
Abstract
Disorder of long non-coding RNAs (LncRNAs) is found in various types of cancers and demonstrated to be associated with tumor occurrence and development. Our study found that lncRNA insulin growth factor 2 antisense (IGF2-AS) is up-regulated in gastric adenocarcinoma (GAC) tissues and correlated with poor prognosis in patients with GAC. Cell counting kit-8 (CCK8), colony formation, wound healing and transwell assays revealed that knockdown of IGF2-AS in BGC823 and SGC7901 cells significantly suppressed cell proliferation, migration and invasion. While, overexpression of IGF2-AS in AGS and MGC803 cells exhibited the opposite effects. RNA-FISH and subcellular fractionation assay found that most IGF2-AS was distributed in the cytoplasm, suggesting that IGF2-AS functioned as a potential ceRNA. RNA binding protein immunoprecipitation (RIP) assays further confirmed this assumption. By informatics prediction and luciferase reporter assay, we found that IGF2-AS functioned as an efficient miR-503 sponge and the level of miR-503 showed an inverse correlation with IGF2-AS. Short stature homeobox 2 (SHOX2) is predicted and verified as a target of miR-503. Moreover, IGF2-AS expression exhibited a negative correlation with miR-503 and a positive correlation with IGF2-AS. Subsequent rescue assay revealed that down-regulation of miR-503 or restoration of SHOX2 canceled IGF2-AS depletion-induced depression in proliferation and motility of BGC823 and SGC7901 cells. Meanwhile, up-regulation of miR-503 or down-regulation of SHOX2 decreased IGF2-AS overexpression induced promotion in proliferation and motility of AGS and MGC803 cells. In vivo tumorigenicity assay showed that knockdown of IGF2-AS significantly reduced tumor volume. Taken together, our results demonstrated that IGF2-AS takes important regulatory parts in GAC development by functioning as a ceRNA to regulate SHOX2 via sponging miR-503.
Similar content being viewed by others


Introduction
Gastric cancer (GC) is one of the most common cancers and a leading cause of cancer mortality [1]. Gastric adenocarcinoma (GAC) occupied the vast majority of GC and could be further divided into intestinal and diffuse types [2]. Although current studies have gained new progress in uncovering the mechanism of GC [3, 4], it still remains elusive, which hinders us from discovering more effective treatments. Therefore, it is of great significance to elucidate the mechanism of GC and carried out novel as well as effective therapeutics for GC.
Long non-coding RNAs (LncRNAs) are noncoding transcripts greater than 200 nucleotides in length [5]. Recently, increasing studies have speculated the involvement of lncRNAs in many complicated cancers, including GC [6,7,8]. Several lncRNAs (such as FENDRR, GAPLINC and H19) have been evidenced to be correlated with drug resistance metastasis, tumorigenesis, and prognosis of GC [9,10,11]. However, lncRNAs in GC is still an emerging field and quantities of novel lncRNAs that may closely related to GC progression remain to be explored [12].
Through bioinformatics analysis based on The Cancer Genome Atlas (TCGA) database, we found that the expression of insulin growth factor 2 antisense (IGF2-AS) was significantly up-regulated in GAC tissues and high expression of IGF2-AS is correlated with worse prognosis in patients with GAC. IGF2-AS, one of human lncRNAs, was originally identified as an antisense transcript of the IGF2 locus in the Wilms’ tumor [13]. Subsequently, IGF2-AS was characterized as a regulator in anesthetic-damaged mouse neural stem cell derived neurons [14]. Suppression of IGF2-AS has been found to facilitate angiogenesis in type 2 diabetes [15]. Currently, the down-regulation of IGF2-AS was found in non-small cell lung cancer and prostate cancer, and was suggested as a tumor suppressor in prostate cancer [16, 17]. However, in hepatocellular carcinoma tissues and cells, IGF2-AS was up-regulated [18]. On the basis of our bioinformatics analysis results and the published data, we speculated that IGF2-AS may be implicated in GAC progression. Therefore, in our present study, we aimed to investigate the function of IGF2-AS in GAC and the underlying regulatory mechanism. We demonstrated for the first time that IGF2-AS promoted GAC cell proliferation and migration possibly through sponging miR-503 to up-regulate short stature homeobox 2 (SHOX2), suggesting IGF2-AS acts as an oncogene in GAC development.
