninvolved skin without prior destruction of the epidermal barrier by tape stripping or other procedures [13].
A few years ago, key aspects of the molecular basis of epidermal barrier dysfunction were elucidated. Two loss-of-function mutations of the filaggrin gene were found to cause ichthyosis vulgaris [14]. Filaggrin is a filament-aggregating protein important in the formation of an intact stratum corneum. The two loss-of-function mutations, as well as another rare mutation in the filaggrin gene, were found to be strong predisposing factors for AD [15–17]. The fact that more than one third of all ichthyosis vulgaris patients suffer from AD and about 2–4% of all AD patients are diagnosed with ichthyosis vulgaris at the same time [18] implicated a connection between both diseases.
Environmental exposure to allergens, proteases, soaps, and detergents may further impair epidermal barrier function [8]. The IL-4- and IL-13-dominated inflammatory cytokine milieu in atopic skin further reduces filaggrin production in AD lesions, thus, contributing to epidermal barrier dysfunction in AD [19]. These findings indicate that epidermal barrier defects may have a leading role and is probably a primary event in AD, also predisposing to secondary infections of the skin [5]. This is mirrored by the well-established clinical relevance of daily emollient application to non-lesional skin of AD patients [20].
Pathogen-Associated Molecular Patterns and Pathogen Recognition Receptors
In innate immunity, pathogen-associated molecular patterns (PAMP), such as specific motifs of bacterial or viral DNA, RNA, or proteins, are recognized through a pattern recognition system provided by a limited number of inborn pathogen recognition receptors (PRR) [21]. These pathogen recognition receptors comprise 12 members of the Toll-like receptor (TLR) family which have been identified in mammals [22]. Moreover, the mannose receptor (CD206), formyl peptide receptor (FPRL-1), and complement receptors may also be regarded as pathogen receptors of innate immune system [23, 24]. Patterns recognized by the innate immune system involve double-stranded viral RNA (TLR-3), single-stranded viral RNA (TLR-7), CpG motifs from bacterial DNA (TLR-9), LPS from gram-negative bacterial membranes (TLR-4), FMLP from bacterial proteins (FPR-1), mannose-rich glycans from microbial glycoproteins and glycolipids (CD206), bacterial lipoteichoic acid (TLR-2), and flagellin from bacterial flagella (TLR 5) [25]. Therapeutic activation of selected TLR pathways may be achieved by small molecules such as imiquimod or by specific CpG motifs in bacterial-like DNA fragments to modulate or initiate specifically desired immune responses [26, 27]. Binding of PAMPs to PRRs leads to activation of intracellular signaling cascades such as the NfκB pathway and secretion of innate effector molecules such as cytokines or antimicrobial peptides.
Antimicrobial Peptides
Antimicrobial peptides (AMP) function as endogenous antibiotics and play an important part in cutaneous innate immunity [28]. The production of AMP is a mechanism used by many species for fighting invading microorganisms such as bacteria, fungi, and viruses or immediate defense after damage of the epithelial surface [29]. A single cathelicidin AMP and a number of defensin AMPs have been characterized in man [30]. Whereas the human beta-defensin (HBD)-1 is constitutively expressed by normal human keratinocytes, skin inflammation induces the expression of HBD-2, HBD-3, and the cathelicidin peptide LL-37. The expression of inducible AMP in AD lesions was significantly reduced if compared to psoriasis [7]. Therefore, it has been suggested that a lack of AMP contributes to the increased susceptibility of AD patients for disseminated viral skin infections, which is not the case for psoriasis patients [31, 32]. However, this view has been challenged by several groups in the meantime, as an increased expression and secretion of AMP, and especially cathelicidin, has been demonstrated in AD lesions and superficial skin injury in AD [33, 34]. Dysfunctional induction of cathelicidin has been demonstrated in injured lesional skin in AD in another study, which could contribute to the increased susceptibility of AD patients to skin infections [35]. Clinical and experimental data support the view that AD patients with a history of eczema herpeticum show a defective upregulation of AMP [32, 36].
The cutaneous regulation of AMP expression has been extensively studied during the last years. While defensin expression is controlled by proinflammatory mediators such as TNFα, the factors controlling cathelicidin expression remained unknown for a long time. Unexpectedly, vitamin D3 was identified as the sole factor inducing cathelicidin in keratinocytes, and in the meantime, critical elements involved in cathelidicin control in the skin have been identified [29, 37–40]. These observations stimulated experimental and clinical trials, which investigated whether improved vitamin D3 metabolism would affect cutaneous innate immunity. Indeed, keratinocytes produce and activate vitamin D3 under the influence of UVB irradiation in sufficient concentration to activate cathelicidin expression in an autocrine fashion [41]. Narrow-band UVB irradiation increases vitamin D3 and cathelicidin levels in the skin of AD patients [42]. Oral supplementation of vitamin D3 increases cathelicidin expression in AD lesions [43]. Further studies are needed to establish a firm link between AMP expression and infectious complications in AD patients and the use of vitamin D3 to prevent these [29].
