
Abstract
The association between coagulation and cancer development has been observed for centuries. However, the connection between inflammation and malignancy is also well-recognized. The plethora of evidence indicates that among multiple hemostasis components, platelets play major roles in cancer progression by providing surface and granular contents for several interactions as well as behaving like immune cells. Therefore, the anticancer potential of anti-platelet therapy has been intensively investigated for many years. Anti-platelet agents may prevent cancer, decrease tumor growth, and metastatic potential, as well as improve survival of cancer patients. On the other hand, there are suggestions that antiplatelet treatment may promote solid tumor development in a phenomenon described as “cancers follow bleeding.” The controversies around antiplatelet agents justify insight into the subject to establish what, if any, role platelet-directed therapy has in the continuum of anticancer management.
Similar content being viewed by others

1 Introduction
The idea that platelets contribute to tumor growth and progression is not new, and multiple mechanisms have been described that explain these complex platelet/tumor cell interactions [1,2,3]. Advanced knowledge about the molecular and functional aspects of platelet-mediated tumor dissemination motivated scientists to search for drugs with anticancer potential. The experimental models demonstrated that inhibitors of platelet-evoked processes as well as thrombocytopenia reduce tumor metastasis, while clinical data indicate that anticoagulant (AC) therapy may influence cancer-specific mortality [4,5,6].
The most important processes contributing to cancer progression regulated by platelets and their granular contents are adhesion and tumor cell-induced platelet aggregation (TCIPA), which result in tumor emboli in microvasculature, tumor cell migration, intra-/extravasation and angiogenesis [3, 7]. Additionally, platelets are critical for survival of tumor cells within solid mass as they regulate intratumoral vasculature and hemostasis, and they protect circulating tumor cells (CTCs) in the blood stream from NK cell lysis [8,9,10]. Moreover, platelets are able to recognize pathogens and secrete active cytokines and so should be regarded as an important component of the immune system [11].
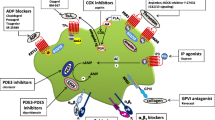
Platelet activation pathways are associated mainly with their membrane receptors, which may be activated during TCIPA and inhibited by their respective inhibitors (Fig. 1) [1, 12]. Platelet membranes contain integrins (e.g., glycoproteins Ib-IX-V (GP Ib-IX-V), glycoprotein VI (GP VI), and glycoprotein IIb-IIIa (GP IIb-IIIa), integrin αIIbβ3), thromboxane (TxA2) receptor (TPR), purigenic P2 receptors for nucleotides (adenosine diphosphate (ADT) and adenosine triphosphate (ATP)) as well as protease-activated receptors for thrombin (PAR-1 and PAR-4) that are intimately involved in cancer progression [13]. The newly discovered C-type lectin-like receptor-2 (CLEC-2 receptor) and its activator, podoplanin, a key protein in platelet aggregation, have attracted the interest of the scientific community [14, 15]. Additional attractive targets for antiplatelet treatment involve arachidonic acid (AA) metabolism, cyclooxygenase (COX) and lipoxygenase (LOX) enzymes, and many other compounds exhibiting antiplatelet activity [16].

Platelet receptors and molecules contributed to cancer dissemination. CLEC-2 C-type lectin-like receptor-2, GPIb-IX-V glycoproteins Ib-IX-V, VI, IIb-IIIa, PAR protease-activated receptors for thrombin, P2Y purigenic P2 receptors for nucleotides, TCIPA tumor cell-induced platelet aggregation
Aspirin is an antiplatelet drug whose anticancer activity has been thoroughly investigated both in vitro and in vivo using cancer cell lines, animal models as well as clinical trials [17,18,19]. Targeting platelet cyclooxygenases (COX-1, COX-2) with low-dose aspirin exerts antimetastatic and antiproliferative effects [18, 19], and in vivo analyses indicate that aspirin may even reduce distant metastases rates (DMR), disease-free survival (DFS), and/or overall survival in cancer patients [20,21,22,23,24]. Moreover, a population-based historical cohort study and randomized trials have shown that aspirin prevents cancer incidence [25, 26]. New trials are underway [27]. Aspirin derivatives with less gastrointestinal effects are currently being investigated as therapeutics as well [28].
There is new and somewhat alarming data that prolonged anti-platelet/coagulant treatment may promote cancer development [29,30,31]. This effect has been described for prasugrel (TRITON trial), vorapaxar (TRACER trial, Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome), and for 30-month therapy with prasugrel and clopidogrel (Dual Antiplatelet Therapy, DAPT trial). Long-term antiplatelet therapy was associated with cancer development that contributed to about half of the noncardiovascular deaths (NCVD) in these trials. However, the population-based cohort study among colorectal, breast, and prostate cancer patients did not confirm an increased risk of cancer-specific mortality in this group of patients using clopidogrel [32]. Obviously, targeting platelet receptors is an approach that can be associated with other serious side effects related to suppression of prothrombotic pathways such as an increased risk for bleeds in treated patients.
To dispel concerns and address controversies surrounding antiplatelet therapy, we have gathered the newest data on platelet receptors and focused on potential modes of inhibition (Fig.1).
2 Markers of platelet activation in cancer patients
2.1 Thrombocytosis
An increase in platelet number (thrombocytosis) and activity is seen in patients with malignancies and was first noticed by Reiss et al. in 1872 [33, 34]. Moreover, a large number of platelets are found in the tumor microenvironment outside of the blood vessels inducing angiogenesis and facilitating cancer cell dissemination [35]. The concept of using an antiplatelet approach for cancer treatment originated in 1968 when Gasic et al. [36] demonstrated that intravenous injection of neuraminidase resulting in thrombocytopenia was associated with decreased metastasis in a mouse model. Thrombocytosis is a poor prognostic indicator for epithelial ovarian carcinoma [37]. Further experiments on animal models and analysis in cancer patients confirmed these associations [38,39,40,41,42]. Moreover, platelet transfusion in orthotopic models of human ovarian cancer resulted in significantly greater tumor weight than in untreated mice or platelet-depleted mice, where in the latter condition, mean tumor weight was diminished by 70% [43]. Lower platelet counts and antiplatelet therapy independently predicted better outcomes in patients with head and neck squamous cell carcinoma, invasive ductal breast carcinoma, gastric cancer, and renal cancer [39, 40, 44]. Patients with rectal adenocarcinoma presenting lower platelet counts were more likely to answer with good or complete response to neo-adjuvant treatment than patients with higher platelet counts [41]. Similarly, reduced pre-operative platelet levels related to better histopathological features and improved overall survival in gastric cancer patients [41].
Recent data support the prognostic value of the platelet-lymphocyte ratio (PLR) in cancer patients (e.g. gastric, colorectal, esophageal, ovarian, lung cancer) [45,46,47]. Namely high peripheral blood PLR was related to poor tumor differentiation, local staging (T – tumor), recurrence of the disease, and decreased overall survival. Therefore, PLR may be used as a predictor of overall survival (OS) in association with clinicopathological parameters in cancer patients.
Platelet activation is observed in cancer patients and is detected by increased secretion of platelet content and release of microparticles and exosomes [2]. Biomarkers of platelet activation in cancer patients are soluble P-selectin, thrombospondin 1, TSP-1, β-thromboglobulin, CD40 ligand, transforming growth factor-β (TGF-β), platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), angiopoetin-1 (AP-1), matrix proteins, CCL17, CCXL1, CXCL5, and others [2, 48, 49]. Among the proteins identified on platelet exosomes is GAPDH, which can function as a membrane fusion protein and can serve as a plasmin/ogen receptor along with histone H2B that is also found on exosomes of different cell types [50,51,52,53]. This represents an underexplored source of immune suppression, as plasmin can potentially suppress NK cells through the release of fibrin and excessive exosome release could exhaust an NK response [10].
Seminal in vitro and in vivo studies have demonstrated that platelet activation contributes to the metastatic potential of cancer cells [1,2,3]. Intriguingly, the level of platelet reactivity may also contribute to prognosis of cancer patients [54]. The observation that decreased platelet reactivity, as measured by platelet surface expression of P-selectin, related to high risk of death in patients with cancer is most likely a consequence of continuous platelet activation.
It should be noted that there are no prospective studies assessing cancer incidence in healthy individuals with elevated platelet levels versus normal platelet levels. The results from mouse animal models where large numbers of malignant cells are injected directly into circulation cannot explain all aspects of cancer dissemination, which is a complex and multi-signal process in the human body. The elevated number of platelets in some cancer patients may be simply a paraneoplastic syndrome aroused by growth factors secreted by tumor cells.
2.2 PMPs
Circulating tumor cells (e.g., of gastric, ovarian, oral, and prostate cancer) may activate platelets resulting in a production of small microparticles (MPs) that contain membrane receptors as well as cytoplasmic constituents [55,56,57]. Platelet-derived microparticles (PMPs) play a role in tumor growth and invasiveness (at least in part by stimulation of MMP-2 production), and angiogenesis [by releasing vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF)], as well as induce metastasis [58]. The level of PMPs is an indicator for patient survival [59].
2.3 TCIPA
The effect of platelet activation induced by tumor cells is associated with platelet aggregation (TCIPA) mediated by multiple agents: tissue factor (TF), thrombin, adenosine diphosphate (ADP), TxA2, and matrix metalloproteinases (MMPs) [60, 61]. Additionally, factors secreted by activated platelets elicit signaling within tumor cells and the tumor microenvironment so that the activation of cancer cells and platelets is reciprocal [62]. TCIPA is regarded as crucial for cancer dissemination. Studies based on three-dimensional visualization demonstrated clearly that tumor cells arrest in the pulmonary vasculature of mice in a complex with platelets and fibrinogen [63]. Tumor cell-platelet interactions in the pulmonary microvasculature were also studied in vivo following tail vein injection of murine Lewis lung carcinoma (LLC), 16C mammary adenocarcinoma, and B16 amelanotic melanoma tumor cells [64].
Importantly, tumor cells can be found surrounded by platelets in blood stream, which may afford protection from lysis by natural killer cells (NKs), demonstrating that TCIPA increases CTC survival. Moreover, activation of platelet-derived TGF-β decreases immunoreceptor NKG2D expression on the surface of NKs thus inhibiting their antitumor reactivity [65]. There is ample evidence that some antiplatelet drugs exert their antimetastatic effects by TCIPA inhibition (presented later).
3 Platelet-associated molecules and therapy
3.1 AA metabolites, COX, and LOX inhibitors
It is widely recognized that inflammation may trigger cancer initiation and progression [66,67,68,69]. Arachidonic acid is metabolized enzymatically via COX and LOX generating bioactive lipids termed eicosanoids: 2-series prostaglandins (PGs) and thromboxanes (Txs) (COX pathway) or 4-series leukotrienes (LTs) and hydroxyeicosatetraenoic acids (HETEs) (LOX pathway) [70,71,72]. Tumor cell-platelet interaction may activate COX individually or COX and LOX in concert resulting in production of the above mentioned eicosanoids. These in turn are involved in the growth and progression of cancers by regulating such processes as apoptosis, angiogenesis, and tumor cell invasiveness [68,69,70,71,72,73]. Thrombin-activated platelets (activation dependent on PARs) are a source of 12- and 15-HETE, as well as TxA2 [2, 74, 75]. The release of TxA2 into the microenvironment activates prostanoid receptors (TP) on platelets resulting in increased expression of GPIIb/IIIa (integrin αIIbβ3) and platelet aggregation. Preincubation of platelet-rich plasma with TxA2 receptor inhibitor SQ-29,548 decreased platelet aggregation induced by tumor osteogenic sarcoma cells without affecting TxA2 release [76]. TCIPA in carcinosarcoma cells resulted in 12-HETE activation and a subsequent increase in platelet expression of GPIIb/IIIa [77].
Due to the activities of COX and LOX pro-inflammatory metabolites, these enzymes are relevant targets for anticancer therapies [68,69,70,71,72,73].
3.1.1 Cyclooxygenase inhibitors
Compelling evidence exists for antitumor activity of the COX-1 and -2 inhibitor, aspirin, which disrupts platelet function by inhibiting prostaglandin and thromboxane formation [78,79,80]. Expression of COX-1 and/or COX-2 is cell-dependent. ECs and tumor cells express COX-2, but platelets express COX-1 [11]. It is difficult to distinguish which effects observed in experimental studies are attributable to which isoform of the enzyme, and the exact anticancer mechanisms of aspirin remain unexplained. There were hypotheses that aspirin benefits from COX-2 inhibition were from its antiproliferative and proapoptosis potential. However, it has been argued that aspirin at low doses could not result in the blood concentrations necessary to impact those processes [17]. Thus, the hypothesis that a dominant mechanism for the cancer preventive and anti-metastasis property of aspirin is associated with its antiplatelet activity (COX-1 inhibition) is plausible [78]. Platelets have been proven to impact metastases by direct and indirect interactions with tumor cells [1,2,3]. As aspirin can induce platelet apoptosis via caspase-3 activation [81], its cancer preventive role may be explained by thus interfering with platelet-induced epigenetic modifications of tumor cells during early phases of colorectal tumorigenesis, for example. Platelet activation in intestine may lead to local inflammation and cellular transformation [82].
Experiments with cancer cell lines in vitro provided proof that the anticancer effect of aspirin is associated with its influence on cross-talk between platelets and cancer cells (Table 1). Incubation of HT29 human colon carcinoma cells with human platelets induced their transformation from epithelial to mesenchymal-like features of cancer cells that is an indispensable step for cancer dissemination [3, 13]. Enhanced cancer cell mobility was the result of E-cadherin downregulation and Twist1 upregulation. There was also a proaggregatory effect on the platelets by the cancer cells. Similar results have been described for ovarian cancer cell line SK-OV-3, where platelets increased their invasive potential by inducing the epithelial-to-mesenchymal transition phenotype [85], but that could be inhibited by aspirin. The anticancer potential of aspirin when administered in low, antiplatelet doses to colon and pancreatic cancer cells was associated with inhibition of oncoprotein c-MYC expression and a reduction in proliferative potential of cancer cells [19].
The injection of platelet-exposed HT29 cells into the tail vein of humanized, immunodeficient mice resulted in higher incidence of lung metastasis. Inhibition of platelet COX-1 by aspirin prevented the in vitro changes as well as reduced the metastatic rate in vivo [1, 17]. The inhibitory effect of aspirin was also observed on B16-F0 melanoma cell proliferation and tumor growth in a mouse model [86].
Preclinical experiments also demonstrated the vital role of platelet activation in pathogenesis of hepatitis B virus (HBV)-associated hepatocellular carcinoma (HCC) as well as the anticancer effectiveness of dual platelet inhibitors (aspirin plus clopidogrel) [87, 90]. Platelet activation in HBV-HCC mouse models led to accumulation of virus-specific CD8 T cells and virus-non-specific inflammatory cells in the liver, increased hepatocellular proliferation, liver fibrosis, and hepatocarcinogenesis. Platelet inhibition prevented injury of liver cells and tumor development most likely due to the reduction of immune-mediated pathological effects in the liver.
Aspirin has also been shown to reduce mediastinal lymph node metastasis in a mouse model after injection of LLC cells [66]. Aspirin had no effect on primary tumor in the lung but significantly reduced mouse mortality. There is heterogeneity in the human response to aspirin in the setting of lung cancer. An English study found no survival benefit [91], while a German study suggested that there are benefits to specific subgroups that warrant careful scrutiny [92].
Aspirin is also an important antiangiogenic agent [88]. Human platelets activated with thrombin secrete both VEGF and endostatin, ultimately promoting tubule-like formation and increased proliferation of endothelial cells. Surprisingly, these proangiogenic actions were only slightly suppressed by VEGF receptor-neutralizing antibody. The other platelet-induced signaling inhibitors of protein kinase C (PKC), p38, ERK1/2, Src kinases, or PI3K/Akt also exerted only partial inhibitory effects. In contrast, aspirin fully blocked the pro-angiogenic activity of the platelet releasate.
A novel derivative of aspirin, nitric oxide- and hydrogen sulfide-releasing hybrid or NOSH-aspirin (NBS-1120), has been developed that is stronger than classic aspirin in its potential to inhibit tumor growth and decrease tumor size [28]. Moreover, it is suggested that aspirin does not prevent TCIPA while the aspirin prodrug, 5-nicotinate salicylate (ST0702 salicylate), is effective in TCIPA inhibition [93].
Another inhibitor of platelet COX and PDGF, trapidil, does prevent spontaneous pulmonary metastases formation in mice, but only when administered after excision of the primary tumor [94]. Non-steroidal anti-inflammatory drugs, e.g., flurbiprofen, prostacyclin analogues, and thromboxane A2 synthase inhibitors, also significantly decrease spontaneous metastases formation [95, 96].
An antagonist of the prostaglandin(PG)E2 EP3 receptor, DG-041, prevented induction of mesenchymal-like changes in colon cancer cells by lowering E-cadherin expression and elevating Twist1 protein resulting in disrupted metastatic potential. Cancer cell motility and proaggregatory action on platelets were also abrogated by this drug [18].
Solid-phase von Willebrand Factor (sVWF) is known to mediate TCIPA. Unstimulated platelets can adhere irreversibly to sVWF and stimulation by ADP, thrombin, or ristocetin increases adherence [97]. However, aggregation can be blocked by S-nitroso-glutathione (GSNO) and prostacyclin (PGI(2)). Moreover, pre-incubation of platelets with PGI(2) inhibits sVWF-tumor cell-stimulated platelet surface expression of GPIIb/IIIa essential for tumor cell/platelet interactions [98].