Materials and methods
Data collection
The RNA-Seq expression data of 375 GAC samples and 32 normal samples were downloaded from TCGA database (https://tcga-data.nci.Nih.gov/tcga) and used to identify differential expressed lncRNAs. Among these 375 samples, 306 samples have complete clinical data. These data were downloaded and used to analyze the association between IGF2-AS or SHOX2 expression and the clinical factors of GAC patients, as well as to draw the Kaplan–Meier survival plot. These 306 samples were dived into high and low expression groups according to the median expression level of IGF2-AS or SHOX2. The advantage of using the median as the cutoff is that the effect of outliers is negligible and could divide the samples into high and low groups of virtually the same size which enables the drawing of robust Kaplan–Meier plot.
Clinical specimens
GAC tissues and the corresponding para-cancer tissues were collected from 62 patients who underwent a GAC surgery in the Affiliated Yantai Yuhuangding Hospital of Qingdao University. The percentage of tumor cells in GAC tissue samples was greater than 65%. The para-cancer tissues we resected were 5 cm or more from the cancerous site (most were dysplasia or intestinal metaplasia) and were verified by pathological examination that they included no tumor cells. All patients did not receive any radiotherapy and chemotherapy before surgery. All these samples were maintained in liquid nitrogen and stored at – 80 °C. These tissues were used to analyze the expression of IGF2-AS. Informed consents were signed by all patients. Our study was approved by the ethics committee of the Affiliated Yantai Yuhuangding Hospital of Qingdao University.
Cell culture and transfection
Human gastric normal epithelial mucosa cell line GES1, human GAC cell lines (BGC823, SGC7901, MGC803 and AGS) and GC cell lines (HGC27 and MKN45) were bought from Shanghai Institute of Biosciences Cell Resource Center, Chinese Academy of Sciences (Shanghai, China). Cells were cultivated in Dulbecco’s minimal essential medium (DMEM) (Hyclone) containing serum (10%), penicillin (100 U/ml), and streptomycin (0.1 mg/ml) at 37 °C with 5% CO2. Si-IGF2-AS 1#, si-IGF2-AS 2#, si-SHOX2 and the control siRNA were synthesized by Shenzhen Huada Gene Research Institute (Guangdong, China). pcDNA3.1-IGF2-AS and pcDNA3.1-SHOX2 plasmids were constructed and used to overexpress IGF2-AS in AGS and MGC803 cells or overexpress SHOX2 in BGC823 and SGC7901 cells. The siRNAs and plasmids were transfected into BGC823, SGC7901, AGS and MGC803 cells using Lipofectamine 2000 (Invitrogen, Shanghai, China) following the supplier’s instructions. The sequences of the siRNAs were shown in Table 1.
Luciferase reporter assay
The binding sites between miR-503 and IGF2-AS or SHOX2 were predicted on miRcode or miRWalk website. The reporter vectors were constructed by cloning the sequences which contained the wide type (WT) or mutant type (MT) binding sites into pmirGLO reporter vectors (Promega, USA). The constructed vectors were co-transfected with miR-503 agomir or control into HRK 293T cells. Forty-eight hours later, luciferase activity was evaluated using a Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA).
qRT-PCR
Total RNA of the tissue samples, GES1, BGC823, SGC7901, HGC27, AGS, MGC803, MKN45 cells and the transfected cells were isolated utilizing the Trizol reagent (Invitrogen, Carlsbad, CA, USA). The primers used for qRT-PCR were all exhibited in Table 1. The ABI PRISM 7000 Fluorescent Quantitative PCR System (Applied Biosystems, Foster City, CA, USA) was used to carry out the qRT-PCR following manufacturer’s instruction. The relative expression of IGF2-AS, miR-503 miR-113a, miR150, miR-142-3p and SHOX2 were calculated by 2−∆∆Ct method. The expression of GAPDH or U6 was used for normalization.