Dendritic Cell Subsets in AD Lesions
Dendritic cells (DCs) are a morphologically and functionally defined family of antigen-presenting cells (APCs) which are found in small percentages in most organs of the human body. DCs initiate primary and secondary adaptive immune responses. Cell lineage, localization, and immunophenotype determine nomenclature of DC [2, 44]. Skin dendritic cells are subdivided after their immunophenotype into normal Langerhans cells, inflammatory dendritic epidermal cells, and plasmacytoid dendritic cells (PDCs) [45]. Langerhans cells (LCs) are DCs of the normal epidermis and defined as bone marrow-derived, epidermally located, dendritic APCs that contain Birbeck granules and express CD1a and MHC class II molecules [46]. Topical glucocorticoid therapy as well as UV light exposure may deplete LCs from the epidermis. Inflammatory dendritic epidermal cells (IDECs) are defined as epidermally located dendritic cells that do not contain Birbeck granules but express CD1a, CD11b, and class II molecules [47]. These IDECs have been demonstrated in AD, psoriasis vulgaris, allergic contact eczema, mycosis fungoides, lichen planus, Dorfman–Chanarin syndrome, Netherton syndrome, Sulzberger–Garbe disease [48–50]. As the cytokines granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4/IL-13 are abundant in AD lesions [51–54], it can be assumed that peripheral blood monocytic cells differentiate into IDECs whilst migrating into the AD lesions. IDECs express more costimulatory molecules CD80 and CD86 than LC, supporting the role of IDECs in the presentation of antigens in AD skin. [55]. The mannose receptor CD206 is also expressed on IDECs and is functional in terms of antigen uptake of mannosylated antigens by means of mannose receptor-mediated endocytosis [56]. Immature dendritic cells with a higher expression of high-affinity IgE receptor (FcεRI), thereby resembling the IDEC phenotype, can be generated in vitro under reducing cell culture conditions [57]. Treatment of AD with the topical calcineurin inhibitor tacrolimus selectively depletes IDECs from the epidermis, reduces the FcεRI expression and the stimulatory capacity of LCs and IDECs [58]. This selective IDEC depletion phenomenon can also be induced by the topical calcineurin inhibitor pimecrolimus [59]. Topical hydrocortisone treatment increases the rate of early apoptotic DCs in situ, whereas topical tacrolimus does not [60]. Both agents decrease the expression of the costimulatory molecules CD80 and CD86 on epidermal DCs [60].
Plasmacytoid Dendritic Cells
Plasmacytoid dendritic cells (PDCs), a dendritic cell subset first described in the blood (CD123+++, BDCA-2+, HLA-DR+++), comprise 0.1% of peripheral blood mononuclear cells [61]. As PDCs produce large amounts of type I interferon (IFN-α and IFN-β) upon viral infection [62], they are effector cells of innate immunity. However, PDCs exhibit an intrinsic activity to induce Th2 responses [63] and regulatory T cells [64], and therefore also play a role in adaptive immunity. Skin PDCs have been demonstrated in cutaneous lupus erythematosus [65], in psoriasis [45], and in contact dermatitis [45] in relatively high numbers, while in AD lesions, only few PDCs can be detected [45]. While the number of peripheral blood PDCs is increased in AD [66], the lack of PDCs in AD lesions seems to contribute to the high susceptibility of AD patients to viral skin infections such as eczema herpeticum [4]. Crosslinking of FcεRI on PDCs reduces type I interferon production induced by TLR-9 induction [67] and the stimulatory capacity of PDCs [68]. Immunostimulatory CpG oligonucleotides and imiquimod work by direct stimulation of PRR expressed on PDCs [3].
Natural Killer Cells
Natural killer cells (NK cells) account for 10–15% of all peripheral blood lymphocytes. These important effector cells of the innate immune system may lyse microbial-infected or malignant target cells without prior activation. The lack of MHC-I expression is picked up by NK cells as a “missing self” signal and starts a cascade of cell lysis [69]. Target cell lysis is mediated by release of cytoplasmic granules containing the membrane-attacking perforin and the protease granzyme. NK cells produce a variety of cytokines such as IFN-γ, TNF-α, GM-CSF, IL-5, and IL-8 [70]. The number of circulating NK cells is reduced in AD [70, 71], whereas in AD lesions, NK cells have been detected in close contact to DCs [72]. Coculture of Malassezia antigen-stimulated DCs with NK cells may increase DC numbers in vitro [73], whereas cell-contact-dependent preferential apoptosis of NK cells by activated monocytes has also been described in AD patients [71]. This may help in deviating immune responses towards a Th2 pattern and contribute to the susceptibility to infection in AD [71].
Innate Immunity and Staphylococcus aureus Colonization
Colonization with Staphylococcus aureus is a phenomenon observed in most AD patients [1]. S. aureus may be cultured from skin lesions and, to a lesser extent, also from non-lesional AD skin. The nasal vestibulum is the most important reservoir for recolonization of the skin [74]. An impaired epidermal barrier, an altered innate immune system, a decreased bacterial clearance, and an increased bacterial adhesion are important pathogenic factors for staphylococcal colonization [5, 75]. The levels of the antimicrobial defensin HBD-2 and the cathelicidin LL-37, and also of IL-8 and inducible NO synthases (iNOS) were found to be reduced in AD compared to psoriasis patients [7, 76]. Differential AMP expression in AD and psoriasis has been confirmed for HBD-2 and other antimicrobially active substances like elafin, calgranulin C, iNOS, psoriasin, and calgranulin A [77]. Although HBD-2 and LL-37 exert a synergistic antimicrobial activity, the concentration of both in AD might be too low to effectively kill S. aureus [7]. In extrinsic AD skin, high concentrations of IL-4 and IL-13 were shown to additionally inhibit HBD-2, IL-8, and iNOS gene expression [7, 76]. Elevated IL-10 gene expression in extrinsic and intrinsic AD was associated with a decreased HBD-2 expression [78], but here, neutralizing antibodies to IL-10 not only augmented production of HBD-2 and LL-37 but also of TNF-α and IFN-γ [78].