The majority of clinical data relates to aspirin use in colorectal cancer (CRC) which is well recognized to be associated with inflammation (colitis–dysplasia–carcinoma) [78]. Although the Physicians’ Health Study reported in 1998 that they found no association between the use of aspirin and the incidence of CRC [99], subsequent studies reported more promising results [24, 100]. Long-term effects of aspirin on CRC incidence and mortality during the more than 20-year follow-up were presented in the analysis of their randomized trials [24, 100]. Aspirin taken daily for 5 years or longer at a minimal dose of 75 mg reduced both incidence and mortality due to CRC with the greatest preventive effect for cancers of the proximal colon. Importantly, aspirin reduced the incidence of CRC and benign polyps (adenomas) in the general population and individuals with a history of adenomas or CRC [100, 101]. In the general population, the CRC incidence was decreased by 26% (RR 0.74, 95% CI 0.57 to 0.97), while in patients with a history of adenomas or CRC, risk of adenoma recurrence was reduced by 21% [relative risk (RR) 0.79, 95% confidence interval (CI) 0.68 to 0.92]. In case-control studies, regular use of aspirin has also been documented to reduce risk of CRC in good agreement with results of the randomized trials [102].
The preventive role of aspirin has now been documented in multiple studies of other cancers [78, 103].
The assessment of short-term effects of aspirin from the analyses of 51 randomized controlled trials that were designed to evaluate the role of aspirin in the primary and secondary prevention of vascular events demonstrated that aspirin reduced the incidence of several cancers from 3 years onwards [104]. In addition to having a preventive role in cancer, depending on the duration of treatment, aspirin also improves the results of treatment among cancer patients [7, 17, 24].
Pooled data for eight randomized controlled trials demonstrated that daily aspirin reduced deaths due to cancer (OR 0.79, 95% CI 0.68 to 0.92) [24]. A 5 years follow-up (data assessed in seven trials) revealed a significant benefit from daily aspirin for all cancers (HR 0.66, 95% CI 0.50 to 0.87) with the best results observed for gastrointestinal cancers (HR 0.46, 95% CI 0.27 to 0.77). Interestingly, three trials that assessed the 20-year risk of cancer death also showed that patients who took aspirin had a lower risk of cancer-associated death (HR 0.78, 95% CI 0.70 to 0.87 and for gastrointestinal cancers HR 0.65, 95% CI 0.54 to 0.78). Taking into consideration all solid cancers, the benefits significantly increased with treatment duration of 7.5 years and longer. The subgroup analysis indicated that for esophageal, pancreatic, brain, and lung cancers, this time may be around 5 years. Another important conclusion by authors was that the effects of aspirin did not depend on dose (75 mg and over 75 mg) and did not differ in relation to gender and smoking status. The final result was that the absolute reduction in 20-year risk of cancer death was 7.08% (95% CI 2.42 to 11.74) in patients 65 years or older at randomization. The decreased risk of death among patients diagnosed with cancer during trials may have resulted from the lower frequency of distant metastases observed in the aspirin group versus control [105], particularly in patients with colorectal cancer (HR 0·26, 95% CI 0·11–0·57, p = 0·0008). Aspirin lessened the frequency of metastasis both at initial diagnosis and during follow-up in patients who did not present with metastasis initially.
At this point, data regarding breast cancer are inconclusive [17, 106]. A 10-year Women’s Health Study Trial with almost 40,000 women did not correlate aspirin use with decreased breast cancer incidence [26], while other meta-analyses have demonstrated less development of breast cancer (9–30%) among women who have been taking aspirin [107, 108]. The differences in results may be associated with the length of time aspirin is taken [17] Shiao]. A prospective observational study of 4164 women with breast cancer showed that decreased risk of distant recurrence and breast cancer death is observed when aspirin is taken at least 12 months [7]. The newest retrospective analysis of 222 stage II and III triple-negative breast cancer (TNBC) patients demonstrates improvement in 5-year DFS (80.1 vs 62.6%, p = 0.04) and 5-year DMR (9.0 vs 31.6%, p = 0.007) in the aspirin group compared with the control group. Intriguingly, aspirin also decreased the rate of central nervous system (CNS) metastases.
The benefits of an aspirin regimen were also documented for lowering prostate cancer incidence [109,110,111,112] and for prostate cancer patients, especially in the high-risk group [4, 113]. The study, comprising nearly 6000 patients after prostatectomy or radiotherapy who received different anticoagulants (AC), showed that aspirin use was associated with lower prostate cancer-specific mortality (PCSM) compared with the non-AC group (adjusted hazard ratio 0.43; 95% CI 0.21 to 0.87; P = 0.02) [4]. Disease recurrence and bone metastasis were also significantly lower in the AC group.
The prevention of carcinogenesis in cell line models and improvement of recurrence-free survival and overall survival (52.8 vs 47.9%; p = 0.021 and 80.3 vs 65.4%; p < 0.001, respectively) were observed among HBV-related hepatocellular carcinoma (HCC) patients after liver resection [20].
Aspirin assessment in cancer patients summarizes Table 2.
The recommendations on the preventive use of aspirin are summarized on the official website of the US preventive services task force (Table 3).
3.1.2 Lipoxygenase inhibitors
The enhanced expression of 5-LOX and 12-lipoxygenase (12-LOX) is associated with carcinoma progression and invasion [114, 115]. Increased expression of 5-LOX was investigated in adenomatous colon polyps and cancer compared with normal colonic mucosa, while 12-LOX expression was higher in more advanced stages of prostate cancer and in high-grade tumors [116].
Overexpression of the platelet 12-LOX in PC-3 prostate cancer cells results in 12(S)-HETE production which leads to the activation of transcription factors such as NF-κB resulting in increased MMP9 expression and in hypoxia inducible factor-1alpha (HIF-1alpha)-mediated in VEGF expression [71, 72, 117]. There is also evidence that platelet 12-LOX-mediated AA metabolism is responsible for radioresistance of prostate cancer cells and metastatic potential of melanoma cells [118]. The experimental in vitro and in vivo studies demonstrated the antiproliferative potential of LOX-inhibitors toward cancer cells [119]. The 5-LOX inhibitor zileuton decreased tumor growth of HT29 and LoVo human colon cancer cells in a heterotopic xenograft model in athymic mice [115]. The LOX inhibitors (nonselective nordihydroguaiaretic acid, 5-LOX inhibitor Rev-5901, and the 12-LOX inhibitor baicalein) suppressed proliferation of pancreatic cancer cells and decreased survival of prostate cancer cell line [120]. The newest approach is based on even dual inhibition of COX and LOX activity which leads to reduced proliferation and decreased tumor burden as well as cell migration and invasion [68, 117].
4 ADP receptors
4.1 ADP receptor P2Y12 antagonists, thienopyridines, and pyrimides
The platelet adenosine diphosphate (ADP) receptor P2Y12 is essential for thrombus formation by amplifying the hemostatic responses activated by a variety of platelet agonists. It stabilizes platelet aggregation and is involved in granule secretion as well as integrin activation. Apart from its role in hemostasis, the P2Y12 receptor has been shown to influence multiple mechanisms leading to cancer progression mainly by regulating tumor cell/platelet interaction and angiogenesis [121, 122]. There is also evidence that P2Y12 receptors mediate tumor cell transendothelial migration [123]. P2Y12 inhibition hampers LLC cells from influencing platelet shape and induces release of active TGFβ1 that disrupts platelet-mediated EMT-like transformation of the LLC. Similar effects were documented for melanoma cells [121].
Thienopyridines are antiplatelet drugs that inhibit the platelet adenosine diphosphate (ADP) receptor, P2Y12, and in that way inhibit ADP-mediated platelet activation and aggregation. The in vitro studies demonstrated the anticancer activity of thienopyridines in multiple cancer cell lines and cancer murine models (e.g. ovarian, breast, hepatocellular cancer, melanoma) [7, 85, 124]. The mechanisms of curtailing the invasive capacity of cancer cells were associated with suppression of platelet aggregation and adhesion, and decreased potential for platelets to create complexes with tumor cells [7, 32, 125]. Moreover, P2Y12 inhibitors influenced platelet proangiogenic potential in cancer patients [7]. Vascular endothelial growth factor and endostatin are the most vital protein modulators of angiogenesis released from platelets by the ADP-dependant pathway [1]. Cangrelor, the in vitro equivalent of clopidogrel, decreased secretion of VEGF, TSP1, and TGF-β1 by platelets to a greater extent when coming from cancer patients than from healthy controls. Thienopyridine SR 25989, an enantiomer of the anti-aggregant clopidogrel, inhibited angiogenesis by increasing the expression of endogenous TSP1 and reduced lung metastasis in metastatic melanoma mouse model [126].
Within the thienopyridine family, the ADP receptor inhibitor ticlopidine exhibited inhibitory effects on pulmonary metastases of B16 melanoma and AH130 ascite hepatoma as well as spontaneous metastases of LLC [94, 127]. The reversible P2Y12 inhibitor ticagrelor, recommended for the prevention of cardiovascular and cerebrovascular events, exerted an inhibitory metastatic effect in intravenous and intrasplenic melanoma and breast cancer mouse models [124]. The reduction of lung, liver, and bone marrow metastasis rate was by 55–84, 86, and 87%, respectively. The decreased interactions between tumor cells/platelets were the consequence of disrupted GPIIbIIIa expression. However, these findings may be context dependent [128].
Additionally, the anticancer/antimetastatic activity of thienopyridines may be associated with their inhibiting action on the interactions between cancer cell-derived tissue factor-positive microparticles (TMP), fibrinogen, and activated platelets [129, 130]. Study with human pancreatic adenocarcinoma cell lines showed that tumor-derived TMPs may activate platelets in vitro and in mice leading to thrombosis. The administration of clopidogrel inhibited thrombus generation and diminished tumor size in a mouse model by preventing the P-selectin and integrin-dependent accumulation of cancer cell-derived microparticles at the site of thrombosis.
The influence of antiplatelet treatment based on clopidogrel and aspirin on number of CTC numbers was investigated in a randomized phase II trial in breast cancer patients [131]. Unfortunately, there was no difference in the number of patients with detectable CTCs between the clopidogrel/aspirin and control groups suggesting that such an approach has no treatment effect.
Aspirin and thienopyridine are an established Dual Antiplatelet Therapy (DAPT) for reduction of stent thrombosis and myocardial infarction after coronary stenting. The alarming data that such an approach may be associated with increased cancer incidence forced re-analysis of the main trials from this point of view [29,30,31]. The adverse effects of antiplatelet therapy were first observed during the TRITON trial in which prasugrel versus clopidogrel was assessed in patients with acute coronary syndromes and for vorapaxar treatment in the TRACER trial [30]. Ticagrelor treatment (reversibly binding oral P2Y12 receptor blocker) assessed in the PEGASUS trial was also associated with significant excess in cancer death [132]. DAPT therapy from a 30-month trial with prasugrel and clopidogrel provided abundant data [30]. This trial revealed that a long-term antiplatelet approach is associated with cancer development that contributes to about half of noncardiovascular deaths (NCVD). The outcomes of the analysis indicated that cancer risk that reflects a class effect and is not drug-specific occurs after 24 months of extended antiplatelet therapy. It is suggested that extensive periods of antiplatelet therapy render platelets unable to aggregate with tumor cells and restrict them locally in situ [31]. For this reason, antiplatelet therapy with thienopyridines is recommended not to exceed 2 years. Such an approach reduces cancer risk, while most vascular benefits are still preserved [30]. Likewise, triple therapy aggressive regimens, lasting more than 1 year, are not recommended and intake of additional NSAIDS in combination with these regimens must be considered carefully for cardiovascular risk [133].
Importantly, there are some controversies in the construction of DAPT trial highlighted by FDA (Food and Drug Agency) which might have affected the final results [31]. There was imbalance between groups enrolled in the study. Namely, 22 more patients were randomly assigned to the thienopyridine group than to the placebo group leading to a difference in the number of patients in whom cancer had been diagnosed before enrollment (8 vs 1). Moreover, the rate of diagnosis of cancer did not differ significantly after randomization, even though there were more cancer-related deaths among patients treated with continued thienopyridine than among those who received placebo. Apart from that there is a lack of precise information about dosages for prasugrel and aspirin and the follow-up details. DAPT trial did not also describe how the follow-up statistics count deaths, allowing for about 5% data being missing. The Agency suspect that the way the enrolled patients were selected for randomization could, in turn, affect the cancer risks. Taking into consideration that the DAPT trial was a large, randomized trial, the initial risks should have been equal in both arms.
The doubts are enhanced by the fact that there are other data not confirming these alarming findings. In direct contrast to derived from the aforementioned clinical trials, a population-based cohort study of 10,359 colorectal, 17,889 breast, and 13,155 prostate cancer patients showed no evidence of an increased cancer-specific mortality among clopidogrel users [32]. The retrospective analysis among patients with hepatocellular cancer after liver resection showed that use of aspirin or clopidogrel was associated with better recurrence-free survival and overall survival among patients with HBV-related HCC [20]. Moreover, the analysis of survival after solid cancer (SASC) rates in patients enrolled in antithrombotic trials showed that survival in these groups of patients does not differ from Surveillance, Epidemiology, and End Result (SEER) or World Health Organization averages [31].
4.2 ADP scavenger apyrase
ADP is a marker of platelet activation stored in dense granules that interacts with 2 P2Y receptors, the Gq coupled P2Y(1) receptor, and the Gi-coupled P2Y(12) receptor leading to platelet shape change, aggregation, and release of TxA2 [134]. ADP is involved in TCIPA. Very early studies showed that two ADP-scavenging agents, apyrase (10 U/ml) and creatine phosphate (CP) (5 mM)/creatine phosphokinase (CPK) (5 U/ml), completely inhibited TCIPA induced by B16-F10 melanoma and lung cancer cell lines [60]. Recent experiments were unable to confirm the inhibitory effect of apyrase on platelet aggregation and secretion [135]. Perhaps the inhibitory potential of ADP scavenger apyrase alone is too weak, as the novel soluble apyrase/ADPase, APT102, given together with aspirin, but not either drug alone, did inhibit TCIPA and lead to significant decreases in breast cancer and melanoma bone metastases in mouse models. Additionally, these effects were accompanied by fewer bleeding complications than observed with GPIIaIIIb inhibitors [136].
5 Integrins
Integrins are transmembrane receptors composed of α- and β subunits [eprenbeck]. Their expression on platelets increases after platelet activation, which promotes integrin interactions with extracellular matrix (ECM) proteins [1, 2].
5.1 Integrin αIIbβ3 (glycoprotein IIb/IIIa, GPIIb/IIIa) antagonists
The main glycoprotein on the surface of platelets and the key factor in carcinogenesis is αIIbβ3-Integrin (GP IIb/IIIa). GP IIb/IIIa contains an arginine-glycine-aspartic acid (RGD) sequence and functions as a receptor for fibrinogen and numerous other adhesion proteins [137]. GP IIb/IIIa regulates platelet aggregation and granule secretion as well as interaction between platelets, ECs, extracellular matrix components (ECM), and tumor cells resulting in platelet adhesion to these structures [138,139,140]. Thus, targeting the GP IIb/IIIa and subsequent impairment of platelet function within the tumor microenvironment provides a therapeutic option to inhibit metastasis.
The role of GP IIb/IIIa in TCIPA was first documented in 1987 by preincubation of human platelets with antibodies to platelet glycoprotein Ib and the IIb/IIIa complex [141]. The inhibitory action of monoclonal antibody against GPIIb/IIIa, 10E5, on pulmonary metastasis was subsequently determined using in vivo models the following year [142]. Since then, many inhibitors of the GP have been shown to prevent ECs, ECM, and cancer cell-platelet interactions, e.g., RGDS peptide, tirofiban (nonpeptide mimetic of the RGD sequence), eptifibatide (cyclic heptapeptide based on the KGD amino acid sequence), and abciximab (monoclonal antibody 7E3 Fab fragment) [143,144,145]. These three inhibitors (abciximab, eptifibatide, and tirofiban) are currently approved for human use in cardiovascular diseases [146].
Glycoprotein IIb/IIIa antagonists exert a strong anti-platelet effect by inhibition of the final pathway of platelet aggregation and reduce platelet adherence to tumor cells, ECM, and ECs. GP IIb/IIIa inhibition resulted in TCIPA blockade in MCF-7 breast cancer, human cervical carcinoma (HeLa), Lewis Lung carcinoma (LL2), and B16-F10 mouse melanoma cell lines resulting in decreased hematogenous metastasis to the lung in a mouse model [147,148,149,150]. Inhibition of TCIPA was also documented for snake venom Arg-Gly-Asp-containing peptides (e.g. triflavin, rhodostomin, trigramin) against osteosarcoma cells, hepatoma cells, and several adenocarcinoma cells (e.g. prostate, breast) [151,152,153]. These drugs exerted their effects by binding to the fibrinogen receptor associated with GP IIb/IIIa complex on the platelet surface.