Western blot
Tissue samples were homogenized with a manual homogenizer. Protein were extracted from the tissue samples, BGC823 and AGS cells using RIPA buffer and then the concentration of total protein was determined by BCA method. The proteins were separated by 10% SDS-PAGE and transferred to a PVDF membrane. Then the membrane was blocked by 5% fat-free milk and incubated with the primary antibodies against SHOX2 and GAPDH. Following that, the membrane was washed by TBS and incubated with the secondary antibody. The results were determined using an enhanced chemiluminescence system. GAPDH was utilized for normalization.
Cell counting Kit-8 (CCK8) assay
The viability of BGC823, SGC7901, AGS and MGC803 cells were determined using Cell Counting Kit-8 (CCK8, Dojindo, Kumamoto, Japan) 24 h after transfection. The cells were inoculated into 96-well plate and cultured under standard conditions. Cell viability was tested at 0, 24, 48, 72 and 96 h time points following the procedure described by the manufacturer.
Colony formation assay
BGC823 and SGC7901 cells transfected with scrambled siRNA or si IGF2-AS, and AGS and MGC803 cells transfected with vector or pcDNA-IGF2-AS were inoculated into 6-well plate and cultivated at 37 °C for two weeks. The culture media was replaced with fresh media every 3 days. The colonies were stained by 0.1% crystal violet and counted under a microscope.
Wound healing assay
After 24 h transfection, BGC823, SGC7901, AGS and MGC803 cells were inoculated into 6-well plates and cultured at 37 °C until reached to 90% confluence. Then a wound was made with a pipette tip. The plates were continued to be maintained in an incubator at 37 °C for 24 h with 5% CO2. The widths of the wounds were captured and measured at 0 and 24 h. The relative wound closure was calculated by regarding the wound closure of BGS823 or SGC7901 cells transfected with scrambled siRNA and AGS or MGC803 cells transfected with vector as 1.0, respectively.
Transwell assay
The migration and invasion abilities of GC cells were determined using transwell chambers. A total of 10,000 cells were added to the upper chamber for migration assays. The lower chamber was added with complete medium. After being incubated overnight, we fixed the migrated cells by 4% paraformaldehyde followed by staining them with 0.1% crystal violet. The number of migrated cells was counted under a microscope (Nikon). The procedure of invasion assay was similar with the migration assay, except that the upper chamber was coated with Matrigel (BD Biosciences).
RNA binding protein immunoprecipitation (RIP) assays
EZ-Magna RNA-binding protein immunoprecipitation (RIP) RNA-binding protein immunoprecipitation kit (Millipore, Billerica, MA) was applied for performing the RIP assay. BGC823 and AGS cells were lysed with lysis buffer. Then cell lysates were incubated with Ago2 antibody and negative control IgG. The precipitation was then digested by proteinase K and RNA was isolated. Finally, the RNA was analyzed by qRT-PCR to examine whether IGF2-AS involves in ceRNA.
Subcellular fractionation assay
The isolation of cytoplasmic and nuclear RNA in BGC823 and AGS cells were performed using Cytoplasmic and Nuclear RNA Purification Kit (Norgenbiotek Corporation, Thorold, ON, Canada) following the supplier’s description. Then the subcellular fractions were subjected to qRT-PCR. U6 (nucleus control) and Actin (cytoplasm control) was used for normalization.
RNA fluorescent in situ hybridization (FISH)
FISH assay was performed to detect and localize IGF2-AS in BGC823 and AGS cells. The probes of IGF2-AS was synthesized by Ribobio Company (China) and labeled with Cy3 fluorescent dye. Ribo™ FISH Kit (Ribobio Company, China) was used to carry out RNA FISH assay according to the procedure provided by the manufacturer.