Another AMP with no homology to other known APM is dermcidin, which is expressed in eccrine sweat glands, secreted into sweat, and transported to the epidermis [79]. Dermcidin has a broad spectrum of activity including S. aureus. AD patients did not show significant reduction of bacterial skin surface colonization after sweating that was observed in healthy subjects [79] as several dermcidin-derived peptides were significantly reduced in AD compared to healthy subjects, and in AD patients with previous bacterial or viral infections, concentrations were found to be lowest [79].
Local production of staphylococcal exotoxins with superantigenic properties is the consequence of staphylococcal colonization permitted by a weak innate immune system impaired by Th2 signals [80]. Antigen-specific immune response to aero- and food allergens involving IgE-mediated, facilitated antigen presentation colocalize with polyclonal activation of T cells driven by superantigenic properties of staphylococcal exotoxins [3], which may also be a target for IgE responses in AD patients [81].
All these findings support the idea that a deficiency in various antimicrobial peptides may cause the high rate of S. aureus impetiginization in AD. Reduction of S. aureus colonization is a well-accepted therapeutic strategy for long-term management of AD, and may be achieved by the addition of antibacterial substances to the daily used emollients recommended since many years [20, 82]. In severe cases of impetiginized AD, oral antibiotic therapy with cephalosporins or penicillins is a useful strategy [82].
Eczema Herpeticum
Patients with compromised cell-mediated immunity, as present in AD, may develop recurrent and severe herpes simplex virus (HSV) infections, which may be caused by either primary or secondary HSV infection [4]. Eczema herpeticum (EH) is defined as the disseminated cutaneous infection of an eczematous skin disease with HSV, which in clinical reality is almost exclusively AD, whereas Kaposi’s varicelliform eruption is the disseminated cutaneous infection of any skin disease (not only AD, but also Darier disease, pemphigus foliaceus, mycosis fungoides, Sézary syndrome, ichthyosis vulgaris, Hailey–Hailey disease, and burns) with HSV or related viruses [83]. EH may be diagnosed based on clinical criteria by disseminated, distinctly monomorphic dome-shaped vesicles, frequently accompanied by fever, malaise, and lymphadenopathy [84]. Head and neck are most commonly affected. Associated complications include not only keratoconjunctivitis but also viremia, leading to multiple organ involvement with meningitis and encephalitis if not treated adequately [83]. The pathogenesis of EH involves an infection of keratinocytes, followed by an antiviral immune response, which must be mounted in a Th2-skewed environment. This underlying predisposition to Th2-type responses may favor HSV overgrowth [85]. Regarding innate immune mechanisms, a pathogenic factor that contributes to HSV infections in AD is the spongiosis and disruption of the compact keratinocyte layer present in AD that leads to an unmasking of the desmosomal protein nectin-1, which acts as one of the relevant entry receptors for HSV [86]. The relevance of AMP dysregulation in the pathogenesis of EH is well established [32]: Physiological concentrations of the cathelicidin LL-37 show antiviral activity against HSV in vitro, but there may be disturbed upregulation of LL-37 in AD lesions in vivo [32, 35]. Interestingly, cathelicidin peptide LL-37 expression is also inversely correlated to the serum IgE level of EH patients [32].
PDCs play a key role in the cascade of antiviral innate immune responses as they produce high amounts of antiviral type I IFN-α and IFN-β upon viral infection. Although the number of peripheral blood PDC is increased in AD [66], in skin lesions of AD, very few PDCs are found compared to other inflammatory skin diseases such as psoriasis, contact dermatitis, or lupus erythematosus [45], which can be explained by dose-dependent PDC apoptosis induction caused by IL-4, an effect that is potentiated by IL-10 [63]. Crosslinking of the high-affinity IgE receptor on PDCs reduces the type I interferon production induced by TLR-9 activation [67, 68]. So, PDC depletion and functional impairment, and therefore diminished production of antiviral type I interferons, may contribute to the predisposition of AD patients to viral skin infections [45, 67].
EH treatment should be started as soon as possible with systemic antiviral chemotherapy with nucleoside analogs such as acyclovir [4]. A 7-day course of intravenous acyclovir (5–10 mg/kg per dose i.v. t.i.d.) is currently recommended and may be prolonged according to the clinical course of the disease [4]. Oral antibiotics may be helpful in fighting bacterial superinfection, topical antiseptics may favor the healing process by drying out vesicles and preventing infectious complications. Topical antiviral agents are usually not applied for cutaneous EH lesions as they carry a significant risk for contact sensitization and drug reactions [87], on the other hand, they offer advantages for prophylaxis and treatment of ocular complications. Lid lesions and reduced corneal sensitivity in EH should be treated prophylactically, patients with keratitis should receive combined systemic and topical therapy [4]. The administration of glucocorticosteroids in acute EH is still controversial. A number of clinicians use topical and systemic glucocorticosteroids once acyclovir has been started to favor healing of the underlying AD.
Eczema Vaccinatum
Eczema vaccinatum is defined as a disseminated eruption of Vaccinia lesions in AD or other pre-existing eczematous skin diseases following immunization with Vaccinia virus [88]. Since eczema vaccinatum is one of the few potentially life-threatening, true emergencies in clinical dermatology, vaccination is contraindicated in all patients currently affected by or having a past medical history of AD [85]. Cathelicidin AMP but not human defensins can inhibit Vaccinia virus growth in vitro [31]. This suggests that the eczema vaccinatum susceptibility of AD patients may be due to cathelicidin deficiency [31]. Vaccinia virus replicates faster in AD skin explants than in normal or psoriasis skin explants [89], and Vaccinia virus-induced expression of cathelicidin LL-37 peptide is reduced in AD skin [89]. This nicely demonstrates that the increased Vaccinia replication corresponds to a decreased expression of LL-37. The underlying cause for both observations seems to be the cytokine milieu in AD as IL-4 and IL-13 enhance Vaccinia virus replication and downregulate LL-37 [89, 90].