More recent in vitro findings have documented the pivotal role of platelet GP IIb/IIIa in the development of bone metastasis by mediating tumor cell/host stroma interactions [154,155,156]. As many as 74% of mice expressing beta3 integrin developed osteolytic bone metastasis after injection of B16 melanoma cells intracardialy, while only 4% of mice depleted of beta3 integrin demonstrated bone lesions [154]. Rhodostomin, a disintigrin, strongly inhibited the adhesion, migration, and invasion of tumor cells in bone extracellular matrices [49].
It was demonstrated that a novel humanized single-chain antibody (scFv Ab; A11) against integrin GPIIIa49–66 could induce platelet lysis within the tumor environment [157]. This compound effectively inhibited multiple interactions between tumor and host cells with the end result being blocked development of pulmonary metastases in the LLC metastatic model. There is also an orally administered non-peptide small molecular peptidomimetic inhibitor of GPIIb-IIIa (XV454). It has been shown to reduce TCIPA in vivo and experimental metastasis in the Lewis lung carcinoma (LL2) mouse model [158].
GPIIb-IIIa inhibitors may also affect angiogenesis by reducing the release of VEGF from platelets as well as decreasing platelet/tumor cell complexes adherence to ECs and ECM an important step in angiogenesis [138, 139]. Additionally, the monoclonal antibody 7E3 F(ab’)2 reduced platelet-stimulated sprouting of ECs and tube formation [159, 160].
New studies have focused on the integrin β subunit plexin-semaphorin-integrin (PSI) domain that has endogenous thiol isomerase activity and is potentially a novel target for anti-platelet therapy [161]. The monoclonal antibodies directed to the PSI domain reduce the interaction of αIIbβ3-integrin with fibrinogen as well as inhibit murine and human platelet aggregation in vitro and ex vivo. This study showed that the PSI domain is an important factor for integrin functions and cell-cell interactions, such that its inhibition should be investigated beginning with cancer cell lines.
The inhibitory potential toward platelet integrins is also exerted by holothurian glycosaminoglycan (hGAG), a sulfated polysaccharide extracted from the sea cucumber, Holothuria leucospilota Brandt [162]. It blocks interactions between breast cancer cells MDA-MB-231 and platelets by suppressing both mRNA and protein levels of β1 and β3 integrins, as well as MMP-2 and -9, while increasing the expression of the MMP inhibitor, tissue inhibitor of metalloproteinase (TIMP)-1. hGAG reduced thrombin-induced platelet activation and aggregation, and disrupted platelet adhesion to cancer cells and fibrinogen to ultimately attenuate platelet-cancer cell complex formation.
Although results coming from experimental studies are promising, there are no strong clinical data for use of β3 integrin blockers in cancer patients.
5.2 Integrin α2β1
The role of platelet α2β1integrin in cancer is much less documented than GPIIbIIIa. However, there are some data that support its role in pancreatic cancer cell adhesion to ECM [163]. Moreover, recent findings demonstrated that β1 integrin-dependant interactions within the tumor microenvironment may modify tumor response to radiation and chemotherapy [164]. Therefore, targeting β1 integrin is also a promising anticancer approach.
6 Glycoproteins
The receptor complex, GP1b/IX/V and platelet collagen receptor, GPVI plays a predominant role in platelet adhesion and activation in the blood stream by interactions with their ligands, von Willebrand factor (VWF) and collagen, respectively, resulting in platelet capture and rolling at the site of vascular injury [165]. Inhibition of platelet GP1b/IX/V or GPVI receptors has been shown to reduce TCIPA and attenuate metastases [166,167,168,169]. GPVI-deficient mice developed 50% less visible metastatic tumor foci in the lung as compared with control mice after injection of LLC (D121) or melanoma (B16 F10.1) cell lines. Intriguingly, the size of subcutaneously implanted tumor cells did not change in relation to presence or absence of platelet GPVI.
However, others have observed that inhibition of GPIbα by monovalent, monoclonal antibodies strongly increased lung metastasis in melanoma animal models [170].
7 P-selectin inhibitors
There are three selectins, P-, L-, and E-selectin, which belong to the C-type lectins that recognize glycans [165]. The most important platelet-membrane integrin for regulating platelet interactions with mononuclear cells and cancer cells is P-selectin that is expressed by both platelets and ECs [1, 3]. P-selectin is implicated in blood coagulation by mediating fibrin generation. Increased expression of P- and L-selectins (expressed on leukocytes) is observed within the metastatic microenvironment of tumor cells, thus regulating thrombi formation [171]. Experimental metastasis models demonstrated that both P- and L-selectin-depleted mice had improved survival [172]. Heparin as well as other compounds (e.g. dermatan sulfates) inhibited P-selectin-based platelet interactions with carcinoma cell-surface mucin ligands resulting in reduced adherence of tumor cells to platelets and decreased metastatic dissemination [173,174,175]. The observed effects were not associated with the anticoagulant potential of heparin, but by its ability to behave as a P-selectin ligand to impair interactions between P-selectin and its cognate ligands [172].
8 Thrombin inhibitors
Tissue factor expressed by multiple cancer cells triggers local generation of thrombin that is a known potent platelet agonist [3, 13]. Thrombin-induced platelet activation leads to secretion and release of TxA2 and proangiogenic molecules, enhanced expression of P-selectin, CD40 ligand, and integrins on platelet membrane, and increased release of fibronectin and VWF from platelet onto their surface. These thrombin-mediated effects promote primary tumor growth, chemotaxis, migration, intravasation, and angiogenesis thereby facilitating prometastatic adhesion events [3, 176]. Additionally, platelets coated with thrombin survive longer, providing a surface for cancer cells for adherence [1]. Thrombin triggers its effects mainly by PAR-1 and PAR-4 receptors (described later) [3]. Thrombin-induced platelet activation may be associated in part with ADP receptors, mainly P2Y(12) [177]. The inhibition of P2Y(12) attenuated the thrombin-mediated response in blood received from healthy individuals. The inhibition of thrombin-associated effects was mostly observed at thrombin concentrations that caused complete PAR-1 proteolysis and secretion of all ADP. The thrombin-dependent response was inhibited by 70–86% [177].
Over the last several decades, numerous studies documented the prometastatic potential of thrombin [reviewed in 3, 13]. Pretreatment of cancer cells with thrombin or preinfusion of thrombin into mice increased tumor size, enhanced invasive potential of several types of cancer cells, caused 4–413-fold increase in the number of experimental lung metastases, and decreased survival rates [149, 178,179,180,181,182,183,184,185]. In contrast, thrombin inhibition by dansylarginine N-(3-ethyl-1,5-pentanediyl) amide blocked TCIPA of colon cancer [186], while administering bivalirudin or oral reversible thrombostatin FM19 decreased prostate cancer cell invasion as well as tumor growth and angiogenesis in mouse model [187].
8.1 Hirudin
Hirudin, a specific inhibitor of thrombin, diminishes invasive and metastatic potential of thrombin [185]. It decreased by almost half the amount of platelet-derived VEGF released at the site of the thrombus formation and inhibited metastasis in several experimental models [173, 174]. Hirudin inhibited TCIPA in non-small-cell lung cancer cell lines (NSCLC) and in SCLC, but in the last case, blocking TCIPA required addition of the ADP scavenger apyrase with hirudin [60]. Recombinant hirudin (rH) has also been shown to inhibit growth and exert anti-metastatic effects in several types of cancers through multiple mechanisms [185, 188]. Experimental studies with laryngeal carcinoma cells (HEp-2 cells) demonstrated that rH inhibited the adhesion, migration, and invasion of the cancer cells. Another study indicated that co-treatment with rH and liposomal vinblastine exerts a more potent anti-metastasic effect against murine B16 melanoma tumor growth compared to chemotherapy alone [189]. However, hirudin is less intensively investigated in preclinical studies due to its inhibitory potential of thrombin-fibrinogen interactions.
8.2 Heparin
Heparin is commonly used in cancer patients to prevent or treat thrombosis. The anticoagulant therapy also improves survival rates in these patients by an unknown mechanism. Inhibition of thrombin generation induced by low molecular weight heparins, LMWHs in cancer cells, depends mainly on the anti-Xa activity [190]. The most probable anticancer mechanisms mediated by heparin are inhibition of tumor cell/platelet/endothelial interactions to suppress cell invasion and angiogenesis [191].
There is evidence that the important antimetastatic potential of heparin and modified heparin (with heparanase inhibitory activity) is due to their influence on selectin-induced interactions between tumor and blood cells (leukocytes, platelets) as well as ECs, thus inhibiting cancer cell colonization in distant tissues [192, 193]. It has been shown that heparin-mediated P-selectin inhibition disrupted interactions of endogenous platelets with sialylated fucosylated mucins on CTCs and reduced tumor cell survival [194]. Experimental metastatic models with LMWHs demonstrated varying potential of these drugs (tinzaparin, nadroparin, dalteparin, or enoxaparin) for inhibiting P-selectin binding and attenuation of metastasis. Namely, tinzaparin and nadroparin were strong inhibitors of P-selectin functions, while fondaparinux, a synthetic heparinoid pentasaccharide, was unable to suppress this protein and decrease the metastasis rate in a murine model [165, 195]. A new and unique heparin sulfate from the bivalve mollusk Nodipecten nodosus has been shown to inhibit P-selectin-mediated colon cancer cell/platelet complex formation and to decrease lung metastasis [196].
Heparin may also attenuate cancer cell dissemination by altering platelet angiogenic potential via decreasing secretion of platelet-derived VEGF [197]. The other mechanism of heparins in inhibition of hematogenous but not lymphatic metastasis [198] may be associated with decreasing tumor-induced VWF fiber formation and diminished platelet aggregation the fibrous net.
8.3 DTIs
There are four parenteral direct inhibitors of thrombin (DTIs) activity that received Food and Drug Administration (FDA) approval in North America: lepirudin, desirudin, bivalirudin, and argatroban [199]. Argatroban has been shown to decrease tumor migration as well as bone metastasis in mouse breast cancer models [200, 201]. Animal models provided promising data about dabigatran etexilate, which administered twice daily at a dose of 45 mg/kg over 4 weeks, and attenuated invasive and metastatic potential of malignant breast tumors [202]. Dabigatran etexilate decreased tumor volume by 50% and led to reduction of tumor cells in the blood and micrometastases in the liver by 50–60%. Importantly, it has been shown that dabigatran etexilate and low-dose cyclosphosphamide (CP) reciprocally potentiate their own effects [203]. Namely, mice being co-treated with both drugs presented significantly smaller mammary tumors and developed fewer lung metastases than mice treated with CP or dabigratran etexilate alone. Similar observations have been made for dabigatran administered with cisplatin or gemcitabine in ovarian or pancreatic cancer, respectively [204, 205]. Dabigatran co-treatment with low-dose cisplatin or gemcitabine in mice was more efficient in decreasing tumor growth than dabigatran or chemotherapeutic given as a single agent. Dabigatran also exerted antiangiogenic potential by decreasing levels of Twist and GRO-α proteins as well as impeding vascular tube formation induced by thrombin in breast and glioblastoma cell lines [206]. The other thrombin inhibitor, FM19, was also found to reduce angiogenesis in prostate tumor models [187]. Additionally, dabigatran etexilate prevented a tumor-induced increase in circulating TF(+) microparticles and decreased the amount of activated platelets by 40% demonstrating thrombin participation in intravascular events [203].
8.4 Targeting thrombin generation
The factor Xa inhibitors apixaban, edoxaban, fondaparinux, and rivaroxaban inhibit thrombin generation and influence thrombin-mediated platelet activation. It has been shown that the histological type of cancer influences the antithrombotic activity of the thrombin inhibitors, and this should be taken into consideration when analyzing the results of experimental studies [207]. Fondaparinux has demonstrated inhibitory impact on platelet angiogenic potential. Decreased thrombin generation, which is the main activator of PAR-1 and PAR-4 present on the platelet surface, may disrupt PAR-mediated activation and in this way diminish angiogenic potential of cancer cells. The first clinical trial with thrombin inhibitor (Rivaroxaban) as an anti-cancer agent is now being established [208]. The efficacy of thrombin inhibition preoperatively (TIP) in supression of tumor proliferation will be investigated in estrogen receptor negative early breast cancer patients. The reduction in tumor Ki67 from pre-treatment to 14 days post-treatment initiation will be controlled.
The new research has provided evidence that V600EB-Raf signaling is involved in thrombin generation in metastatic melanoma via increased expression of TF on cell membranes [209]. BRAF is a component of the RAS–RAF–MEK–ERK signaling pathway that plays a critical role in cell proliferation, differentiation, and survival. Targeting V600EB-Raf-associated intracellular pathways and decreased thrombin generation may be another target for anticancer treatment.
Although clinical trials provided promising data that heparin treatment improved survival in cancer patients, it is still not recommended as anticancer approach [210].
9 PARs inhibitors
Thrombin activates human platelets by triggering a family of G protein-coupled receptors, PARs, in a unique proteolytic mechanism. Thrombin opens the receptor active site by cleavage of a key extracellular domain of the N-terminus of the receptor. After cleavage, the newly formed amino-terminal peptide acts as a tethered ligand that binds to the receptor inducing intracellular signaling [3]. There are four PARs, PAR-1 to PAR-4, but human platelets express only high-affinity PAR-1 and low-affinity PAR-4 receptors that interact with different concentrations of activator [3]. Thrombin-induced cleavage of PAR-4 causes longer activation of Gαq and sustained Ca2+ response, resulting in prolonged secondary signaling, compared to PAR-1, contributing to the late-phase of platelet aggregation. The thrombin-elicited platelet PAR activity also exerts mitogenic activity toward ECs and myocytes of vessels [3]. The evidence for a pivotal role of thrombin-mediated platelet PAR activation in cancer dissemination comes from studies performed on mice depleted of platelets, PAR-4, or fibrinogen—these animals presented significant reduction in experimental pulmonary metastasis [211].
Both receptors activate pleiotropic cellular effects, one of which is stimulation of angiogenesis. Previous findings that thrombin activation of PAR-1 leads to VEGF secretion from platelets, while activation of PAR-4 results in the release of an inhibitor of angiogenesis–endostatin, were recently disputed [212]. Namely, the activation of platelet with either PAR-1 or PAR-4 activated peptides resulted in significant VEGF secretion. Moreover, both platelet PAR-1 and PAR-4 activation promoted angiogenic activities of endothelial progenitor cells. However, PAR-1-mediated platelet releasate acted more potently than PAR-4-stimulated proteins in vasculogenesis [213].
PAR-1 and PAR-4 induced platelet activation leads to synthesis and release of TxA2 and 12(S)-HETE, which, when esterified to phosphatidylethanolamine, acquire unique functions in that context [214, 215].
Importantly, platelet PAR-1 may also be triggered by MMP-1, leading to Rho-GTP as well as MAPK signaling activation, resulting finally in platelet aggregation, and increased motility and cell proliferation [216]. The ProMMP-1 zymogen is converted to MMP-1 on the platelet surface with the presence of collagen fibrils. Inhibition of MMP-1-mediated PAR-1 signaling is efficient to block thrombosis in animal models, giving evidence that the collagen/MMP-1/PAR-1 pathway may stimulate platelet signaling events independent of thrombin. As PARs also stimulate the expression and release of 12(S)-HETE that upregulates MMP9 [72], it seems that bi-directional regulation of MMPs and PARs signaling exists.
PAR-1 also promotes expression of melanoma cell adhesion molecule MCAM/MUC18 (MUC18), a crucial marker of melanoma metastasis. It is of interest that PAR-1 activity increases expression of platelet-activating factor receptor (PAFR) and its ligand and, in this way, not only induces platelet aggregation but also increases MUC18 levels. The PAR1/PAFR/MUC18 pathway regulates melanoma cell adhesion to microvascular ECs and transendothelial migration, and finally enhances metastatic seeding in the lungs [217]. Upregulation of PAFR and its agonists in this context may also impact immune functions against cancer [218,219,220,221,222].
The pleiotropic mechanisms regulated by PARs make inhibitors of these receptors an exciting option for antiplatelet and anticancer treatment. Many compounds, such as RWJ-58259, SCH-79797, or RWJ-56110, were developed that target PAR-1 and exert strong antithrombotic effects in vivo [223,224,225]. Additionally, PAR-1 small-interfering RNA (siRNA) incorporated into neutral liposomes (1,2-dioleoyl-sn-glycero-3-phosphatidylcholine, DOPC) was assessed in experimental melanoma models. Mice treated with the PAR-1 siRNA-DOPC presented a decline in tumor growth, weight, and formation of metastatic lung colonies [226]. siRNA delivery also affected angiogenesis by reducing VEGF, Il-8, and MMP-2 expression levels, and blood vessel density. PAR-1 silencing downregulates expression of the adhesive protein MUC18, which also attenuates the metastatic phenotype of melanoma cells [226].
The first PAR-1 antagonist, vorapaxar, was recently approved for use as an antiplatelet agent to reduce the frequency of ischemic events [227]. However, vorapaxar and other PAR-1 inhibitors may cause serious adverse effects associated with bleeding events or cause increased cancer death (data derived from TRACER trial) [31]. These results have led to recent studies to determine the specific antiplatelet effects of inhibiting PAR-4 function. A rabbit polyclonal antibody against the thrombin cleavage site of PAR-4 alone (without PAR-1 inhibition) effectively inhibited thrombin-induced cellular activities like elevation of cytosolic calcium levels and consequent phosphatidylserine exposure. Unfortunately, and important with respect to anticancer treatment, PAR-4 inhibition did not suppress integrin αIIbβ3 activation nor did it decrease α-granule secretion and platelet aggregation—the mechanisms strongly contributing to cancer progression. Thus, no clear progress in the investigation of antiplatelet/anticancer therapy was made.