In vivo tumorigenicity assay
Animal work was approved by the Affiliated Yantai Yuhuangding Hospital of Qingdao University. BGC823 cells transfected with si-IGF2-AS 2# or scrambled siRNA (2 × 107 cells in 200 μl of PBS) were injected sub-cutaneously into the right flank of five-week-old, female athymic BALB/c nu/nu mice (17–19 g) (Vital River Laboratories, Beijing, China). Tumor volume was measured at different time points. Volume = length × width × width × 0.5. The mice were euthanized after 21 days to take out the tumor.
Statistical analysis
All the experiments data were shown as mean ± SD. The significant difference between 2 groups was compared by Student’s t test. While multiple comparisons was performed by One-way ANOVA followed by Dunnett's post hoc test. Chi-square test was performed to analyze the correlation between IGF2-AS or SHOX2 and the clinical-pathological factors of GAC patients. Kaplan–Meier analysis was performed to analyze the correlation between IGF2-AS or SHOX2 expression and the overall survival of GC patients with log rank test used for comparison. Univariate and multivariate analyses were performed to determine whether IGF2-AS or SHOX2 could be served as independent predictors for GC patients. p value lower than 0.05 was regarded as statistically significant.
Results
IGF2-AS was up-regulated in GAC tissues and predictive of worse prognosis in patients with GAC
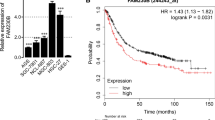
Based on the expression data downloaded from TCGA database, we found that the expression of IGF2-AS was significantly increased in the GAC tissues compared to the normal stomach tissues (p = 0.0000239, Fig. 1a). Then the relationship between IGF2-AS expression and clinical-pathological factors were analyzed. We identified that enhanced expression of IGF2-AS was closely correlated with pathologic-T (p = 0.034), pathologic-M (p = 0.001) and death (p = 0.036) (Table 2). Kaplan–Meier analysis revealed that high expression of IGF2-AS was closely related to lower overall survival rate (p = 0.01), indicating that higher IGF2-AS is predictive for poor prognosis in patients with GAC (Fig. 1b). By univariate analysis, we found that IGF2-AS expression (p = 0.011), clinical-stage (p = 0.001), pathologic-N (p = 0.007) and age (p = 0.11) could be considered as predictors for GAC. Further multivariate analysis suggested that IGF2-AS expression and age can be regarded as independent prognosis factors for patients with GAC (Table 3). Collectively, we supposed that IGF2-AS might be related to the progression and prognosis of GAC.

The expression of IGF2-AS was up-regulated in GAC tissues and cells. a The relative expression of IGF2-AS in gastric adenocarcinoma tissues was analyzed based on the TCGA database. b Kaplan–Meier overall survival (OS) curves according to IGF2-AS expression levels. c The relative expression of IGF2-AS in 62 GAC tissue samples and their corresponding para-cancer tissues was determined by qRT-PCR. d The relative expression of IGF2-AS in GES1, BGC823, SGC7901, HGC27, MGC803, MKN45 and AGS cells was determined by qPCR. N = 6, **p < 0.01 vs. normal tissues or GES1 cells
To verify the results we obtained based on the public data, we collected 62 human GAC tissue samples and their corresponding adjacent normal tissues to detect IGF2-AS expression by qRT-PCR. In accordance with the above data, we observed that the expression of IGF2-AS was significantly higher in the tumor tissues than that in the normal tissues (p < 0.01, Fig. 1c). Moreover, we also tested IGF2-AS expression in GES1 cell line and several GC cell lines. The results showed that IGF2-AS was up-regulated in all tested GC cell lines compared with the GES1 cell line (p < 0.01, Fig. 1d), and BGC823 cells exhibited the highest IGF2-AS level (about 10 folds of that in GES1 cells).