The treatment of choice for eczema vaccinatum is Vaccinia immune globulin, which may be combined with cidofovir and other still experimental drugs [88, 91]. Newly produced Vaccinia immune globulin preparations will allow an intravenous application.
Molluscum contagiosum Virus
Lesional and non-lesional skin of AD patients is susceptible to infection with Molluscum contagiosum virus (MCV), which is the sole member of the Molluscipoxvirus subfamily [4]. MCV replication in the cytoplasm is mediated by distinct enzymes which are not encoded in other DNA viruses. Umbilicated small skin-colored papules are the diagnostic hallmark of molluscum infection [18]. Patients with AD have more frequently MCV infections than non-atopic individuals and also have more widespread disease, with up to several hundred lesions [92]. This disseminated eruption is referred to as eczema molluscatum (EM) [3]. Although the first papules are frequently confined to inflamed skin regions, autoinoculation may produce papules in other areas. MCV infection does not induce clinical symptoms of systemic infection. MCV contains some genes encoding for unique host response-evasion proteins which help it avoid innate and adaptive antiviral defense mechanisms [93]. MCV produces a soluble interleukin-18 binding protein which inhibits the IL-18-mediated induction of IFN-γ [94]. This may explain the absence of T lymphocytes and NK cells at the base of typical MCV lesions [95]. Treatment of EM lesions speeds up the healing process and prevents spreading by auto- and heteroinoculation. Therefore, treatment of EM is recommended in spite of its tendency towards spontaneous regression [4]. The destruction of limited numbers of lesions with a small curved forceps or a ring curette is our treatment of choice. Other destructive treatment measures include cryotherapy or carbon dioxide laser vaporization [4]. Topical application of the TLR-7-agonist imiquimod or other topical immunostimulatory drugs is another treatment alternative [96].
Conclusion
Recent research advances allow a better understanding of the mechanisms underlying innate immune deficiency in AD patients. Several innate immune mechanisms are relevant for AD pathophysiology as these are impaired in AD patients and may contribute to the increased susceptibility of AD patients to skin infections. Also, innate and adaptive immunity do not simply coexist but are linked to one another by complex mechanisms, which are subject to current and future research. Finally, as we begin to understand the mechanisms of innate immune dysregulation in AD, the identification of possible targets for treatment or prevention will lead to novel approaches in the management of AD patients.
The pathogenesis of atopic dermatitis involves an impaired innate as well as a distorted adaptive immune response. Both mechanisms may aggravate a hereditary skin barrier defect.
Innate and adaptive immunities are different in many key features, but linked to one another through various mechanisms. Some of the signals are already known.
Abbreviations
- AD:
-
Atopic dermatitis
- AMP:
-
Antimicrobial peptides
- APC:
-
Antigen-presenting cell
- DC:
-
Dendritic cell
- EH:
-
Eczema herpeticum
- EM:
-
Eczema molluscatum
- FcεRI:
-
High-affinity IgE receptor
- FPR-1:
-
Formyl peptide receptor
- HBD:
-
Human beta-defensin
- HSV:
-
Herpes simplex virus
- IDEC:
-
Inflammatory dendritic epidermal cell
- IFN:
-
Interferon
- IL18-BP:
-
Interleukin-18 binding protein
- iNOS:
-
Inducible NO synthase
- KVE:
-
Kaposi’s varicelliform eruption
- LC:
-
Langerhans cell
- MCV:
-
Molluscum contagiosum virus
- NK:
-
Natural killer cell
- PAMP:
-
Pathogen-associated molecular pattern
- PDC:
-
Plasmacytoid dendritic cell
- PRR:
-
Pathogen recognition receptor
- S. aureus :
-
Staphylococcus aureus
- TLR:
-
Toll-like receptor
References
Bieber T (2008) Atopic dermatitis. N Engl J Med 358:1483–1494
Wollenberg A, Bieber T (2000) Atopic dermatitis: from the genes to skin lesions. Allergy 55:205–213
Wollenberg A, Klein E (2007) Current aspects of innate and adaptive immunity in atopic dermatitis. Clin Rev Allergy Immunol 33:35–44
Wollenberg A, Wetzel S, Burgdorf WH, Haas J (2003) Viral infections in atopic dermatitis: pathogenic aspects and clinical management. J Allergy Clin Immunol 112:667–674
Baker BS (2006) The role of microorganisms in atopic dermatitis. Clin Exp Immunol 144:1–9
Guiducci C, Gong M, Xu Z, Gill M, Chaussabel D, Meeker T, Chan JH, Wright T, Punaro M, Bolland S, Soumelis V, Banchereau J, Coffman RL, Pascual V, Barrat FJ (2010) TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature 465:937–941
Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, Gallo RL, Leung DY (2002) Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med 347:1151–1160