Although PARs-targeted drugs have been implemented in the treatment of cardiovascular disease (vorapaxar, Zontivity) in cancer disease, these have not made it to clinic for treatment of cancer [228]. As platelet activation is mediated by multiple molecular signaling pathways, inhibition of only one may not be effective. A study showing that both ADP inhibitor and antagonists of PAR-1 (SCH 19197), or its activator kallikrein, should have been used to inhibit platelet activation suggested that bi-modal inhibition may need to be applied to achieve clinical benefit [229].
10 CLEC-2 inhibitors and podoplanin-targeting cancer therapy
There is a newly discovered surface receptor on platelets, C-type lectin-like receptor 2 (CLEC-2), that is activated by the mucin-type sialoglycoprotein podoplanin (aggrus), which is a known platelet aggregation-inducing factor present on multiple tumor cells (squamous cell carcinoma, mesothelioma, glioblastoma, sarcoma) and lymphatic endothelial cells [230,231,232]. Physiologically, podoplanin takes part in lymphatic vessel development and thrombus formation but in the presence of tumor cells may promote tumor growth and hematogenous tumor metastasis by inducing secretion of growth factors and inducing tumor emboli formation in the microvasculature [232]. The disialyl-core1-attached glycopeptide as well as the stereostructure of the podoplanin protein was found to be critical for the CLEC-2-binding activity of podoplanin [231]. There are platelet aggregation-inducing domains on human podoplanin, PLAG4 (81-EDLPT-85), and PLAG3 that contribute to the binding of platelet CLEC-2. Studies showed that simultaneous inhibition of PLAG3/4 is indispensable for complete suppression of podoplanin-mediated procancer activity. CLEC-2 induces intraplatelet signaling in conjunction with a single YxxL motif in its cytoplasmic tail, Src and Syk kinases, and phospholipase Cγ2 [232].
B16F10 melanoma cells co-cultured with wild-type platelets, but not with CLEC-2-deficient platelets inhibited by anti-mouse CLEC-2 monoclonal antibody 2A2B10, demonstrated increased proliferation. Hematogenous metastases were also significantly inhibited in CLEC-2-depleted mice giving the rationale for targeted inhibition of CLEC-2 as a new strategy for preventing hematogenous tumor metastasis. The monoclonal antibody NZ-1 directed against podoplanin disrupted both the podoplanin-CLEC-2 interaction and podoplanin-induced pulmonary metastasis [231]. Another non-cytotoxic 5-nitrobenzoate compound, 2CP, specifically inhibited the podoplanin-CLEC-2 interaction and TCIPA [233] resulting in anti-cancer metastatic activity in vivo in the experimental animal model. Importantly, inhibition of CLEC-2 receptor potentiated the therapeutic efficacy of cisplatin without increasing the risk of bleeding. 2CP directly blocks the region for podoplanin on the CLEC-2 receptor, affects Akt1/PDK1 and PKCμ signaling pathways, and by this mechanism inhibits platelet activation. The fact that absence or reduction of CLEC-2 receptor activity on platelets decreases the hematogeneous metastasis rate without significantly increasing bleeding tendency indicates that CLEC-2 may be a good target for anti-platelet/anti-metastatic drugs.
High expression of podoplanin was demonstrated in primary brain tumors and contributed to platelet aggregation and hypercoagulability state, suggesting the potential antitumor role of podoplanin inhibitors in glioblastoma [234].
11 Microbial proteases
Platelets are now regarded as components of the immune system that are able, similar to leukocytes, to tether microbial pathogens via Toll-like receptors (TLRs) [11]. It is estimated that over 300 proteins and molecules (e.g. CD40, CD154) may be released to the vasculature due to platelet activation elicited by direct contact with pathogens. Platelets may also adhere to endothelial walls and leukocytes in the place of inflammation forming thrombi with microbial pathogens, and this way separate them in the blood. Considering the fact that tumors also promote inflammation, the influence of platelets on immune system is meaningful.
On the other hand, the role of microbial proteases in inflammation and carcinogenesis has now been widely investigated [reviewed in 3, 239]. It was demonstrated that arginine-specific gingipains from Porphyromonas gingivalis may lead to platelet activation through PAR cleavage and thus cause platelet activation and aggregation [235]. Additionally, it has been shown that the same bacteria may secrete proteases that activate PAR and proteases with inhibitory potential. Pseudomonas aeruginosa secretes protease LepA, which is able to activate PARs-1, -2, and 4 leading to stimulation of the NF-κB pathway and the elastolytic metalloprotease that inactivates PAR-2 by proteolysis of the extracellular part of this receptor [236, 237]. Antiplatelet activity has been described for a serine protease with fibrin(ogen)olytic properties named Bacethrombase that is produced by Bacillus cereus strain FF01 [238]. Bacethrombase inhibited ADP-induced platelet aggregation in a dose-dependent manner (Table 4).
12 Microparticles inhibitors
Platelets are known to release microparticles (MP) containing compounds that promote invasiveness of cancer cells and metastatic potential [239]. It is well recognized that glioblastoma increases risk of thrombotic events that activate platelets and may induce MP-mediated interactions between tumor cells, platelets, and the vasculature. Platelet-derived MPs are responsible for immunosuppression in the glioma microenvironment inducing glioma invasiveness and neovascularization [240]. Platelets may take up MPs of tumor cells and carry EGFRvIII transcript. Platelet MPs also contain microRNAs that have been shown to be released after platelet activation with thrombin. It has been demonstrated that MPs laden with miRNA may be internalized by human umbilical vein endothelial cells (HUVEC). MicroRNA in complex with Argonaute 2 (Ago2), Ago2 miR-223, regulates transcription and translation of endothelial cell genes, e.g., promoting advanced glycation end product-induced vascular endothelial cell apoptosis via targeting insulin-like growth factor 1 receptor [241]. In that case, platelets serve as carriers of genetic information between different types of cells.
Several compounds that interfere with MP-mediated intracellular actions, e.g., vaccines or immunomodulators, are now being investigated [reviewed in 243].
13 Other targets and inhibitors
13.1 CD151 inhibitors
The tetraspanin CD151, primarily identified as a molecular organizer of interacting proteins (integrins, growth factors, MMPs) into tetraspanin-enriched microdomains, is also involved in tumor progression and is thus being seriously considered as a potential anticancer target [160, 242]. There is evidence that CD151 regulates tumor cell adhesion, migration and invasion/intravasation, and dissemination. This protein is also engaged in angiogenesis. Reportedly, platelet-expressed CD151 is important for the enhancement of ECFC tube formation, as a CD151 antagonist attenuated this effect [160].
13.2 β-Nitrostyrene derivatives
β-Nitrostyrene derivatives are promising in the antiplatelet/anticancer approach due to their wide influence on platelet functions [135]. Studies revealed that β-nitrostyrene derivatives (4-methylene-dioxy-β nitrostyrene, MNS and 4-O-benzoyl-3-methoxyl-β-nitrostyrene, BMNS) prevented not only TCIPA, P-selectin expression, and PDGF secretion but also platelet adhesion to cancer cells. Incubation of platelets, activated previously by tumor cells, with MNS and BMNS resulted in inhibition of initial interactions [135].
13.3 Curcumin
Curcumin (diferuloylmethane) is a polyphenol derived from the Curcuma longa plant that may exert antiplatelet and anticancer effects by cell cycle regulation and proapoptotic activities against multiple cancers, e.g., of the liver, skin, pancreas, prostate, ovary, lung, and head/neck [243, 244]. The antimetastatic actions of curcumin result from inhibition of transcription factors (e.g. NF-κB), inflammatory cytokines (e.g. CXCL1, CXCL2, IL-6, IL-8), proteases (e.g. MMPs), multiple protein kinases (MAPKs), and/or regulation of miRNAs (e.g. miR21, miR181b) [244].
13.4 AMP activator
Adenylate cyclase/cyclic AMP activation leads to platelet inhibition both ex vivo and in vivo. CME-1, a novel polysaccharide from the mycelia of Cordyceps sinensis, activated cAMP resulting in inhibition of intracellular ATP release, decreased intracellular [Ca(+2)] mobilization and phosphorylation of phospholipase C (PLCγ2) and protein kinase C (PKC), ultimately inhibiting platelet aggregation. Cyclin AMP activation also suppressed signaling mediated by thrombin and AA, such as phosphorylation of p38 MAPK, extracellular signal-regulated kinase 2, c-Jun N-terminal kinase 1, and Akt [245, 246].
The anticancer effect of CME-1 was demonstrated on B16-F10 murine melanoma cell lines [247]. CME-1 inhibited MAPK signaling, thereby disrupting tumor cell migration and dissemination. However, there is no information available about the anticancer effect of CME-1 against other malignancies and its inhibitory effect on platelet/tumor cell interactions.
13.5 Hinokitol
Hinokitiol is a natural bioactive compound found in Chamacyparis taiwanensis, widely used in cosmetics and food for its antimicrobial potential. Some studies revealed its antiplatelet activity [248, 249]. Hinokitiol inhibited PLCγ2, PKC, MAPKs, and Akt in collagen-activated human platelets. Unfortunately, it did not exert antiplatelet activity induced by other agents like thrombin, AA, and ADP. However, taking into consideration the fact that hinokitol may inhibit B16-F10 melanoma cell migration, it is of interest whether some form of tumor cell/platelet interaction could be affected by this compound. Possible mechanisms of hinokitiol action on migration of highly metastatic melanoma cell line should be investigated with respect to its inhibitory effect toward platelets, e.g., as an antagonist of collagen GP VI, or a factor that reduces the expression of MMP-1 that may be produced by platelets [160, 249].
13.6 Extract of Caulis Spatholobi
Ethanol extract of Caulis Spatholobi has been found to block TCIPA and breast cancer metastasis by disrupting the formation of tumor cell/platelet complexes in mouse models after mouse tail vein injection of 4T1 cells [250]. Decreased tumor metastasis development due to dissociation of the tumor cell/platelet complex improved survival in a mouse breast cancer model.
13.7 Tyrosine kinase inhibitors
Tyrosine kinases are essential for platelet signaling so that inhibitors of TKs used in cancer patient treatment may affect platelet functions to increase bleeding risk [251]. Blood of renal cancer patients treated with sunitinib as well as from healthy donors were analyzed after preincubation with sunitinib. Treatment with sunitinib decreased platelet number and their activity.
13.8 PKC inhibitors
PKCdelta plays a crucial role in transducing the invasion-promoting effects of platelets in breast cancer cells, and the specific inhibition of PKCdelta may be a strategy to decrease platelet-mediated cancer cell invasion [252].
13.9 STS
Staurosporine (STS) is a microbial alkaloid and potent PKC inhibitor that has been shown to induce platelet apoptosis and thus has been suggested to be a promising anti-cancer drug [253]. The experimental results are controversial though, as enzastaurin, another PKC inhibitor with known antiproliferative and proapoptotic effects in a variety of cancers has been shown to potentiate platelet aggregation and VEGF secretion thereby counteracting its anticancer potential [254].
13.10 NF-κB-mediated platelet signaling inhibitors
The inhibition of NF-κB transcription factor should also be considered as antiplatelet/anticancer treatment, as this factor regulates platelet activation even though platelets lack a nucleus. NF-κB-regulated events include IKKβ phosphorylation, IκBα degradation, and p65 phosphorylation [255]. Experiments with washed platelets revealed that sesamol activated cAMP-PKA signaling, followed by inhibition of the NFκB-PLC-PKC cascade, ultimately leading to inhibition of [Ca(2+)] mobilization and platelet aggregation [256]. NF-κB is a transcription factor that regulates many important processes responsible for cellular homeostasis. Studies proved that although platelets are anucleated cells, they also express NF-κB that may influence platelet functions in a non-genomic/transcriptional way.
13.11 Calcium channel-blocking agents: nifedipine, diltiazem, verapamil, or amlodopine
Calcium channels are mobilized during platelet activation, and so their inhibitors were analyzed for antiplatelet potential [257]. Platelet-rich plasma (PRP) derived from normal subjects was incubated with one of four drugs: nifedipine, diltiazem, verapamil, or amlodopine. These agents administered in high concentration inhibited osteogenic sarcoma-induced platelet aggregation.
13.12 Blockers of collagen binding sites and galectin-3 inhibitors
Platelets may induce COX-2 expression in HT29 colon carcinoma cells resulting in increased expression of PGE2, upregulation of cyclin B1, and expression of proteins involved in epithelial-mesenchymal transition (EMT). Galectin-3 is a member of a family of carbohydrate-binding proteins with a unique collagen-like domain that plays a role in platelet-cancer cell crosstalk through interaction with platelet collagen receptors, e.g., GPVI. Inhibition of platelet-cancer cell interaction by a novel antiplatelet drug revacept, which blocks collagen binding sites, prevented platelet-induced mRNA changes of EMT markers in colon carcinoma cells [258].
13.13 PDGFR inhibitors
Thrombin-activated platelets secrete α-granules containing high levels of platelet-derived growth factor receptor (PDGFR). PDGFR influences tumor invasion by increasing CTC survival by maintaining CTCs in an epithelial-mesenchymal transition (EMT) state within the blood vessel [259]. PDGFR activity is documented in the pathogenesis of glioblastoma (GBM) [260]. There is evidence that insulin-mediated signaling (insulin receptor, IR/insulin growth-like factor receptor, IGF1R) may trigger resistance to PDGFR inhibition. Co-targeting IR/IGF1R and PDGFR decreased the number of resistant clones in vitro. The enhanced growth inhibition was observed in rhabdomyosarcoma cell lines when cells were treated with dual IGF-1R and PDGFR-β agents in vitro [261]. Furthermore, PDGFR tyrosine phosphorylation may be inhibited by Compound C resulting in decreased proliferation of A172 glioblastoma cells [262].
13.14 Platelet aggregation inhibitors
Platelet aggregation inhibitors (ditazole, bencyclan, cyproheptadine, dipyridamole, pentoxyfilline) showed no significant influence on metastases formation in animal tumor models [reviewed in 1]. Mopidamole was demonstrated to inhibit significantly spontaneous lung metastasis in syngeneic Wilms’ tumor of the rat, the C1300-neuroblastoma of the mouse, the HM-Kim mammary carcinoma of the rat, and Lewis lung carcinoma of the mouse.
13.15 Tamoxifen
There is also evidence that tamoxifen, a selective estrogen receptor modulator used for the treatment of breast cancer, and its metabolite 4-hydroxytamoxifen exert antitumor activity through inhibition of platelet-dependant angiogenesis and metastasis formation [263]. Tamoxifen-treated platelets released lower levels of proangiogenic factors like VEGF, angiogenin, CXCL1, CCL5, EGF, CXCL5, and PDGF-BB, resulting in decreased capillary tube formation and diminished endothelial migration. The laboratory tests were confirmed in vivo in cancer patients treated with tamoxifen. Their platelets secreted lower levels of VEGF compared with patients not being treated with tamoxifen, and these were characterized as having less angiogenic and metastatic potential.
14 Conclusions
Evidence for the contribution of platelets to cancer progression is substantial and continues to mount. However, despite the discovery of new platelet surface receptors and molecular mechanisms involved in carcinogenesis, there is still an unmet need for effective antiplatelet/anticancer therapy. There is an established preventive role for aspirin in colorectal cancer, but there are no breakthroughs in treatment procedures. The majority of data come from laboratory studies, and numerous clinical questions remain unanswered. There is insufficient information on how specific types of cancer and advancement of disease progression depend on platelet function. Additionally, chronic administration of antiplatelet agents and thrombocytopenia relate to risk of bleeding complications or even increased cancer incidence that must be considered in clinical practice. There are still questions as to whether single or dual antiplatelet regimens should be applied to cancer patients, how long it is safe to block platelets, and whether antiplatelet treatment should be connected with classic chemotherapy. Laboratory studies have demonstrated superior results with the co-treatment approach. Although several doubts related to antiplatelet therapy as anticancer treatment exist, the experimental results justify continuing an effort to discover new drugs and anti-platelet approaches.
References
Sierko, E., & Wojtukiewicz, M. Z. (2007). Inhibition of platelet function: does it offer a chance of better cancer progression control? Seminars in Thrombosis and Hemostasis, 33(7), 712–721.
Menter, D. G., Tucker, S. C., Kopetz, S., Sood, A. K., Crissman, J. D., & Honn, K. V. (2014). Platelets and cancer: a casual or causal relationship: revisited. Cancer and Metastasis Reviews, 33(1), 231–269.
Wojtukiewicz, M. Z., Hempel, D., Sierko, E., Tucker, S. C., & Honn, K. V. (2016). Thrombin - unique coagulation system protein with multifaceted impacts on cancer and metastasis. Cancer and Metastasis Reviews, 35, 213–233.
Choe, K. S. C. J., Chan, J. M., Carroll, P. R., D’Amico, A. V., & Liauw, S. L. (2012). Aspirin use and the risk of prostate cancer mortality in men treated with prostatectomy or radiotherapy. Journal of Clinical Oncology, 30(28), 3540–3544.
Gerotziafas, G. T. P. C., Hatmi, M., Samama, M. M., & Elalamy, I. (2008). Clinical studies with anticoagulants to improve survival in cancer patients. Pathophysiology of Haemostasis and Thrombosis, 36(3–4), 204–211.