Altering the expression of IGF2-AS affects the proliferation, migration and invasion of GAC cells
To better understand the effect of IGF2-AS on GAC development, we then investigated whether knockdown or overexpression of IGF2-AS affects the proliferation, migration and invasion of GAC cells. Our results showed that both si- IGF2-AS 1# and si-IGF2-AS 2# decreased IGF2-AS expression in BGC823 and SGC7901 cells significantly, with si- IGF2-AS 2# exhibits higher knockdown efficiency (Fig. 2a). By CCK8 assay, we found that IGF2-AS knockdown by si-IGF2-AS 1# or si-IGF2-AS 2# significantly suppressed the viability of BGC823 and SGC7901 cells at 48, 72 and 96 h time points (p < 0.05, Fig. 2b, c). Similar with the knockdown efficiency, si-IGF2-AS 2# showed a greater influence on cell viability than si- IGF2-AS 1#. The results of colony formation assay showed that the colony formation ability of BGC823 and SGC7901 cells decreased dramatically after down-regulating IGF2-AS by si-IGF2-AS 1# or si-IGF2-AS 2# (p < 0.01, Fig. 2d, e). Wound healing assay revealed that the relative wound closure was significantly lower in the si-IGF2-AS 1# and si-IGF2-AS 2# groups than that in the scrambled group (p < 0.01, Fig. 2f, g). In migration and invasion assays, the numbers of migrated and invaded BGC823 and SGC7901 cells in the si-IGF2-AS 1# and si-IGF2-AS 2# groups were markedly less than that in the scrambled group (p < 0.01, Fig. 2h, i).

Knockdown of IGF2-AS in BGC823 and SGC7901 cells suppressed cell proliferation, migration and invasion. a The expression of IGF2-AS decreased significantly in BGC823 and SGC7901 cells after transfected with si-IGF2-AS 1# or si-IGF2-AS 2# compared with the scrambled group. b, c CCK8 assay found that knockdown of IGF2-AS inhibited BGC823 and SGC7901 cell proliferation. d Representative images of colony formation assay. e Knockdown of IGF2-AS significantly decreased the relative number of colonies. f Representative images of wound healing assays which were performed to explore the effect of IGF2-AS depletion on cell migration. g Relative wound closure of BGC823 and SGC7901 cells transfected with si-IGF2-AS 1# or 2# compared with that transfected with scrambled siRNA. h Representative images of transwell migration and invasion assays. i The mean number of migrated and invaded BGC823 and SGC7901 cells transfected with si-IGF2-AS 1# or 2# was significantly lower than that of cells transfected with scrambled siRNA. N = 6, **p < 0.01 vs. scrambled group
In addition, we overexpressed IGF2-AS by transfecting pcDNA-IGF2-AS into AGS and MGC803 cells and the level of IGF2-AS increased to about 6-fold of the vector group (p < 0.01, Fig. 3a). Overexpression of IGF2-AS in AGS and MGC803 cells yields the opposite effect on cell proliferation, migration and invasion as observed above. In briefly, up-regulation of IGF2-AS dramatically improved cell viability at 72 and 96 h time points (p < 0.01, Fig. 3b, c), and increased the number of colonies compared with the vector group (p < 0.01, Fig. 3d, e). Furthermore, overexpression of IGF2-AS in AGS and BGC803 cells significantly increased the relative wound closure (p < 0.01, Fig. 3f, g), the numbers of migrated and invaded cells (p < 0.01, Fig. 3h, i). Collectively, these data suggested that down-regulation of IGF2-AS inhibited GC cell proliferation and motility, while overexpression of IGF2-AS enhanced cell viability and motility.