Cork MJ, Danby SG, Vasilopoulos Y, Hadgraft J, Lane ME, Moustafa M, Guy RH, Macgowan AL, Tazi-Ahnini R, Ward SJ (2009) Epidermal barrier dysfunction in atopic dermatitis. J Invest Dermatol 129:1892–1908
Wollenberg A, Bieber T (2009) Proactive therapy of atopic dermatitis—an emerging concept. Allergy 64:276–278
Proksch E, Folster-Holst R, Jensen JM (2006) Skin barrier function, epidermal proliferation and differentiation in eczema. J Dermatol Sci 43:159–169
Mihm MC, Soter NA, Dvorak HF, Austen KF (1976) The structure of normal skin and the morphology of atopic eczema. J Invest Dermatol 67:305–312
Traidl-Hoffmann C, Mariani V, Hochrein H, Karg K, Wagner H, Ring J, Mueller MJ, Jakob T, Behrendt H (2005) Pollen-associated phytoprostanes inhibit dendritic cell interleukin-12 production and augment T helper type 2 cell polarization. J Exp Med 201:627–636
Darsow U, Laifaoui J, Kerschenlohr K, Wollenberg A, Przybilla B, Wüthrich B, Borelli Sj, Giusti F, Seidenari S, Drzimalla K, Simon D, Disch R, Borelli S, Devillers ACA, Oranje AP, De Raeve L, Hachem JP, Dangoisse C, Blondeel A, Song M, Breuer K, Wulf A, Werfel T, Roul S, Taieb A, Bolhaar S, Bruijnzeel-Koomen C, Brönnimann M, Braathen LR, Didierlaurent A, André C, Ring J (2004) The prevalence of positive reactions in the atopy patch test with aeroallergens and food allergens in subjects with atopic eczema: a European multicenter study. Allergy 59:1318–1325
Smith FJ, Irvine AD, Terron-Kwiatkowski A, Sandilands A, Campbell LE, Zhao Y, Liao H, Evans AT, Goudie DR, Lewis-Jones S, Arseculeratne G, Munro CS, Sergeant A, O’Regan G, Bale SJ, Compton JG, DiGiovanna JJ, Presland RB, Fleckman P, McLean WH (2006) Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet 38:337–342
Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, Goudie DR, Sandilands A, Campbell LE, Smith FJ, O’ Regan GM, Watson RM, Cecil JE, Bale SJ, Compton JG, Di Giovanna JJ, Fleckman P, Lewis-Jones S, Arseculeratne G, Sergeant A, Munro CS, El Houate B, McElreavey K, Halkjaer LB, Bisgaard H, Mukhopadhyay S, McLean WH (2006) Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet 38:441–446
Weidinger S, Illig T, Baurecht H, Irvine AD, Rodriguez E, Diaz-Lacava A, Klopp N, Wagenpfeil S, Zhao Y, Liao H, Lee SP, Palmer CN, Jenneck C, Maintz L, Hagemann T, Behrendt H, Ring J, Nothen MM, McLean WH, Novak N (2006) Loss-of-function variations within the filaggrin gene predispose for atopic dermatitis with allergic sensitizations. J Allergy Clin Immunol 118:214–219
Sandilands A, O’regan GM, Liao H, Zhao Y, Terron-Kwiatkowski A, Watson RM, Cassidy AJ, Goudie DR, Smith FJ, McLean WH, Irvine AD (2006) Prevalent and rare mutations in the gene encoding filaggrin cause ichthyosis vulgaris and predispose individuals to atopic dermatitis. J Invest Dermatol 126(8):1770–1775
Braun-Falco O, Plewig G, Wolff H, Burgdorf W (2000) Dermatology, 3rd edn. Springer, New York
Howell MD, Kim BE, Gao P, Grant AV, Boguniewicz M, Debenedetto A, Schneider L, Beck LA, Barnes KC, Leung DY (2007) Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol 120:150–155
Darsow U, Lubbe J, Taieb A, Seidenari S, Wollenberg A, Calza A, Giusti F, Ring J (2005) Position paper on diagnosis and treatment of atopic dermatitis. J Eur Acad Dermatol Venereol 19:286–295
Kaisho T, Shizuo A (2006) Toll-like receptor function and signaling. J Allergy Clin Immunol 117:979–987
Balamayooran T, Balamayooran G, Jeyaseelan S (2010) Review: toll-like receptors and NOD-like receptors in pulmonary antibacterial immunity. Innate Immun 16:201–210
Gordon S (2002) Pattern recognition receptors: doubling up for the innate immune response. Cell 111:927–930
Wollenberg A, Mommaas M, Oppel T, Schottdorf EM, Gunther S, Moderer M (2002) Expression and function of the mannose receptor CD206 on epidermal dendritic cells in inflammatory skin diseases. J Invest Dermatol 118:327–334
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124:783–801
Hengge UR, Cusini M (2003) Topical immunomodulators for the treatment of external genital warts, cutaneous warts and molluscum contagiosum. Br J Dermatol 149(Suppl 66):15–19
Kerkmann M, Rothenfusser S, Hornung V, Towarowski A, Wagner M, Sarris A, Giese T, Endres S, Hartmann G (2003) Activation with CpG-A and CpG-B oligonucleotides reveals two distinct regulatory pathways of type I IFN synthesis in human plasmacytoid dendritic cells. J Immunol 170:4465–4474
Schauber J, Gallo RL (2008) The vitamin D pathway: a new target for control of the skin’s immune response? Exp Dermatol 17:633–639
Schauber J, Gallo RL (2008) Antimicrobial peptides and the skin immune defense system. J Allergy Clin Immunol 122:261–266
Braff MH, Bardan A, Nizet V, Gallo RL (2005) Cutaneous defense mechanisms by antimicrobial peptides. J Invest Dermatol 125:9–13
Howell MD, Jones JF, Kisich KO, Streib JE, Gallo RL, Leung DY (2004) Selective killing of vaccinia virus by LL-37: implications for eczema vaccinatum. J Immunol 172:1763–1767