Sekiguchi, T., Takemoto, A., Takagi, S., Takatori, K., Sato, S., Takami, M., & Fujita, N. (2016). Targeting a novel domain in podoplanin for inhibiting platelet-mediated tumor metastasis. Oncotarget, 7(4), 3934–3946.
Holmes, C. E., Levis, J. E., Schneider, D. J., Bambace, N. M., Sharma, D., Lal, I., et al. (2016). Platelet phenotype changes associated with breast cancer and its treatment. Platelets, 27(7), 703–711.
Ho-Tin-Noé, B., Goerge, T., & Wagner, D. D. (2009). Platelets: guardians of tumor vasculature. Cancer Research, 69(14), 5623–5626.
Ho-Tin-Noé, B., Goerge, T., Cifuni, S. M., Duerschmied, D., & Wagner, D. D. (2008). Platelet granule secretion continuously prevents intratumor hemorrhage. Cancer Research, 68(16), 6851–6858.
Palumbo, J. S., Talmage, K. E., Massari, J. V., La Jeunesse, C. M., Flick, M. J., Kombrinck, K. W., et al. (2005). Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood, 105, 178.
Franco, A. T., Corken, A., & Ware, J. (2015). Platelets at the interface of thrombosis, inflammation, and cancer. Blood, 126(5), 582–588.
Lian, L., Li, W., Li, Z. Y., Mao, Y. X., Zhang, Y. T., Zhao, Y. M., et al. (2013). Inhibition of MCF-7 breast cancer cell-induced platelet aggregation using a combination of antiplatelet drugs. Oncology Letters, 5(2), 675–680.
Wojtukiewicz, M. Z., Hempel, D., Sierko, E., Tucker, S. C., & Honn, K. V. (2015). Protease-activated receptors (PARs)--biology and role in cancer invasion and metastasis. Cancer and Metastasis Reviews, 34(4), 775–796.
Takemoto, A., Okitaka, M., Takagi, S., Takami, M., Sato, S., Nishio, M., et al. (2017). A critical role of platelet TGF-β release in podoplanin-mediated tumour invasion and metastasis. Scientific Report, 7, 42186. doi:10.1038/srep42186.
Lowe, K. L., Navarro-Nunez, L., & Watson, S. P. (2012). Platelet CLEC-2 and podoplanin in cancer metastasis. Thrombosis Research, 129(Suppl 1), S30–S37.
Umar, A., Steele, V. E., Menter, D. G., & Hawk, E. T. (2016). Mechanisms of nonsteroidal anti-inflammatory drugs in cancer prevention. Seminars in Oncology, 43(1), 65–77.
Shiao, J., Thomas, K. M., Rahimi, A. S., Rao, R., Yan, J., Xie, X. J., et al. (2017). Aspirin/antiplatelet agent use improves disease-free survival and reduces the risk of distant metastases in stage II and III triple-negative breast cancer patients. Breast Cancer Research and Treatment, 161(3), 463–471.
Guillem-Llobat, P., Dovizio, M., Bruno, A., Ricciotti, E., Cufino, V., Sacco, A., et al. (2016). Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget, 7(22), 32462–32477.
Mitrugno, A., Sylman, J. L., Ngo, A. T., Pang, J., Sears, R. C., Williams, C. D., & McCarty, O. J. (2017). Aspirin therapy reduces the ability of platelets to promote colon and pancreatic cancer cell proliferation: implications for the oncoprotein c-MYC. American Journal of Physiology. Cell Physiology, 312(2), C176–C189.
Lee, P. C., Yeh, C. M., Hu, Y. W., Chen, C. C., Liu, C. J., Su, C. W., et al. (2016). Antiplatelet therapy is associated with a better prognosis for patients with hepatitis B virus-related hepatocellular carcinoma after liver resection. Annals of Surgical Oncology, 23(Suppl 5), 874–883.
Furlan, C., Steffan, A., Polesel, J., Trovo, M., Gobitti, C., Vaccher, E., et al. (2015). Lower platelet counts and antiplatelet therapy independently predict better outcomes in patients with head and neck squamous cell carcinoma: a retrospective analysis. Biomarker Research, 3(25), 1–3.
Downer, M.K., Allard, C.B., Preston, M.A., Gazian, J.M., Stampfer, M.J., Mucci, L.A. et al. (2017). Regular Aspirin Use and the Risk of Lethal Prostate Cancer in the Physicians’ Health Study. European Urology Feb 8. pii: S0302–2838(17)30069–6. doi:10.1016/j.eururo.2017.01.044.
Holmes, M. D., Chen, W. Y., Li, L., Hertzmark, E., Spiegelman, D., & Hankinson, S. E. (2010). Aspirin intake and survival after breast cancer. Journal of Clinical Oncology, 28(9), 1467–1472.
Rothwell, P. M. F. F., Belch, J. F., Ogawa, H., Warlow, C. P., & Meade, T. W. (2011). Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet, 377(9759), 31–41.
Leader, A., Zelikson-Saporta, R., Pereg, D., Spectre, G., Rozovski, U., Raanani, P. et al. (2017). The Effect of Combined Aspirin and Clopidogrel Treatment on Cancer Incidence. American Journal Medicine, Feb 14. doi:10.1016/j.amjmed.2017.01.022.
Cook, N. R., Lee, I. M., Gaziano, J. M., Gordon, D., Ridker, P. M., Manson, J. E., et al. (2005). Low-dose aspirin in the primary prevention of cancer: the Women’s Health Study: a randomized controlled trial. Journal of the American Medical Association, 294(1), 47–55.
Drew, D. A., Chin, S. M., Gilpin, K. K., Parziale, M., Pond, E., Schuck, M. M., et al. (2017). ASPirin intervention for the REDuction of colorectal cancer risk (ASPIRED): a study protocol for a randomized controlled trial. Trials, 18(1), 50. doi:10.1186/s13063-016-1744-z.
Kodela, R., Chattopadhyay, M., Velázquez-Martínez, C. A., & Kashfi, K. (2015). NOSH-aspirin (NBS-1120), a novel nitric oxide- and hydrogen sulfide-releasing hybrid has enhanced chemo-preventive properties compared to aspirin, is gastrointestinal safe with all the classic therapeutic indications. Biochemical Pharmacology, 98(4), 564–572.
Serebruany, V. L., Cherepanov, V., Cabrera-Fuentes, H. A., & Kim, M. H. (2015). Solid cancers after antiplatelet therapy: confirmations, controversies, and challenges. Thrombosis and Haemostasis, 114(6), 1104–1112.
Serebruany, V. L., Cherepanov, V., Golukhova, E. Z., & Kim, M. H. (2015). The dual antiplatelet therapy trial after the FDA update: noncardiovascular deaths, cancer and optimal treatment duration. Cardiology, 132(2), 74–80.
Serebruany, V. L., Tomek, A., & Kim, M. H. (2015). Survival after solid cancers in antithrombotic trials. American Journal of Cardiology, 116(6), 969–972.
Hicks, B. M., Murray, L. J., Hughes, C., & Cardwell, C. R. (2015). Clopidogrel use and cancer-specific mortality: a population-based cohort study of colorectal, breast and prostate cancer patients. Pharmacoepidemiology and Drug Safety, 24(8), 830–840.
Tranum, B. L., & Haut, A. (1974). Thrombocytosis: platelet kinetics in neoplasia. Journal of Laboratory and Clinical Medicine, 84(5), 615–619.
Sharma, D., Brummel-Ziedins, K. E., Bouchard, B. A., & Holmes, C. E. (2014). Platelets in tumor progression: a host factor that offers multiple potential targets in the treatment of cancer. Journal of Cellular Physiology, 229(8), 1005–1115.
Li, R., Ren, M., Chen, N., Luo, M., Deng, X., Xia, J., et al. (2014). Presence of intratumoral platelets is associated with tumor vessel structure and metastasis. BioMed Center Cancer, 14, 167. doi:10.1186/1471-2407-14-167.
Gasic, G. J., Gasic, T. B., & Stewart, C. C. (1968). Antimetastatic effects associated with platelet reduction. Proceedings of the National Academy of Sciences, 61, 46–52.
Allensworth, S. K., Langstraat, C. L., Martin, J. R., Lemens, M. A., McGree, M. E., Weaver, A. L., et al. (2013). Evaluating the prognostic significance of preoperative thrombocytosis in epithelial ovarian cancer. Gynecologic Oncology, 130(3), 499–504.
Karpatkin, S., Pearlstein, E., Salk, P. L., & Yogeeswaran, G. (1981). Role of platelets in tumor cell metastases. Annals of the New York Academy of Sciences, 370, 101–118.
Rachidi, S., Wallace, K., Day, T. A., Alberg, A. J., & Li, Z. (2014). Lower circulating platelet counts and antiplatelet therapy independently predict better outcomes in patients with head and neck squamous cell carcinoma. Journal of Hematology & Oncology, 27(7), 65. doi:10.1186/s13045-014-0065-5.
Jacobsen, J., Grankvist, K., Rasmuson, T., & Ljungberg, B. (2002). Prognostic importance of serum vascular endothelial growth factor in relation to platelet and leukocyte counts in human renal cell carcinoma. European Journal of Cancer Prevention, 11(3), 245–252.
Steele, M., & Voutsadakis, I. A. (2017). Pre-treatment platelet counts as a prognostic and predictive factor in stage II and III rectal adenocarcinoma. World Journal Gastrointestinal Oncology, 9(1), 42–49.
Mantas, D., Kostakis, I. D., Machairas, N., & Markopoulos, C. (2016). White blood cell and platelet indices as prognostic markers in patients with invasive ductal breast carcinoma. Oncology Letters, 12(2), 1610–1614.
Bottsford-Miller, J., Choi, H. J., Dalton, H. J., Stone, R. L., Cho, M. S., Haemmerle, M., et al. (2015). Differential platelet levels affect response to taxane-based therapy in ovarian cancer. Clinical Cancer Research, 21(3), 602–610.
Shen, X. M., Xia, Y. Y., Lian, L., Zhou, C., Li, X. L., Han, S. G., et al. (2016). Mean platelet volume provides beneficial diagnostic and prognostic information for patients with resectable gastric cancer. Oncology Letters, 12(4), 2501–2506.
Tan, D., Fu, Y., Su, Q., & Wang, H. (2016). Prognostic role of platelet-lymphocyte ratio in colorectal cancer: a systematic review and meta-analysis. Medicine (Baltimore), 95(24), e3837. doi:10.1097/MD.0000000000003837.
Zhao, Q. T., Yuan, Z., Zhang, H., Zhang, X. P., Wang, H. E., Wang, Z. K., & Duan, G. C. (2016). Prognostic role of platelet to lymphocyte ratio in non-small cell lung cancers: a meta-analysis including 3,720 patients. International Journal of Cancer, 139(1), 164–170.
Zhou, X., Du, Y., Huang, Z., Xu, J., Qiu, T., Wang, J., Wang, T., et al. (2014). Prognostic value of PLR in various cancers: a meta-analysis. PloS One, 9(6), e101119.
http://www.genengnews.com/gen-news-highlights/aspirin-prevents-platelets-from-suppressing-anticancer-t-cells/81254316. Accessed 1 June 2017.
Mezouar, S., Frère, C., Darbousset, R., Mege, D., Crescence, L., Dignat-George, F., Panicot-Dubois, L., et al. Role of platelets in cancer and cancer-associated thrombosis: experimental and clinical evidences. Thrombosis Research, 139, 65–76.
Bergmann, S., & Hammerschmidt, S. (2007). Fibrinolysis and host response in bacterial infections. Thrombosis and Haemostasis, 98(3), 512–520.
Chauhan, A.S., Kumar, M., Chaudhary, S., Patidar, A., Dhiman, A., Sheokand, N., et al. (2017). Moonlighting glycolytic protein glyceraldehyde-3-phosphate dehydrogenase (GAPDH): an evolutionarily conserved plasminogen receptor on mammalian cells. Federation of American Societies for Experimental Biology, Mar 15. pii: fj.201600982R.
Cicero, L. A., Raposo, G., & Zhang, H. G. (2013). The cell biology of exosomes: historical and perspectives, Emerging concepts of tumor exosome–mediated cell-cell communication. New York: Springer.
Ortiz, A. (2017). Not all extracellular vesicles were created equal: Clinical implications. Annals of Translational Medicine, 5(5), 111.
Riedl, J., Kaider, A., Marosi, C., Prager, G. W., Eichelberger, B., Assinger, A., et al. (2017). Decreased platelet reactivity in patients with cancer is associated with high risk of venous thromboembolism and poor prognosis. Thrombosis and Haemostasis, 117(1), 90–98.
Dashevsky, O., Varon, D., & Brill, A. (2009). Platelet-derived microparticles promote invasiveness of prostate cancer cells via upregulation of MMP-2 production. International Journal of Cancer, 124(8), 1773–1777.
Hu, Q., Wang, M., Cho, M. S., Wang, C., Nick, A. M., Thiagarajan, P., et al. (2016). Lipid profile of platelets and platelet-derived microparticles in ovarian cancer. Biochimica et Biophysica Acta Clinical, 6, 76–81.
Ren, J. G., Zhang, W., Liu, B., Man, Q. W., Xiong, X. P., Li, C., et al. (2016). Clinical significance and roles in angiogenesis of circulating microparticles in oral cancer. Journal of Dental Research, 95(8), 860–867.
Żmigrodzka, M., Guzera, M., Miśkiewicz, A., Jagielski, D., & Winnicka, A. (2016). The biology of extracellular vesicles with focus on platelet microparticles and their role in cancer development and progression. Tumor Biology, 37(11), 14391–14401.
Helley, D., Banu, E., Bouziane, A., Banu, A., Scotte, F., Fischer, A. M., & Oudard, S. (2009). Platelet microparticles: a potential predictive factor of survival in hormone-refractory prostate cancer patients treated with docetaxel-based chemotherapy. European Urology, 56(3), 479–484.
Heinmöller, E., Weinel, R. J., Heidtmann, H. H., Salge, U., Seitz, R., Schmitz, I., et al. (1996). Studies on tumor-cell-induced platelet aggregation in human lung cancer cell lines. Cancer Research and Clinical Oncology, 122(12), 735–744.
Alonso-Escolano, D., Strongin, A. Y., Chung, A. W., Deryugina, E. I., & Radomski, M. W. (2004). Membrane type-1 matrix metalloproteinase stimulates tumor cell-induced platelet aggregation: role of receptor glycoproteins. British Journal of Pharmacology, 141(2), 241–252.
Yan, M., & Jurasz, P. (2016). The role of platelets in the tumor microenvironment: from solid tumors to leukemia. Biochimica et Biophysica Acta, 1863(3), 392–400.
Im, J. H., Fu, W., Wang, H., Bhatia, S. K., Hammer, D. A., Kowalska, M. A., & Muschel, R. J. (2004). Coagulation facilitates tumor cell spreading in the pulmonary vasculature during early metastatic colony formation. Cancer Research, 64(23), 8613–8619.
Menter, D. G., Hatfield, J. S., Harkins, C., Sloane, B. F., Taylor, J. D., Crissman, J. D., et al. (1987). Tumor cell-platelet interactions in vitro and their relationship to in vivo arrest of hematogenously circulating tumor cells. Clinical and Experimental Metastasis, 5(1), 65–78.
Kopp, H. G., Placke, T., & Salih, H. R. (2009). Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Research, 69(19), 7775–7783.
Ogawa, F., Amano, H., Ito, Y., Matsui, Y., Hosono, K., Kitasato, H., et al. (2014). Aspirin reduces lung cancer metastasis to regional lymph nodes. Biomedicine and Pharmacotherapy, 68(1), 79–86.
Gautam, S., Roy, S., Ansari, M. N., Saeedan, A. S., Saraf, S. A., & Kaithwas, G. (2017). DuCLOX-2/5 inhibition: a promising target for cancer chemoprevention. Breast Cancer, 24(2), 180–190.
Hall, Z., Ament, Z., Wilson, C. H., Burkhart, D. L., Ashmore, T., Koulman, A., et al. (2016). Myc expression drives aberrant lipid metabolism in lung cancer. Cancer Research, 76(16), 4608–4618.
Knab, L. M., Grippo, P. J., & Bentrem, D. J. (2014). Involvement of eicosanoids in the pathogenesis of pancreatic cancer: the roles of cyclooxygenase-2 and 5-lipoxygenase. World Journal Gastroenterology, 20(31), 10729–10739.
Tuncer, S., & Banerjee, S. (2015). Eicosanoid pathway in colorectal cancer: recent updates. World Journal of Gastroenterology, 21(41), 11748–11766.
Dilly, A. K., Tang, K., Guo, Y., Joshi, S., Ekambaram, P., Maddipati, K. R., et al. (2017). Convergence of eicosanoid and integrin biology: role of Src in 12-LOX activation. Experimental Cell Research, 351(1), 1–10.
Dilly, A. K., Ekambaram, P., Guo, Y., Cai, Y., Tucker, S. C., Fridman, R., Kandouz, M., et al. (2013). Platelet-type 12-lipoxygenase induces MMP9 expression and cellular invasion via activation of PI3K/Akt/NF-κB. International Journal of Cancer, 133(8), 1784–1791.
Nie, D., & Honn, K. V. (2002). Cyclooxygenase, lipoxygenase and tumor angiogenesis. Cellular and Mollecular Life Sciences, 59(5), 799–807.