Overexpression of IGF2-AS in AGS and MGC803 cells promoted cell proliferation, migration and invasion. a The expression of IGF2-AS increased significantly in AGS and MGC803 cells after transfected with PCDNA3.1-IGF2-AS plasmid compared with that transfected with vector. b, c CCK8 assay found that overexpression of IGF2-AS promoted AGS and MGC803 cell proliferation. d Representative images of colony formation assay. e Overexpression of IGF2-AS significantly increased the relative number of colonies. f Representative images of wound healing assays of AGS cells transfected with PCDNA3.1- IGF2-AS or vector. g Relative wound closure of AGS and MGC803 cells transfected with PCDNA3.1- IGF2-AS compared with that transfected with vector. h Representative images of transwell migration and invasion assays. i The mean number of migrated and invaded AGS and MGC803 cells transfected with PCDNA3.1- IGF2-AS was significantly larger than that of cells transfected with vector. N = 6, **p < 0.01 vs. vector group
IGF2-AS promotes GAC proliferation by involving in ceRNA mechanism
The subcellular localization of IGF2-AS was detected by subcellular fractionation assay and RNA-FISH. The outcomes showed that most of the IGF2-AS located in the cytoplasm, a small part in the nucleus (Fig. 4a, b). Then RIP experiments were carried out using anti-Ago2 antibody to determine whether IGF2-AS interact with RNA-induced silencing complexes (RISCs). The results showed that endogenous IGF2-AS in both BGC823 and AGS cells were specifically enriched in Ago2 immunoprecipitates compared with IgG (Fig. 4c). These data suggested that IGF2-AS functioned as a ceRNA. However, IGF2-AS could bind to which miRNAs and involve in the ceRNA process remains unclear. Hence, we next predicted the relative miRNAs using the miRcode website (https://www.mircode.org/). We selected the top 10 possible miRNAs (miR-503, miR-133abc, miR-142-3p, miR-150, miR-5127, miR-17, miR-17-5p, miR-20ab, miR-20b-5p and miR-93) for bioinformatics analysis. By analysis and reading literatures, we found that miR-503 and miR-142-3p were down-regulated in GC [19, 20], and miR-503 has been demonstrated to be related to the survival of GC patients [19]. We then explored the effect of down-regulation or overexpression of IGF2-AS on the expression of the top 4 miRNAs (miR-503, miR-133a, miR-142-3p and miR-150). We found that knockdown of IGF2-AS in BGC823 cells significantly increased the level of miR-503 and miR-142-3p while decreased the level of miR-150 (Fig. 4d). Up-regulation of IGF2-AS in the AGS cells exhibited the opposite results (Fig. 4e). Since miR-503 changed the most and has been found to be associated with GC development, hence we chose it for the following experiments. And the results suggested that miR-503 was negatively regulated by IGF2-AS.

IGF2-AS functioned as a ceRNA in BGC823 and AGS cells. a, b FISH and subcellular fractionation assay illustrated that IGF2-AS was distributed mostly in the cytoplasm in BGC823 and AGS cells. Bars = 50 μM. c The relative level of IGF2-AS in RIP was determined by qRT-PCR. **p < 0.01 vs. IgG group. d The relative expressions of miR-503, miR-113a, miR-150 and miR-142-3p in BGC823 cells transfected with si-IGF2-AS or scrambled siRNA were determined by qRT-PCR. e The relative expressions of miR-503, miR-113a, miR-150 and miR-142-3p in AGS cells transfected with PCDNA3.1-IGF2-AS or vector were determined by qRT-PCR. N = 6, *p < 0.05, **p < 0.01 vs. vector group
To verify miR-503 could directly interact with IGF2-AS, we performed luciferase assay. Figure 5a showed the predicted binding sites between miR-503 and IGF2-AS. Luciferase assay showed that up-regulation of miR-503 significantly reduced the luciferase activity of pmirGLO- IGF2-AS WT while exhibited no significant effect on the activity of pmirGLO- IGF2-AS MUT, indicating that it could direct bind to the IGF2-AS through the binding sites (p < 0.01, Fig. 5b). Then we found that silencing IGF2-AS in BGC823 cells significantly increased the level of miR-503 (Fig. 5c), while overexpression of IGF2-AS in AGS cells noticeably decreased the level of miR-503 (Fig. 5d). Our results confirmed the above hypothesis that IGF2-AS negatively regulates the expression of miR-503 in GAC cells.

IGF2-AS could direct bind to miR-503. a The predicted binding sites between IGF2-AS and miR-503. b miR-503 agomir reduced the relative luciferase activity of WT-IGF2-AS, while showed no significant effect on MUT-IGF2-AS. **p < 0.01 vs. control group. c Knockdown of IGF2-AS in BGC823 cells significantly increased the level of miR-503. **p < 0.01 vs. scrambled group. d Overexpression of IGF2-AS in AGS cells significantly decreased the level of miR-503. N = 6, **p < 0.01 vs. vector group
SHOX2 is a target of miR-503
We predicted the potential targets of miR-503 using the miRWalk website (https://mirwalk.umm.uni-heidelberg.de/). Combine the prediction results with the differential expressed mRNA obtained based on TCGA database, we obtained 60 mRNA for further analysis (Table 4).