Howell MD, Wollenberg A, Gallo RL, Flaig M, Streib JE, Wong C, Pavicic T, Boguniewicz M, Leung DY (2006) Cathelicidin deficiency predisposes to eczema herpeticum. J Allergy Clin Immunol 117:836–841
Ballardini N, Johansson C, Lilja G, Lindh M, Linde Y, Scheynius A, Agerberth B (2009) Enhanced expression of the antimicrobial peptide LL-37 in lesional skin of adults with atopic eczema. Br J Dermatol 161:40–47
Harder J, Dressel S, Wittersheim M, Cordes J, Meyer-Hoffert U, Mrowietz U, Folster-Holst R, Proksch E, Schroder JM, Schwarz T, Glaser R (2010) Enhanced expression and secretion of antimicrobial peptides in atopic dermatitis and after superficial skin injury. J Invest Dermatol 130:1355–1364
Mallbris L, Carlen L, Wei T, Heilborn J, Nilsson MF, Granath F, Stahle M (2010) Injury downregulates the expression of the human cathelicidin protein hCAP18/LL-37 in atopic dermatitis. Exp Dermatol 19:442–449
Hata TR, Kotol P, Boguniewicz M, Taylor P, Paik A, Jackson M, Nguyen M, Kabigting F, Miller J, Gerber M, Zaccaro D, Armstrong B, Dorschner R, Leung DY, Gallo RL (2010) History of eczema herpeticum is associated with the inability to induce human beta-defensin (HBD)-2, HBD-3 and cathelicidin in the skin of patients with atopic dermatitis. Br J Dermatol 163(3):659–661
Schauber J, Dorschner RA, Coda AB, Buchau AS, Liu PT, Kiken D, Helfrich YR, Kang S, Elalieh HZ, Steinmeyer A, Zugel U, Bikle DD, Modlin RL, Gallo RL (2007) Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. J Clin Investig 117:803–811
Schauber J, Dorschner RA, Yamasaki K, Brouha B, Gallo RL (2006) Control of the innate epithelial antimicrobial response is cell-type specific and dependent on relevant microenvironmental stimuli. Immunology 118:509–519
Schauber J, Oda Y, Buchau AS, Yun QC, Steinmeyer A, Zugel U, Bikle DD, Gallo RL (2008) Histone acetylation in keratinocytes enables control of the expression of cathelicidin and CD14 by 1, 25-dihydroxyvitamin D3. J Invest Dermatol 128:816–824
Peric M, Koglin S, Dombrowski Y, Gross K, Bradac E, Buchau A, Steinmeyer A, Zugel U, Ruzicka T, Schauber J (2009) Vitamin D analogs differentially control antimicrobial peptide/“alarmin” expression in psoriasis. PLoS ONE 4:e6340
Peric M, Lehmann B, Vashina G, Dombrowski Y, Koglin S, Meurer M, Ruzicka T, Schauber J (2010) UV-B-triggered induction of vitamin D3 metabolism differentially affects antimicrobial peptide expression in keratinocytes. J Allergy Clin Immunol 125:746–749
Vahavihu K, Ala-Houhala M, Peric M, Karisola P, Kautiainen H, Hasan T, Snellman E, Alenius H, Schauber J, Reunala T (2010) Narrow-band ultraviolet B treatment improves vitamin D balance and alters antimicrobial peptide expression in skin lesions of psoriasis and atopic dermatitis. Br J Dermatol 163(2):321–328
Hata TR, Kotol P, Jackson M, Nguyen M, Paik A, Udall D, Kanada K, Yamasaki K, Alexandrescu D, Gallo RL (2008) Administration of oral vitamin D induces cathelicidin production in atopic individuals. J Allergy Clin Immunol 122:829–831
von Bubnoff D, Koch S, Bieber T (2003) Dendritic cells and atopic eczema/dermatitis syndrome. Curr Opin Allergy Clin Immunol 3:353–358
Wollenberg A, Wagner M, Günther S, Towarowski A, Tuma E, Moderer M, Rothenfusser S, Wetzel S, Endres S, Hartmann G (2002) Plasmacytoid dendritic cells: a new cutaneous dendritic cell subset with distinct role in inflammatory skin diseases. J Invest Dermatol 119:1096–1102
Wollenberg A, Schuller E. Langerhans Zellen und Immunantwort. In: Plewig G, Wolff H (eds) Fortschritte der praktischen Dermatologie und Venerologie. Berlin: Springer: 41–48
Wollenberg A, Kraft S, Hanau D, Bieber T (1996) Immunomorphological and ultrastructural characterization of Langerhans cells and a novel, inflammatory dendritic epidermal cell (IDEC) population in lesional skin of atopic eczema. J Invest Dermatol 106:446–453
Wollenberg A, Bieber T (2002) Antigen-presenting cells. In: Bieber T, Leung DYM (eds) Atopic dermatitis. Marcel Dekker, New York, pp 267–283
Wollenberg A, Geiger E, Schaller M, Wolff H (2000) Dorfman–Chanarin syndrome in a Turkish kindred: conductor diagnosis requires analysis of multiple eosinophils. Acta Derm Venereol 80:39–43
Wollenberg A, Wen S, Bieber T (1999) Phenotyping of epidermal dendritic cells: clinical applications of a flow cytometric micromethod. Cytometry 37:147–155
Pastore S, Fanales Belasio E, Albanesi C, Chinni LM, Giannetti A, Girolomoni G (1997) Granulocyte macrophage colony-stimulating factor is overproduced by keratinocytes in atopic dermatitis. Implications for sustained dendritic cell activation in the skin. J Clin Investig 99:3009–3017
Horsmanheimo L, Harvima IT, Jarvikallio A, Harvima RJ, Naukkarinen A, Horsmanheimo M (1994) Mast cells are one major source of interleukin-4 in atopic dermatitis. Br J Dermatol 131:348–353