Maskrey, B. H., Bermúdez-Fajardo, A., Morgan, A. H., Stewart-Jones, E., Dioszeghy, V., Taylor, G. W., et al. (2007). Activated platelets and monocytes generate four hydroxyphosphatidylethanolamines via lipoxygenase. Journal of Biological Chemistry, 282(28), 20151–20163.
Kandouz, M., Nie, D., Pidgeon, G. P., Krishnamoorthy, S., Maddipati, K. R., & Honn, K. V. (2003). Platelet-type 12-lipoxygenase activates NF-kappaB in prostate cancer cells. Prostaglandins and Other Lipid Mediators, 71(3–4), 189–204.
Mehta, P., Lawson, D., Ward, M. B., Lee-Ambrose, L., & Kimura, A. (1986). Effects of thromboxane A2 inhibition on osteogenic sarcoma cell-induced platelet aggregation. Cancer Reserch, 46(10), 5061–5063.
Steinert, B. W., Tang, D. G., Grossi, I. M., Umbarger, L. A., & Honn, K. V. (1993). Studies on the role of platelet eicosanoid metabolism and integrin alpha IIb beta in tumor-cell-induced platelet aggregation. International Journal of Cancer, 54(1), 92–101.
Thun, M. J., Jacobs, E. J., & Patrono, C. (2012). The role of aspirin in cancer prevention. Nature Reviews. Clinical Oncology, 9(5), 259–267.
Sutcliffe, P., Connock, M., Gurung, T., Freeman, K., Johnson, S., Kandala, N. B., et al. (2013). Aspirin for prophylactic use in the primary prevention of cardiovascular disease and cancer: a systematic review and overview of reviews. Health Technology Assessments, 17(43), 1–253.
Coppinger, J. A., O'Connor, R., Wynne, K., Flanagan, M., Sullivan, M., Maguire, P. B., et al. (2007). Moderation of the platelet releasate response by aspirin. Blood, 109(11), 4786–4792.
Zhao, L., Zhang, W., Chen, M., Zhang, J., Zhang, M., & Dai, K. (2013). Aspirin induces platelet apoptosis. Platelets, 24(8), 637–642.
Di Francesco, L., López Contreras, L. A., Sacco, A., & Patrignani, P. (2015). New insights into the mechanism of action of aspirin in the prevention of colorectal neoplasia. Current Pharmaceutical Design, 21(35), 5116–5126.
Ding, J. H., Yuan, L. Y., Huang, R. B., & Chen, G. A. (2014). Aspirin inhibits proliferation and induces apoptosis of multiple myeloma cells through regulation of Bcl-2 and Bax and suppression of VEGF. European Journal of Haematology, 93(4), 329–339.
Ding, J. H., Yuan, L. Y., & Chen, G. A. (2017). Aspirin enhances the cytotoxic activity of bortezomib against myeloma cells via suppression of Bcl-2, survivin and phosphorylation of AKT. Oncology Letters, 13(2), 647–654.
Cooke, N. M., Spillane, C. D., Sheils, O., O'Leary, J., & Kenny, D. (2015). Aspirin and P2Y12 inhibition attenuate platelet-induced ovarian cancer cell invasion. BioMed Central Cancer, 15, 627–637.
Vad, N. M., Kudugunti, S. K., Wang, H., Bhat, G. J., & Moridani, M. Y. (2014). Efficacy of acetylsalicylic acid (aspirin) in skin B16-F0 melanoma tumor-bearing C57BL/6 mice. Tumor Biology, 35(5), 4967–4976.
Sitia, G., Iannacone, M., & Guidotti, L. G. (2013). Anti-platelet therapy in the prevention of hepatitis B virus-associated hepatocellular carcinoma. Journal of Hepatology, 59(5), 1135–1138.
Etulain, J., Fondevila, C., Negrotto, S., & Schattner, M. (2013). Platelet-mediated angiogenesis is independent of VEGF and fully inhibited by aspirin. British Journal of Pharmacology, 170(2), 255–265. doi:10.1111/bph.12250.
He, Y., Huang, H., Farischon, C., Li, D., Du, Z., Zhang, K., et al. (2017). Combined effects of atorvastatin and aspirin on growth and apoptosis in human prostate cancer cells. Oncology Reports, 37(2), 953–960.
Aiolfi, R., & Sitia, G. (2014). Emerging role of dual antiplatelet therapy in the prevention of hepatitis B virus-associated hepatocellular carcinoma. Journal of Hepatocellular Carcinoma, 1, 183–186.
Mc Menamin, Ú. C., Cardwell, C. R., Hughes, C. M., & Murray, L. M. (2015). Low-dose aspirin and survival from lung cancer: a population-based cohort study. BioMed Central Cancer, 15, 911. doi:10.1186/s12885-015-1910-9.
Hochmuth, F., Jochem, M., & Schlattmann, P. (2016). Meta-analysis of aspirin use and risk of lung cancer shows notable results. European Journal of Cancer Prevention, 25(4), 259–268.
Medina, C., Harmon, S., Inkielewicz, I., Santos-Martinez, M. J., Jones, M., Cantwell, P., et al. (2012). Differential inhibition of tumor cell-induced platelet aggregation by the nicotinate aspirin prodrug (ST0702) and aspirin. British Journal of Pharmacology, 166(3), 938–949.
Bando, H., Yamashita, T., & Tsubura, E. (1984). Effects of antiplatelet agents on pulmonary metastases. Gann, 75, 284–291.
Mamytbeková, A., Rezábek, K., Kacerovská, H., Grimová, J., & Svobodová, J. (1986). Antimetastatic effect of flurbiprofen and other platelet aggregation inhibitors. Neoplasma, 33, 417–421.
Mehta, P., Lawson, D., Ward, M. B., Lee-Ambrose, L., & Kimura, A. (1986). Effect of tromboxane A2 inhibition on osteogenic sarcoma cell-induced platelet aggregation. Cancer Research, 46, 5061–5063.
Olson, J. D., Zaleski, A., Herrmann, D., & Flood, P. A. (1989). Adhesion of platelets to purified solid-phase von Willebrand factor: effects of wall shear rate, ADP, thrombin, and ristocetin. Journal of Laboratory and Clinical Medicine, 114(1), 6–18.
Jurasz, P., Stewart, M. W., Radomski, A., Khadour, F., Duszyk, M., & Radomski, M. W. (2001). Role of von Willebrand factor in tumour cell-induced platelet aggregation: differential regulation by NO and prostacyclin. British Journal of Pharmacology, 134, 1104–1112.
Stürmer, T., Glynn, R. J., Lee, I. M., Manson, J. E., Buring, J. E., & Hennekens, C. H. (1998). Aspirin use and colorectal cancer: post-trial follow-up data from the Physicians’ Health Study. Annuals of Internal Medicine, 128(9), 713–720.
Cooper, K., Squires, H., Carroll, C., Papaioannou, D., Booth, A., Logan, R. F., Maguire, C., et al. (2010). Chemoprevention of colorectal cancer: systematic review and economic evaluation. Health Technology Assessment, 14(32), 1–206.
Rothwell, P. M., Wilson, M., Elwin, C. E., Norrving, B., Algra, A., Warlow, C. P., & Meade, T. W. (2010). Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet, 376(9754), 1741–1750.
Algra, A. M., & Rothwell, P. M. (2012). Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncology, 13(5), 518–527.
Bosettik, C., Gallus, S., & La Vecchia, C. (2009). Aspirin and cancer risk: a summary review to 2007. Recent Results in Cancer Research, 181, 231–251.
Rothwell, P. M., Price, J. F., Fowkes, F. G., Zanchetti, A., Roncaglioni, M. C., Tognoni, G., et al. (2012). Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet, 379(9826), 1602–1612.
Rothwell, P. M., Wilson, M., Price, J. F., Belch, J. F., Meade, T. W., & Mehta, Z. (2012). Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet, 379(9826), 1591–1601.
Gierach, G. L., Lacey, J. V., Schatzkin, A., Leitzmann, M. F., Richesson, D., Hollenbeck, A. R., & Brinton, L. A. (2008). Nonsteroidal anti-inflammatory drugs and breast cancer risk in the National Institutes of Health-AARP diet and health study. Breast Cancer Research, 10(2), R38.
Khuder, S. A., & Mutgi, A. B. (2001). Breast cancer and NSAID use: a meta-analysis. British Journal of Cancer, 84(9), 1188–1192.
Takkouche, B., Regueira-Mendez, C., & Etminan, M. (2008). Breast cancer and use of nonsteroidal anti-inflammatory drugs: a metaanalysis. Journal National Cancer Institute, 100(20), 1439–1447.
Dasgupta, K., Di Cesar, D., Ghosn, J., Rajan, R., Mahmud, S., & Rahme, E. (2006). Association between nonsteroidal anti-inflammatory drugs and prostate cancer occurrence. Cancer Journal, 12(2), 130–135.
Habel, L. A., Zhao, W., & Stanford, J. L. (2002). Daily aspirin use and prostate cancer risk in a large, multiracial cohort in the US. Cancer Causes & Control, 13(5), 427–434.
Jacobs, E. J., Rodriguez, C., Mondul, A. M., Connell, C. J., Henley, S. J., Calle, E. E., & Thun, M. J. (2005). A large cohort study of aspirin and other nonsteroidal anti-inflammatory drugs and prostate cancer incidence. Journal of National Cancer Institute, 97(13), 975–980.
Mahmud, S. M., Franco, E. L., & Aprikian, A. G. (2010). Use of nonsteroidal anti-inflammatory drugs and prostate cancer risk: a meta-analysis. International Journal of Cancer, 127, 1680–1691.
Jacobs, C. D., Chun, S. G., Yan, J., Xie, X. J., Pistenmaa, D. A., Hannan, R., Lotan, Y., et al. (2014). Aspirin improves outcome in high risk prostate cancer patients treated with radiation therapy. Cancer Biology & Therapy, 15(6), 699–706.
Pidgeon, G. P., Lysaght, J., Krishnamoorthy, S., Reynolds, J. V., O'Byrne, K., Nie, D., & Honn, K. V. (2007). Lipoxygenase metabolism: roles in tumor progression and survival. Cancer Metastasis Review, 26(3–4), 503–524.
Rásó, E., Döme, B., Somlai, B., Zacharek, A., Hagmann, W., Honn, K. V., & Tímár, J. (2004). 12-lipoxygenase in human melanoma progression, under experimental and clinical conditions. Melanoma Research, 14(4), 245–250.
Melstrom, L. G., Bentrem, D. J., Salabat, M. R., Kennedy, T. J., Ding, X. Z., Strouch, M., et al. (2008). Overexpression of 5-lipoxygenase in colon polyps and cancer and the effect of 5-LOX inhibitors in vitro and in a murine model. Clinical Cancer Research, 14(20), 6525–6530.
Ye, Y. N., Wu, W. K., Shin, V. Y., Bruce, I. C., Wong, B. C., & Cho, C. H. (2005). Dual inhibition of 5-LOX and COX-2 suppresses colon cancer formation promoted by cigarette smoke. Carcinogenesis, 26(4), 827–834.
Lövey, J., Nie, D., Tóvári, J., Kenessey, I., Tímár, J., Kandouz, M., et al. (2013). Radiosensitivity of human prostate cancer cells can be modulated by inhibition of 12-lipoxygenase. Cancer Letters, 335(2), 495–501.
Tong, W. G., Ding, X. Z., Witt, R. C., & Adrian, T. E. (2002). Lipoxygenase inhibitors attenuate growth of human pancreatic cancer xenografts and induce apoptosis through the mitochondrial pathway. Molecular Cancer Therapy, 1(11), 929–935.
Pidgeon, G. P., Tang, K., Cai, Y. L., Piasentin, E., & Honn, K. V. (2003). Overexpression of platelet-type 12-lipoxygenase promotes tumor cell survival by enhancing alpha(v)beta(3) and alpha(v)beta(5) integrin expression. Cancer Research, 63(14), 4258–4267.
Wang, Y., Sun, Y., Li, D., Zhang, L., Wang, K., Zuo, Y., et al. (2013). Platelet P2Y12 is involved in murine pulmonary metastasis. PloS One, 8(11), e80780.
Bambace, N. M., Levis, J. E., & Holmes, C. E. (2010). The effect of P2Y-mediated platelet activation on the release of VEGF and endostatin from platelets. Platelets, 21(2), 85–93.
Schumacher, D., Strilic, B., Sivaraj, K. K., Wettschureck, N., & Offermanns, S. (2013). Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell, 24(1), 130–137.
Gebremeskel, S., LeVatte, T., Liwski, R. S., Johnston, B., & Bezuhly, M. (2015). The reversible P2Y12 inhibitor ticagrelor inhibits metastasis and improves survival in mouse models of cancer. International Journal of Cancer, 136(1), 234–240.
Bastida, E., Escolar, G., Almirall, L., & Ordinas, A. (1986). Platelet activation induced by a human neuroblastoma tumor cell line is reduced by prior administration of ticlopidine. Thrombosis and Haemostasis, 55(3), 333–337.
Mah-Becherel, M. C., Céraline, J., Deplanque, G., Chenard, M. P., Bergerat, J. P., Cazenave, J. P., & Klein-Soyer, C. (2002). Anti-angiogenic effects of the thienopyridine SR 25989 in vitro and in vivo in a murine pulmonary metastasis model. British Journal of Cancer, 86(5), 803–810.
Kohga, S., Kinjo, M., Tanaka, K., Ogawa, H., Ishihara, M., & Tanaka, N. (1981). Effects of 5-(2-chlorobenzyl)-4,5,6,7,-tetrahydrothieno[3,2-C]pyridine hydrochloride (ticlopidine), a platelet aggregation inhibitor, on blood-borne metastasis. Cancer Research, 41, 4710–4714.
Fabra, A., de Castellarnau, C., Carretero, F., Martinez, E., Sancho, M. J., & Rutllant, M. L. (1987). Effects of ticlopidine on metastasis production in mice bearing Lewis lung carcinoma. Invasion & Metastasis, 7, 53–60.
Mezouar, S., Darbousset, R., Dignat-George, F., Panicot-Dubois, L., & Dubois, C. (2015). Inhibition of platelet activation prevents the P-selectin and integrin-dependent accumulation of cancer cell microparticles and reduces tumor growth and metastasis in vivo. International Journal of Cancer, 136(2), 462–475.
Geddings, J. E., Hisada, Y., Boulaftali, Y., Getz, T. M., Whelihan, M., Fuentes, R., et al. (2016). Tissue factor-positive tumor microvesicles activate platelets and enhance thrombosis in mice. Journal of Thrombosis and Haemostasis, 14(1), 153–166.
Roop, R. P., Naughton, M. J., Van Poznak, C., Schneider, J. G., Lammers, P. E., Pluard, T. J., et al. (2013). A randomized phase II trial investigating the effect of platelet function inhibition on circulating tumor cells in patients with metastatic breast cancer. Clinical Breast Cancer, 13(6), 409–415.
Serebruany, V. L. (2015). Ticagrelor shift from PLATO to PEGASUS: vanished mortality benefit, excess cancer deaths, massive discontinuations, and overshooting target events. International Journal of Cardiology, 201, 508–512.
Li, X., Fries, S., Li, R., Lawson, J. A., Propert, K. J., Diamond, S. L., et al. (2014). Differential impairment of aspirin-dependent platelet cyclooxygenase acetylation by nonsteroidal antiinflammatory drugs. Proceedings of the National Academy of Sciences, 111(47), 16830–16835.
Jin, J., Quinton, T. M., Zhang, J., Rittenhouse, S. E., & Kunapuli, S. P. (2002). Adenosine diphosphate (ADP)-induced thromboxane a(2) generation in human platelets requires coordinated signaling through integrin alpha(IIb)beta(3) and ADP receptors. Blood, 99(1), 193–198.
Wei, C. K., Chang, F. R., Hsieh, P. W., & Wu, C. C. (2015). Inhibition of the interactions between metastatic human breast cancer cells and platelets by β-nitrostyrene derivatives. Life Sciences, 143, 147–155.
Uluçkan, O., Eagleton, M. C., Floyd, D. H., Morgan, E. A., Hirbe, A. C., Kramer, M., et al. (2008). APT102, a novel adpase, cooperates with aspirin to disrupt bone metastasis in mice. Journal of Cellular Biochemistry, 104(4), 1311–1323.
Giordano, A., Musumeci, G., D'Angelillo, A., Rossini, R., Zoccai, G. B., Messina, S., et al. (2016). Effects of glycoprotein IIb/IIIa antagonists: anti-platelet aggregation and beyond. Current Drug Metabolism, 17(2), 194–203.
Dardik, R., Savion, N., Kaufmann, Y., & Varon, D. (1998). Thrombin promotes platelet-mediated melanoma cell adhesion to endothelial cells under flow conditions: role of platelet glycoproteins P-selectin and GPIIb-IIIA. British Journal of Cancer, 77(12), 2069–2075.
Yu, Y., Zhou, X. D., Liu, Y. K., Ren, N., Chen, J., & Zhao, Y. J. (2002). Platelets promote the adhesion of human hepatoma cells with a highly metastatic potential to extracellular matrix protein: involvement of platelet P-selectin and GP IIb-IIIa. Journal Cancer Research and Clinical Oncology, 128(5), 283–287.