Akdis M, Simon HU, Weigl L, Kreyden O, Blaser K, Akdis CA (1999) Skin homing (cutaneous lymphocyte-associated antigen-positive) CD8+ T cells respond to superantigen and contribute to eosinophilia and IgE production in atopic dermatitis. J Immunol 163:466–475
van der Ploeg I, Matuseviciene G, Fransson J, Wahlgren CF, Olsson T, Scheynius A (1999) Localization of interleukin-13 gene-expressing cells in tuberculin reactions and lesional skin from patients with atopic dermatitis. Scand J Immunol 49:447–453
Schuller E, Teichmann B, Haberstok J, Moderer M, Bieber T, Wollenberg A (2001) In situ-expression of the costimulatory molecules CD80 and CD86 on Langerhans cells and inflammatory dendritic epidermal cells (IDEC) in atopic dermatitis. Arch Dermatol Res 293:448–454
Wollenberg A, Mommaas M, Oppel T, Schottdorf EM, Günther S, Moderer M (2002) Expression and function of the mannose receptor CD206 on epidermal dendritic cells in inflammatory skin diseases. J Invest Dermatol 118:327–334
Novak N, Kraft S, Haberstok J, Geiger E, Allam P, Bieber T (2002) A reducing microenvironment leads to the generation of FcepsilonRI high inflammatory dendritic epidermal cells (IDEC). J Invest Dermatol 119:842–849
Wollenberg A, Sharma S, von Bubnoff D, Geiger E, Haberstok J, Bieber T (2001) Topical tacrolimus (FK506) leads to profound phenotypic and functional alterations of epidermal antigen-presenting dendritic cells in atopic dermatitis. J Allergy Clin Immunol 107:519–525
Hoetzenecker W, Ecker R, Kopp T, Stuetz A, Stingl G, Elbe-Burger A (2005) Pimecrolimus leads to an apoptosis-induced depletion of T cells but not Langerhans cells in patients with atopic dermatitis. J Allergy Clin Immunol 115:1276–1283
Schuller E, Oppel T, Bornhövd E, Wetzel S, Wollenberg A (2004) Tacrolimus ointment causes inflammatory dendritic epidermal cell depletion but no Langerhans cell apoptosis in patients with atopic dermatitis. J Allergy Clin Immunol 114:137–143
Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, Colonna M (1999) Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med 5:919–923
Krug A, Rothenfusser S, Selinger S, Bock C, Kerkmann M, Battiany J, Sarris A, Giese T, Speiser D, Endres S, Hartmann G (2003) CpG-A oligonucleotides induce a monocyte-derived dendritic cell-like phenotype that preferentially activates CD8 T cells. J Immunol 170:3468–3477
Rissoan MC, Soumelis V, Kadowaki N, Grouard G, Briere F, de Waal Malefyt R, Liu YJ (1999) Reciprocal control of T helper cell and dendritic cell differentiation. Science 283:1183–1186
Gilliet M, Liu YJ (2002) Human plasmacytoid-derived dendritic cells and the induction of T-regulatory cells. Hum Immunol 63:1149–1155
Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, Jahnsen FL (2001) Plasmacytoid dendritic cells (natural interferon- alpha/beta-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol 159:237–243
Uchida Y, Kurasawa K, Nakajima H, Nakagawa N, Tanabe E, Sueishi M, Saito Y, Iwamoto I (2001) Increase of dendritic cells of type 2 (DC2) by altered response to IL-4 in atopic patients. J Allergy Clin Immunol 108:1005–1011
Wollenberg A, Pavicic T, Wetzel S, Hartmann G (2005) Expression of high affinity IgE receptors on skin- and blood-derived plasmacytoid dendritic cells in inflammatory skin diseases. Allergy Clin Immunol Int: J World Allergy Org Supplement 2:42–44
Novak N, Allam JP, Hagemann T, Jenneck C, Laffer S, Valenta R, Kochan J, Bieber T (2004) Characterization of FcepsilonRI-bearing CD123 blood dendritic cell antigen-2 plasmacytoid dendritic cells in atopic dermatitis. J Allergy Clin Immunol 114:364–370
Sola C, Andre P, Lemmers C, Fuseri N, Bonnafous C, Blery M, Wagtmann NR, Romagne F, Vivier E, Ugolini S (2009) Genetic and antibody-mediated reprogramming of natural killer cell missing-self recognition in vivo. Proc Natl Acad Sci USA 106:12879–12884
Aktas E, Akdis M, Bilgic S, Disch R, Falk CS, Blaser K, Akdis C, Deniz G (2005) Different natural killer (NK) receptor expression and immunoglobulin E (IgE) regulation by NK1 and NK2 cells. Clin Exp Immunol 140:301–309
Katsuta M, Takigawa Y, Kimishima M, Inaoka M, Takahashi R, Shiohara T (2006) NK cells and gamma delta + T cells are phenotypically and functionally defective due to preferential apoptosis in patients with atopic dermatitis. J Immunol 176:7736–7744
Buentke E, Heffler LC, Wilson JL, Wallin RP, Lofman C, Chambers BJ, Ljunggren HG, Scheynius A (2002) Natural killer and dendritic cell contact in lesional atopic dermatitis skin—Malassezia-influenced cell interaction. J Invest Dermatol 119:850–857
Buentke E, D’Amato M, Scheynius A (2004) Malassezia enhances natural killer cell-induced dendritic cell maturation. Scand J Immunol 59:511–516
Abeck D, Mempel M (1998) Staphylococcus aureus colonization in atopic dermatitis and its therapeutic implications. Br J Dermatol 139(Suppl 53):13–16