Stouffer, G. A., & Smyth, S. S. (2003). Effects of thrombin on interactions between beta3-integrins and extracellular matrix in platelets and vascular cells. Arteriosclerosis, Thrombosis, and Vascular Biology, 23(11), 1971–1978.
Grossi, I. M., Fitzgerald, L. A., Kendall, A., Taylor, J. D., Sloane, B. F., & Honn, K. V. (1987). Inhibition of human tumor cell induced platelet aggregation by antibodies to platelet glycoproteins Ib and IIb/IIIa. Proceedings of the Society for Experimental Biology and Medicine, 186(3), 378–383.
Karpatkin, S., Pearlstein, E., Ambrogio, C., & Coller, B. S. (1988). Role of adhesive proteins in platelet tumor interaction in vitro and metastasis formation in vivo. Journal of Clinical Investigation, 81(4), 1012–1019.
Coller, B. S. (2001). Anti-GpIIb/IIIa drugs: current strategies and future directions. Thrombosis and Haemostasis, 86, 427–443.
Huang, F., & Hong, E. (2004). Platelet glycoprotein IIb/IIIa inhibition and its clinical use. Cardiovascular & Hematological Agents in Medicinal Chemistry, 2(3), 187–196.
Kononczuk, J., Surazynski, A., Czyzewska, U., Prokop, I., Tomczyk, M., Palka, J., & Miltyk, W. (2015). αIIbβ3-integrin ligands: abciximab and eptifibatide as proapoptotic factors in MCF-7 human breast cancer cells. Current Drug Targets, 16(13), 1429–1437.
Hagemeyer, C. E., & Peter, K. (2010). Targeting the platelet integrin GPIIb/IIIa. Current Pharmaceutical Design, 16(37), 4119–4133.
Sheu, J. R., Lin, C. H., Peng, H. C., Teng, C. M., & Huang, T. F. (1993). Triflavin, an Arg-Gly-Asp-containing peptide, inhibits tumor cell-induced platelet aggregation. Japanese Journal of Cancer Research, 84(10), 1062–1071.
Lonsdorf, A. S., Krämer, B. F., Fahrleitner, M., Schönberger, T., Gnerlich, S., Ring, S., et al. (2012). Engagement of αIIbβ3 (GPIIb/IIIa) with ανβ3 integrin mediates interaction of melanoma cells with platelets: a connection to hematogenous metastasis. The Journal of Biological Chemistry, 287(3), 2168–2178.
Nierodzik, M. L., Klepfish, A., & Karpatkin, S. (1995). Role of platelets, thrombin, integrin IIb-IIIa, fibronectin and von Willebrand factor on tumor adhesion in vitro and metastasis in vivo. Thrombosis and Haemostasis, 74, 282–290.
Amirkhosravi, A., Amaya, M., Siddiqui, F., Biggerstaff, J. P., Meyer, T. V., & Francis, J. L. (1990). Blockade of GpIIb/IIIa inhibits the release of vascular endothelial growth factor (VEGF) from tumor-cell-activated platelets and experimental metastasis. Platelets, 10, 285–292.
Chiang, H. S., Swaim, M. W., & Huang, T. F. (1994). Characterization of platelet aggregation induced by human colon adenocarcinoma cells and its inhibition by snake venom peptides, trigramin and rhodostomin. British Journal of Haematology, 87, 325–331.
Chiang, H. S., Swaim, M. W., & Huang, T. F. (1995). The Arg-Gly-Asp-containing peptide, rhodostomin, inhibits in vitro cell adhesion to extracellular matrices and platelet aggregation caused by saos-2 human osteosarcoma cells. British Journal of Cancer, 71, 265–270.
Swaim, M. W., Chiang, H. S., & Huang, T. F. (1996). Characterisation of platelet aggregation induced by PC-3 human prostate adenocarcinoma cells and inhibited by venom peptides, trigramin and rhodostomin. European Journal of Cancer, 32A(4), 715–721.
Bakewell, S. J., Nestor, P., Prasad, S., Tomasson, M. H., Dowland, N., Mehrotra, M., et al. (2003). Platelet and osteoclast beta3 integrins are critical for bone metastasis. Proceedings of the National Academy of Sciences of the United States of America, 100(24), 14205–14210.
Schneider, J. G., Amend, S. R., & Weilbaecher, K. N. (2011). Integrins and bone metastasis: integrating tumor cell and stromal cell interactions. Bone, 48(1), 54–65.
Esposito, M., & Kang, Y. (2014). Targeting tumor-stromal interactions in bone metastasis. Pharmacology Therapy, 141(2), 222–233.
Zhang, W., Dang, S., Hong, T., Tang, J., Fan, J., Bu, D., et al. (2012). A humanized single-chain antibody against beta 3 integrin inhibits pulmonary metastasis by preferentially fragmenting activated platelets in the tumor microenvironment. Blood, 120(14), 2889–2898.
Amirkhosravi, A., Mousam, S. A., Amaya, M., Blaydes, S., Desai, H., Meyer, T., & Francis, J. L. (2003). Inhibition of tumor cell-induced platelet aggregation and lung metastasis by the oral GpIIb/IIIa antagonist XV454.Thrombosis and. Haemostasis, 90(3), 549–554.
Trikha, M., Zhou, Z., Jordan, J., & Nakada, M. T. (2000). ReoPro and m7E3 F(ab’)2 inhibit β3 integrin mediated tumor growh and angiogenesis. Proceedings of the American Association for Cancer Research, 42, 824 [Abstract].
Huang, Z., Miao, X., Patarroyo, M., Nilsson, G. P., Pernow, J., & Li, N. (2016). Tetraspanin CD151 and integrin α6β1 mediate platelet-enhanced endothelial colony forming cell angiogenesis. Thrombosis and Haemostasis, 14(3), 606–618.
Zhu, G., Zhang, Q., Reddy, E.C., Carrim, N., Chen, Y., Xu, X.R., et al. (2017). Integrin PSI domain has endogenous thiol isomerase function and is a novel target for anti-platelet therapy. Blood, 2017 Jan 25. doi: 10.1182/blood-2016-07-729400.
Qian, W., Tao, L., Wang, Y., Zhang, F., Li, M., Huang, S., et al. (2015). Downregulation of Integrins in cancer cells and anti-platelet properties are involved in holothurian glycosaminoglycan-mediated disruption of the interaction of cancer cells and platelets in Hematogenous metastasis. Journal of Vascular Research, 52(3), 197–209.
Hall, C. L., Dubyk, C. W., Riesenberger, T. A., Shein, D., Keller, E. T., & van Golen, K. L. (2008). Type I collagen receptor (alpha2beta1) signaling promotes prostate cancer invasion through RhoC GTPase. Neoplasia, 10(8), 797–803.
Eke, I., Zscheppang, K., Dickreuter, E., Hickmann, L., Mazzeo, E., Unger, K., et al. (2015). Simultaneous β1 integrin-EGFR targeting and radiosensitization of human head and neck cancer. Journal of the National Cancer Institute, 107(2). doi:10.1093/jnci/dju419.
Erpenbeck, L., & Schön, M. P. (2010). Deadly allies: the fatal interplay between platelets and metastasizing cancer cells. Blood, 115(17), 3427–3436.
Bambace, N. M., & Holmes, C. E. (2011). The platelet contribution to cancer progression. Journal Thrombosis and Haemostasis, 9(2), 237–249.
Jain, S., Russell, S., & Ware, J. (2009). Platelet glycoprotein VI facilitates experimental lung metastasis in syngenic mouse models. Journal Thrombosis and Haemostasis, 7(10), 1713–1717.
Jain, S., Zuka, M., Liu, J., Russell, S., Dent, J., Guerrero, J. A., et al. (2007). Platelet glycoprotein Ib alpha supports experimental lung metastasis. Proceedings of the National Academy of Sciences, 104(21), 9024–9028.
Nieswandt, B., & Watson, S. P. (2003). Platelet-collagen interaction: is GPVI the central receptor? Blood, 102, 449–461.
Erpenbeck, L., Nieswandt, B., Schön, M., Pozgajova, M., & Schön, M. P. Inhibition of platelet GPIbalpha and promotion of melanoma metastasis. Journal of Investigative Dermatology, 130(2), 576–586.
Läubli, H., & Borsig, L. (2010). Selectins as mediators of lung metastasis. Cancer Microenvironment, 3(1), 97–105.
Stevenson, J. L., Varki, A., & Borsig, L. (2007). Heparin attenuates metastasis mainly due to inhibition of P- and L-selectin, but non-anticoagulant heparins can have additional effects. Thrombosis Research, 120(suppl2), S107–S111.
Borsig, L., Wong, R., Feramisco, J., Nadeau, D. R., Varki, N. M., & Varki, A. (2001). Heparin and cancer revised: mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. PNAS, 98, 3352–3357.
Ludwig, R. J., Beohme, B., Podda, M., Henschler, R., Jager, E., Tandi, C., et al. (2004). Endothelial P-selectin as a target of heparinb action in experimental melanoma lung metastasis. Cancer Research, 64, 2743–2750.
Kozlowski, E. O., Pavao, M. S., & Borsig, L. (2011). Ascidian dermatan sulfates attenuate metastasis, inflammation and thrombosis by inhibition of P-selectin. Journal Thrombosis and Haemostasis, 9(9), 1807–1815.
Di Vito, C., Navone, S. E., Marfia, G., Abdel Hadi, L., Mancuso, M. E., Pecci, A., et al. (2016). Platelets from glioblastoma patients promote angiogenesis of tumor endothelial cells and exhibit increased VEGF content and release. Platelets, 29, 1–10. doi:10.1080/09537104.2016.1247208.
Nylander, S., Mattsson, C., Ramström, S., & Lindahl, T. L. (2003). The relative importance of the ADP receptors, P2Y12 and P2Y1, in thrombin-induced platelet activation. Thrombosis Research, 111(1–2), 65–73.
Wojtukiewicz, M.Z., Ciarelli, J.J., Walz, D.A., Honn, K.V. (1990). Thrombin enhances cancer cell expression of an integrin receptor and increases adhesion. 81st Annual Meeting of the Americsan Association for Cancer Research, Washington, Proceedings of AACR, 31, Abstract 476.
Wojtukiewicz, M.Z., Ciarelli, J.J., Snyder, D.A., Nelson, K.K., Walz, D.A., Honn, K.V.. (1990). Increased tumor cell adhesiveness and experimental metastasis following exposure to alpha-thrombin, its precursor and analogues. American Cancer Society Michigan Division Inc., 1990 Cancer Research Conference, Ypsilanti, MI, USA, Poster 22.
Wojtukiewicz, M.Z., Ciarelli, J.J., Snyder, D., Nelson, K.K., Walz, D.A., Honn, K.V. (1990). Thrombin increases tumor cell adhesiveness via a non-proteolytic pathway. First Regional Meeting of the American Society for Cell Biology, Chicago, IL, USA, 1990, Abstract 91.
Wojtukiewicz, M. Z., Tang, D. G., Nelson, K. K., Walz, D. A., Diglio, C. A., & Honn, K. V. (1992). Thrombin enhances tumor cell adhesive and metastatic properties via increased alpha IIb beta 3 expression on the cell surface. Thrombosis Research, 68, 233–245.
Wojtukiewicz, M. Z., Tang, D. G., Ciarelli, J. J., Nelson, K. K., Walz, D. A., Diglio, C. A., et al. (1993). Thrombin increases the metastatic potential of tumor cells. International Journal of Cancer, 54, 793–806.
Nierodzik, M. L., Kajumo, F., & Karpatkin, S. (1992). Effect of thrombin treatment of tumor cells on adhesion of tumor cells to platelets in vitro and tumor metastasis in vivo. Cancer Research, 52(12), 3267–3272.
Nierodzik, M., Plotkin, A., Kajumo, F., & Karpatkin, S. (1991). Thrombin stimulates tumor-platelet adhesion in vitro and metastasis in vivo. Journal of Clinical Investigation, 87(1), 229–236.
Yuan, L., & Liu, X. (2015). Platelets are associated with xenograft tumor growth and the clinical malignancy of ovarian cancer through an angiogenesis-dependent mechanism. Molecular Medicine Reports, 11(4), 2449–2458.
Pearlstein, E., Ambrogio, C., Gasic, G., Karpatkin, S., et al. (1982). Inhibition of the platelet-aggregating activity of two human adenocarcinomas of the colon and an anaplastic murine tumor with a specific thrombin inhibitor: dansylarginine N-(3-ethyl-1, 5-pentanediyl)amide. Progress in Clinical and Biological Research, 89, 479–502.
Nieman, M. T., LaRusch, G., Fang, C., Zhou, Y., & Schmaier, A. H. (2010). Oral thrombostatin FM19 inhibits prostate cancer. Thrombosis and Haemostasis, 104(5), 1044–1048.
Lu, Q., Lv, M., Xu, E., Shao, F., Feng, Y., Yang, J., & Shi, L. (2015). Recombinant hirudin suppresses the viability, adhesion, migration and invasion of Hep-2 human laryngeal cancer cells. Oncology Reports, 33(3), 1358–1364.
Guo, R. R., Liu, Y., Lu, W. L., Zhao, J. H., Wang, X. Q., Zhang, H., et al. (2008). A recombinant peptide, hirudin, potentiates the inhibitory effects of stealthy liposomal vinblastine on the growth and metastasis of melanoma. Biological and Pharmacological Bulletin, 31(4), 696–702.
Sassi, M., Chakroun, T., Mbemba, E., Van Dreden, P., Elalamy, I., Larsen, A. K., & Gerotziafas, G. T. (2017). The antithrombotic potential of tinzaparin and enoxaparin upon thrombin generation triggered in vitro by human ovarian cancer cells IGROV1. Clinical and Applied Thrombosis/Hemostasis, 23(2), 155–163.
Niers, T. M., Klerk, C. P., DiNisio, M., Van Noorden, C. J., Büller, H. R., Reitsma, P. H., & Richel, D. J. (2007). Mechanisms of heparin induced anti-cancer activity in experimental cancer models. Critical Reviews in Oncology and Hematology, 61(3), 195–207.
Laubli, H., & Borsig, L. (2009). Heparins attenuate cancer metastasis: are selectins the link? Cancer Investment, 27(5), 474–481.
Borsig, L. (2007). Antimetastatic activities of modified heparins: selectin inhibition by heparin attenuates metastasis. Seminars in Thrombosis and Hemostasis, 33(5), 540–546.
Varki, N. M., & Varki, A. (2002). Heparin inhibition of selectin-mediated interactions during the hematogenous phase of carcinoma metastasis: rationale for clinical studies in humans. Seminars in Thrombosis and Hemostasis, 28(1), 53–66.
Stevenson, J. L., Choi, S. H., & Varki, A. (2005). Differential metastasis inhibition by clinically relevant levels of heparins: correlation with selectin inhibition, not antithrombotic activity. Clinical Cancer Research, 11(19), 7003–7011.
Gomes, A. M., Kozlowski, E. O., Borsig, L., Teixeira, F. C., Vlodavsky, I., & Pavão, M. S. (2015). Antitumor properties of a new non-anticoagulant heparin analog from the mollusc Nodipecten nodosus: effect on P-selectin, heparanase, metastasis and cellular recruitment. Glycobiology, 25(4), 386–393.
Battinelli, E. M., Markens, B. A., Kulenthirarajan, R. A., Machlus, K. R., Flaumenhaft, R., & Italiano, J. E. (2014). Anticoagulation inhibits tumor cell-mediated release of platelet angiogenic proteins and diminishes platelet angiogenic response. Blood, 123(1), 101–112.
Goertz, L., Schneider, S. W., Desch, A., Mayer, F. T., Koett, J., Nowak, K., et al. (2016). Heparins that block VEGF-A-mediated von Willebrand factor fiber generation are potent inhibitors of hematogenous but not lymphatic metastasis. Oncotarget, 7(42), 68527–68545.
Lee, C. J., & Ansell, J. E. (2011). Direct thrombin inhibitors. British Journal of Clinical Pharmacology, 72(4), 718.
Asanuma, K., Wakabayashi, H., Okamoto, T., Asanuma, Y., Akita, N., Yoshikawa, T., et al. (2013). The thrombin inhibitor, argatroban, inhibits breast cancer metastasis to bone. Breast Cancer, 20(3), 241–246.
Asanuma, K., Wakabayashi, H., Hayashi, T., Okuyama, N., Seto, M., Matsumine, A., Kusuzaki, K., et al. (2004). Thrombin inhibitor, argatroban, prevents tumor cell migration and bone metastasis. Oncology, 67(2), 166–173.
DeFeo, K., Hayes, C., Chernick, M., Ryn, J. V., & Gilmour, S. K. (2010). Use of dabigatran etexilate to reduce breast cancer progression. Cancer Biology and Therapy, 10, 1001–1008.
Alexander, E. T., Minton, A. R., Hayes, C. S., Goss, A., Van Ryn, J., Gilmour, S. K., et al. (2015). Thrombin inhibition and cyclophosphamide synergistically block tumor progression and metastasis. Cancer Biology & Therapy, 16(12), 1802–1811. doi:10.1080/15384047.2015.1078025.
Alexander, E. T., Minton, A. R., Peters, M. C., van Ryn, J., & Gilmour, S. K. (2016). Thrombin inhibition and cisplatin block tumor progression in ovarian cancer by alleviating the immunosuppressive microenvironment. Oncotarget, 7(51), 85291–85305.