Leung D (2005) Superantigens, steroid insensitivity and innate immunity in atopic eczema. Acta Derm Venereol Suppl (Stockh) 215:11–15
Nomura I, Goleva E, Howell MD, Hamid QA, Ong PY, Hall CF, Darst MA, Gao B, Boguniewicz M, Travers JB, Leung DY (2003) Cytokine milieu of atopic dermatitis, as compared to psoriasis, skin prevents induction of innate immune response genes. J Immunol 171:3262–3269
de Jongh GJ, Zeeuwen PL, Kucharekova M, Pfundt R, van der Valk PG, Blokx W, Dogan A, Hiemstra PS, van de Kerkhof PC, Schalkwijk J (2005) High expression levels of keratinocyte antimicrobial proteins in psoriasis compared with atopic dermatitis. J Invest Dermatol 125:1163–1173
Howell MD, Novak N, Bieber T, Pastore S, Girolomoni G, Boguniewicz M, Streib J, Wong C, Gallo RL, Leung DY (2005) Interleukin-10 downregulates anti-microbial peptide expression in atopic dermatitis. J Invest Dermatol 125:738–745
Rieg S, Steffen H, Seeber S, Humeny A, Kalbacher H, Dietz K, Garbe C, Schittek B (2005) Deficiency of dermcidin-derived antimicrobial peptides in sweat of patients with atopic dermatitis correlates with an impaired innate defense of human skin in vivo. J Immunol 174:8003–8010
Breuer K, Wittmann M, Kempe K, Kapp A, Mai U, Dittrich-Breiholz O, Kracht M, Mrabet-Dahbi S, Werfel T (2005) Alpha-toxin is produced by skin colonizing Staphylococcus aureus and induces a T helper type 1 response in atopic dermatitis. Clin Exp Allergy 35:1088–1095
Ide F, Matsubara T, Kaneko M, Ichiyama T, Mukouyama T, Furukawa S (2004) Staphylococcal enterotoxin-specific IgE antibodies in atopic dermatitis. Pediatr Int 46:337–341
Darsow U, Wollenberg A, Simon D, Taieb A, Werfel T, Oranje A, Gelmetti C, Svensson A, Deleuran M, Calza AM, Giusti F, Lubbe J, Seidenari S, Ring J (2010) ETFAD/EADV eczema task force 2009 position paper on diagnosis and treatment of atopic dermatitis. J Eur Acad Dermatol Venereol 24(3):317–328
Wollenberg A, Zoch C, Wetzel S, Plewig G, Przybilla B (2003) Predisposing factors and clinical features of eczema herpeticum: a retrospective analysis of 100 cases. J Am Acad Dermatol 49:198–205
Rerinck HC, Kamann S, Wollenberg A (2006) Eczema herpeticatum: pathogenese und therapie. Hautarzt 57:586–591
Engler RJM, Kenner J, Leung DY (2002) Smallpox vaccination: risk considerations for patients with atopic dermatitis. J Allergy Clin Immunol 110:357–365
Yoon M, Spear PG (2002) Disruption of adherens junctions liberates nectin-1 to serve as receptor for herpes simplex virus and pseudorabies virus entry. J Virol 76:7203–7208
Wollenberg A, Baldauf C, Ruëff F, Przybilla B (2000) Allergische Kontaktdermatitis und Arzneiexanthem auf Aciclovir - Kreuzreaktion auf Ganciclovir. Allergo J 9:96–99
Wollenberg A, Engler R (2004) Smallpox, vaccination and adverse reactions to smallpox vaccine. Curr Opin Allergy Clin Immunol 4:271–275
Howell MD, Gallo RL, Boguniewicz M, Jones JF, Wong C, Streib JE, Leung DY (2006) Cytokine milieu of atopic dermatitis skin subverts the innate immune response to vaccinia virus. Immunity 24:341–348
Harrison JM, Ramshaw IA (2006) Cytokines, skin, and smallpox—a new link to an antimicrobial Peptide. Immunity 24:245–247
Vora S, Damon I, Fulginiti V, Weber SG, Kahana M, Stein SL, Gerber SI, Garcia-Houchins S, Lederman E, Hruby D, Collins L, Scott D, Thompson K, Barson JV, Regnery R, Hughes C, Daum RS, Li Y, Zhao H, Smith S, Braden Z, Karem K, Olson V, Davidson W, Trindade G, Bolken T, Jordan R, Tien D, Marcinak J (2008) Severe eczema vaccinatum in a household contact of a smallpox vaccinee. Clin Infect Dis 46:1555–1561
Solomon L, Telner P (1966) Eruptive molluscum contagiosum in atopic dermatitis. CMAJ 95:978–979
Senkevich TG, Bugert JJ, Sisler JR, Koonin EV, Darai G, Moss B (1996) Genome sequence of a human tumorigenic poxvirus: prediction of specific host response-evasion genes. Science 273:813–816
Xiang Y, Moss B (2003) Molluscum contagiosum virus interleukin-18 (IL-18) binding protein is secreted as a full-length form that binds cell surface glycosaminoglycans through the C-terminal tail and a furin-cleaved form with only the IL-18 binding domain. J Virol 77:2623–2630
Heng MC, Steuer ME, Levy A, McMahon S, Richman M, Allen SG, Blackhart B (1989) Lack of host cellular immune response in eruptive molluscum contagiosum. Am J Dermatopathol 11:248–254
Syed TA, Goswami J, Ahmadpour OA, Ahmad SA (1998) Treatment of molluscum contagiosum in males with an analog of imiquimod 1% in cream: a placebo-controlled, double-blind study. J Dermatol 25:309–313
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wollenberg, A., Räwer, HC. & Schauber, J. Innate Immunity in Atopic Dermatitis. Clinic Rev Allerg Immunol 41, 272–281 (2011). https://doi.org/10.1007/s12016-010-8227-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-010-8227-x