Shi, K., Damhofer, H., Daalhuisen, J., Ten Brink, M., Richel, D. J., & Spek, C. A. (2017). Dabigatran potentiates gemcitabine-induced growth inhibition of pancreatic cancer in mice. Molecular Medicine, 6, 23. doi:10.2119/molmed.2016.00214.
Vianello, F., Sambado, L., Goss, A., Fabris, F., & Prandoni, P. (2016). Dabigatran antagonizes growth, cell-cycle progression, migration, and endothelial tube formation induced by thrombin in breast and glioblastoma cell lines. Cancer Medicine, 5(10), 2886–2898.
Rousseau, A., Van Dreden, P., Mbemba, E., Elalamy, I., Larsen, A., & Gerotziafas, G. T. (2015). Cancer cells BXPC3 and MCF7 differentially reverse the inhibition of thrombin generation by apixaban, fondaparinux and enoxaparin. Thrombosis Research, 136(6), 1273–1279.
Kirwan, C. C., Bundred, N. J., Castle, J., Clarke, R., Dive, C., Morris, J., et al. (2016). PO-36 thrombin inhibition preoperatively (TIP) in early breast cancer, the first clinical trial of NOACs as an anti-cancer agent: trial methodology. Thrombosis Research, 140(Suppl 1), S189–S190. doi:10.1016/S0049-3848(16)30169-4.
Zhang, P., Feng, S., Liu, G., Wang, H., Zhu, H., Ren, Q., et al. (2016). Mutant B-Raf (V600E) promotes melanoma paracellular transmigration by inducing thrombin-mediated endothelial junction breakdown. Journal of Biological Chemistry, 291(5), 2087–2106.
Kakkar AK. Low molecular weight heparin and survival in cancer. (2005). Haematological Report, 1: 27.
Camerer, E., Qazi, A. A., Duong, D., Cornelissen, I., Advincula, R., & Coughlin, S. R. (2004). Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood, 104(2), 397–401.
Etulain, J., Mena, H. A., Negrotto, S., & Schattner, M. (2015). Stimulation of PAR-1 or PAR-4 promotes similar pattern of VEGF and endostatin release and pro-angiogenic responses mediated by human platelets. Platelets, 26(8), 799–804.
Huang, Z., Miao, X., Luan, Y., Zhu, L., Kong, F., Lu, Q., et al. (2015). PAR1-stimulated platelet releasate promotes angiogenic activities of endothelial progenitor cells more potently than PAR4-stimulated platelet releasate. Journal of Thrombosis and Haemostasis, 13(3), 465–476.
Holinstat, M., Boutaud, O., Apopa, P. L., Vesci, J., Bala, M., Oates, J. A., et al. (2011). Protease-activated receptor signaling in platelets activates cytosolic phospholipase A2α differently for cyclooxygenase-1 and 12-lipoxygenase catalysis. Arteriosclerosis, Thrombosis, Vascular, and Biology, 31(2), 435–442.
Thomas, C. P., Morgan, L. T., Maskrey, B. H., Murphy, R. C., Kühn, H., Hazen, S. L., et al. (2010). Phospholipid-esterified eicosanoids are generated in agonist-activated human platelets and enhance tissue factor-dependent thrombin generation. The Journal of Biological Chemistry, 285(10), 6891–6903.
Trivedi, V., Boire, A., Tchernychev, B., Kaneider, N. C., Leger, A. J., O'Callaghan, K., et al. (2009). Platelet matrix metalloprotease-1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell, 137(2), 332–343.
Melnikova, V. O., Balasubramanian, K., Villares, G. J., Dobroff, A. S., Zigler, M., Wang, H., et al. (2009). Crosstalk between protease-activated receptor 1 and platelet-activating factor receptor regulates melanomacell adhesion molecule (MCAM/MUC18) expression and melanoma metastasis. Journal of Biological Chemistry, 284(42), 28845–28855.
Hackler, P. C., Reuss, S., Konger, R. L., Travers, J. B., & Sahu, R. P. (2014). Systemic platelet-activating factor receptor activation augments experimental lung tumor growth and metastasis. Cancer Growth Metastasis, 7, 27–32.
Ferracini, M., Sahu, R. P., Harrison, K. A., Waeiss, R. A., Murphy, R. C., Jancar, S., et al. (2015). Topical photodynamic therapy induces systemic immunosuppression via generation of platelet-activating factor receptor ligands. Journal of Investigative Dermatology, 135(1), 321–323.
Sahu, R. P., Kozman, A. A., Yao, Y., DaSilva, S. C., Rezania, S., Martel, K. C., et al. (2012). Loss of the platelet activating factor receptor in mice augments PMA-induced inflammation and cutaneous chemical carcinogenesis. Carcinogenesis, 33(3), 694–701.
Bihl, J. C., Rapp, C. M., Chen, Y., & Travers, J. B. (2016). UVB generates microvesicle particle release in part due to platelet-activating factor signaling. Photochemistry and Photobiology, 92(3), 503–506.
Sahu, R. P., Rezania, S., Ocana, J. A., DaSilva-Arnold, S. C., Bradish, J. R., Richey, J. D., et al. (2014). Topical application of a platelet activating factor receptor agonist suppresses phorbol ester-induced acute and chronic inflammation and has cancer chemopreventive activity in mouse skin. PloS One, 9(11), e111608.
Domiano, B. P., Derian, C. K., Maryanoff, B. E., Zhang, H. C., & Gordon, P. A. (2003). RWJ-58259: a selective antagonists of protease activated receptor-1. Cardiovascular Drug Review, 21, 313–326.
Hosokawa, K., Ohnishi, T., Miura, N., Sameshima, H., Koide, T., Tanaka, K. A., Maruyama, I., et al. (2014). Antithrombotic effects of PAR1 and PAR4 antagonists evaluated under flow and static conditions. Thrombosis Research, 133(1), 66–72.
Zania, P., Kritikou, S., Flordellis, C. S., Maragoudakis, T., & N.E. (2006). Blockage of angiogenesis by small molecule antagonists to protease-activated receptor-1: association with endothelial cell growth suppression and induction of apoptosis. Journal of Pharmacology and Experimental Therapeutics, 318, 246–254.
Villares, G. J., Zigler, M., Wang, H., Melnikova, V. O., Wu, H., Friedman, R., et al. (2008). Targeting melanoma growth and metastasis with systemic delivery of liposome-incorporated protease-activated receptor-1 small interfering RNA. Cancer Research, 68, 9078–9086.
French, S. L., Arthur, J. F., Lee, H., Nesbitt, W. S., Andrews, R. K., Gardiner, E. E., & Hamilton, J. R. (2016). Inhibition of protease-activated receptor 4 impairs platelet procoagulant activity during thrombus formation in human blood. Journal of Thrombosis and Haemostasis, 14(8), 1642–1654.
Cunningham, M., McIntosh, K., Bushell, T., Sloan, G., & Plevin, R. (2016). Proteinase-activated receptors (PARs) as targets for antiplatelet therapy. Biochemical Society Transations, 44(2), 606–612.
Ottaiano, T. F., Andrade, S. S., de Oliveira, C., Silva, M. C., Buri, M. V., Juliano, M. A., et al. (2017). Plasma kallikrein enhances platelet aggregation response by subthreshold doses of ADP. Biochimie, 135, 72–81.
Shirai, T., Inoue, O., Tamura, S., Tsukiji, N., Sasaki, T., Endo, H., et al. (2017). C-type lectin-like receptor 2 promotes hematogenous tumor metastasis and prothrombotic state in tumor-bearing mice. Journal of Thrombosis and Haemostasis, 15(3), 513–525.
Kato, Y., Kaneko, M. K., Kunita, A., Ito, H., Kameyama, A., Ogasawara, S., Matsuura, N., et al. (2008). Molecular analysis of the pathophysiological binding of the platelet aggregation-inducing factor podoplanin to the C-type lectin-like receptor CLEC-2. Cancer Science, 99(1), 54–61.
Suzuki-Inoue, K. (2011). Essential in vivo roles of the platelet activation receptor CLEC-2 in tumor metastasis, lymphangiogenesis and thrombus formation. Journal of Biochemistry, 150(2), 127–132.
Chang, Y. W., Hsieh, P. W., Chang, Y. T., Lu, M. H., Huang, T. F., Chong, K. Y., et al. (2015). Identification of a novel platelet antagonist that binds to CLEC-2 and suppresses podoplanin-induced platelet aggregation and cancer metastasis. Oncotarget, 6(40), 42733–42748.
Riedl, J., Preusser, M., Nazari, P. M., Posch, F., Panzer, S., Marosi, C., et al. (2017). Podoplanin expression in primary brain tumors induces platelet aggregation and increases risk of venous thromboembolism. Blood. doi:10.1182/blood-2016-06-720714.
Lourbakos, A., Yuan, Y. P., Jenkins, A. L., Travis, J., Andrade-Gordon, P., Santulli, R., & Potempa, J. (2001). Activation of protease-activated receptors by gingipains from Porphyromonas gingivalis leads to platelet aggregation: a new trait in microbial pathogenicity. Blood, 97(12), 3790–3797.
Kida, Y., Higashimoto, Y., Inoue, H., Shimizu, T., & Kuwano, K. (2008). A novel secreted protease from Pseudomonas Aeruginosa activates NF-kappaB through protease-activated receptors. Cellular Microbiology, 10, 1491–1504.
Dulon, S., Leduc, D., Cottrell, G. S., D'Alayer, J., Hansen, K. K., Bunnett, N. W., & Hollenberg, M. D. (2005). Pseudomonas Aeruginosa elastase disables proteinase-activated receptor 2 in respiratory epithelial cells. American Journal of Respiratory Cell and Molecular Biology, 32, 411–419.
Majumdar, S., Dutta, S., Das, T., Chattopadhyay, P., & Mukherjee, A. K. (2015). Antiplatelet and antithrombotic activity of a fibrin(ogen)olytic protease from Bacillus Cereus strain FF01. International Journal of Biological Macromolecules, 79, 477–489.
D'Asti, E., Chennakrishnaiah, S., Lee, T. H., & Rak, J. (2016). Extracellular vesicles in brain tumor progression. Cellular and Molecular Neurobiology, 36(3), 383–407.
Wurdinger, T., Deumelandt, K., van der Vliet, H. J., Wesseling, P., & de Gruijl, T. D. (2014). Mechanisms of intimate and long-distance cross-talk between glioma and myeloid cells: how to break a vicious cycle. Biochimica et Biophysica Acta, 1846(2), 560–575.
Laffont, B., Corduan, A., Plé, H., Duchez, A. C., Cloutier, N., Boilard, E., & Provost, P. (2013). Activated platelets can deliver mRNA regulatory Ago2•microRNA complexes to endothelial cells via microparticles. Blood, 122(2), 253–261.
Sadej, R., Grudowska, A., Turczyk, L., Kordek, R., & Romanska, H. M. (2014). CD151 in cancer progression and metastasis: a complex scenario. Laboratory Investigation, 94(1), 41–51.
Perrone, D., Ardito, F., Giannatempo, G., Dioguardi, M., Troiano, G., Lo Russo, L., et al. (2015). Biological and therapeutic activities, and anticancer properties of curcumin. Experimental and Therapeutic Medicine, 10(5), 1615–1623.
Deng, Y. I., Verron, E., & Rohanizadeh, R. (2016). Molecular mechanisms of anti-metastatic activity of curcumin. Anticancer Research, 36(11), 5639–5647.
Lu, W. J., Chang, N. C., Jayakumar, T., Liao, J. C., Lin, M. J., Wang, S. H., et al. (2014). Ex vivo and in vivo studies of CME-1, a novel polysaccharide purified from the mycelia of Cordyceps sinensis that inhibits human platelet activation by activating adenylate cyclase/cyclic AMP. Thrombosis Research, 134(6), 1301–1310.
Chang, Y., Hsu, W. H., Lu, W. J., Jayakumar, T., Liao, J. C., Lin, M. J., et al. (2015). Inhibitory mechanisms of CME-1, a novel polysaccharide from the mycelia of Cordyceps sinensis, in platelet activation. Current Pharmaceutical Biotechnology, 16(5), 451–461.
Jayakumar, T., Chiu, C. C., Wang, S. H., Chou, D. S., Huang, Y. K., & Sheu, J. R. (2014). Anti-cancer effects of CME-1, a novel polysaccharide, purified from the mycelia of Cordyceps sinensis against B16-F10 melanoma cells. Journal of Cancer Research Therapy, 10(1), 43–49.
Lin, K. H., Kuo, J. R., Lu, W. J., Chung, C. L., Chou, D. S., Huang, S. Y., Lee, H. C., et al. (2013). Hinokitiol inhibits platelet activation ex vivo and thrombus formation in vivo. Biochemical Pharmacology, 85(10), 1478–1485.
Lu, W. J., Wu, M. P., Lin, K. H., Lin, Y. C., Chou, H. C., & Sheu, J. R. (2014). Hinokitiol is a novel glycoprotein VI antagonist on human platelets. Platelets, 25(8), 595–602.
Chen, X., Li, Q., Kan, X. X., Wang, Y. J., Li, Y. J., Yang, Q., et al. (2016). Extract of Caulis Spatholobi, a novel blocker targeting tumor cell induced platelet aggregation, inhibits breast cancer metastasis. Oncology Reports, 36(6), 3215–3224.
Sabrkhany, S., Griffioen, A. W., Pineda, S., Sanders, L., Mattheij, N., van Geffen, J. P., Aarts, M. J., et al. (2016). Sunitinib uptake inhibits platelet function in cancer patients. European Journal of Cancer, 66, 47–54.
Alonso-Escolano, D., Medina, C., Cieslik, K., Radomski, A., Jurasz, P., Santos-Martínez, M. J., et al. (2006). Protein kinase C delta mediates platelet-induced breast cancer cell invasion. Journal of Pharmacology and Experimental Therapeutics, 318(1), 373–380.
Zhao, L., Lu, G., Zhao, Q., Zhang, M., Chen, M., Zhang, J., & Dai, K. (2015). Staurosporine induces platelet apoptosis through p38 mitogen-activated protein kinase signaling pathway. Clinical Laboratory, 61(7), 717–726.
Lesyk, G., Fong, T., Ruvolo, P. P., & Jurasz, P. (2015). The potential of enzastaurin to enhance platelet aggregation and growth factor secretion: implications for cancer cell survival. Journal of Thrombosis and Haemostasis, 13(8), 1514–1520.
Fuentes, E., Rojas, A., & Palomo, I. (2016). NF-κB signaling pathway as target for antiplatelet activity. Blood Review, 30(4), 309–315.
Chang, C. C., Lu, W. J., Ong, E. T., Chiang, C. W., Lin, S. C., Huang, S. Y., & Sheu, J. R. (2011). A novel role of sesamol in inhibiting NF-κB-mediated signaling in platelet activation. Journal of Biomedical Science, 18, 93.
Mehta, P., Kimura, A., & Lawson, D. (1990). Effects of calcium channel-blocking agents on platelet-osteogenic sarcoma interaction: platelet aggregation and electron microscopic findings. Journal of Orthopeadic Research, 8(5), 629–634.
Dovizio, M., Maier, T. J., Alberti, S., Di Francesco, L., Marcantoni, E., Münch, G., et al. (2013). Pharmacological inhibition of platelet-tumor cell cross-talk prevents platelet-induced overexpression of cyclooxygenase-2 in HT29 human colon carcinoma cells. Molecular Pharmacology, 84(1), 25–40.
Lou, X. L., Sun, J., Gong, S. Q., Yu, X. F., Gong, R., & Deng, H. (2015). Interaction between circulating cancer cells and platelets: clinical implication. Chinese Journal of Cancer Research, 27(5), 450–460.
Almiron Bonnin, D. A., Ran, C., Havrda, M. C., Liu, H., Hitoshi, Y., Zhang, Z., et al. (2017). Insulin-mediated signaling facilitates resistance to in proneural hPDGFB-driven gliomas. Molecular Cancer Therapeutics. doi:10.1158/1535-7163.MCT-16-0616.
Heske, C. M., Yeung, C., Mendoza, A., Baumgart, J. T., Edessa, L. D., Wan, X., & Helman, L. J. (2016). The role of PDGFR-β activation in acquired resistance to IGF-1R blockade in preclinical models of rhabdomyosarcoma. Translational Oncology, 9(6), 540–547.
Kwon, H. J., Kim, G. E., Lee, Y. T., Jeong, M. S., Kang, I., Yang, D., & Yeo, E. J. (2013). Inhibition of platelet-derived growth factor receptor tyrosine kinase and downstream signaling pathways by Compound C. Cellular Signalling, 25(4), 883–897.
Johnson, K. E., Forward, J. A., Tippy, M. D., Ceglowski, J. R., El-Husayni, S., Kulenthirarajan, R., Machlus, K. R., et al. (2017). Tamoxifen directly inhibits platelet Angiogenic potential and platelet-mediated metastasis. Arteriosclerosis, Thrombosis, and Vascular Biology. doi:10.1161/ATVBAHA.116.308791.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wojtukiewicz, M.Z., Hempel, D., Sierko, E. et al. Antiplatelet agents for cancer treatment: a real perspective or just an echo from the past?. Cancer Metastasis Rev 36, 305–329 (2017). https://doi.org/10.1007/s10555-017-9683-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-017-9683-z