We performed bioinformatics analysis of these 60 genes based on the RNA-seq data and clinical data of GAC patients from the TCGA database. Among these 60 genes, we found that SHOX2 is noticeably differently expressed and exhibited a relative higher expression in GAC tissues compared with the normal tissues (logFC = 2.087; p = 2.59E−08). Clinical relevance analysis presented that the expression of SHOX2 is closely associated with the death of patients with GC (p = 0.01, Table 5). Kaplan–Meier survival analysis and log-rank tests demonstrated that high expression of SHOX2 was significantly correlated with lower overall survival (p = 0.003, data not shown). Univariate analysis revealed that SHOX2 expression, clinical-stage, pathologic-N, and age can be considered as prognostic factors for GC patients. What’s more, multivariate analysis demonstrated that SHOX2 expression and age can be independent predictors for GC patients (Table 6). Searching the published papers, we found that SHOX2 has been found to be disordered in several other tumors [21,22,23] and promoter methylation of SHOX2 has been regarded as diagnostic as well as prognostic biomarker for lung cancer patients [24, 25], suggesting the participation of SHOX2 in tumor progression. Based on the bioinformatics analysis results and the published literatures, we selected SHOX2 as the target of miR-503 for subsequent experiments, suggesting IGF2-AS/miR-503/SHOX2 may constitute the ceRNA mechanism.
We then assessed the protein expression of SHOX2 in 10 GAC tissues using western blot. The results showed that the protein expression levels of SHOX2 in these 10 GAC tissues were all noticeably higher than that in their corresponding normal adjacent tissues (p < 0.01, Fig. 6a, b), which was consistent with the results obtained from TCGA database.

SHOX2 is a target of miR-503 and is negatively regulated by miR-503. a, b The protein expression of SHOX2 in 10 GC tumor tissue samples and their corresponding normal adjacent tissues were detected by western blot. T tumor; N normal adjacent tissues. c The predicted binding sites between SHOX2 and miR-503. d miR-503 agomir reduced the relative luciferase activity of WT-SHOX2, while showed no significant effect on MUT-SHOX2. **p < 0.01 vs. control group. e The mRNA level of SHOX2 was positively correlated with IGF2-AS expression and negatively correlated with miR-503 expression in BGC823 cells. f The effect of IGF2-AS and miR-503 on SHOX2 protein expression in BGC823 cells is determined by western blot. g The relative protein expression level of SHOX2 in figure f. h The mRNA level of SHOX2 was positively correlated with IGF2-AS expression and negatively correlated with miR-503 expression in AGS cells. i The effect of IGF2-AS and miR-503 on SHOX2 protein expression in AGS cells is determined by western blot. j The relative protein expression level of SHOX2 in figure i. N = 6, **p < 0.01 vs. control group
The predicted binding sites between miR-503 and SHOX2 were shown in Fig. 6c. Luciferase assay uncovered that up-regulation of miR-503 by agomir significantly decreased the luciferase activity of pmirGLO-SHOX2-WT whilst showed no obviously effect on pmirGLO-SHOX2-Mut (Fig. 6d), indicating that SHOX2 is indeed a target of miR-503. In BGC823 cells, we identified that knockdown of IGF2-AS lead to a significant decrease of SHOX2 at both mRNA (p < 0.01, Fig. 6e) and protein levels (p < 0.01, Fig. 6f, g). However, down-regulation of miR-503 dramatically increased the mRNA and protein expression levels of SHOX2 (p < 0.01). Down-regulation of IGF2-AS together with miR-503 also enhanced the expression of SHOX2 compared with the control group (p < 0.01, Fig. 6e–g) and showed a similar level with the antagomir group. On the contrary, in AGS cells, we found that the overexpression of IGF2-AS increased mRNA and protein level of SHOX2 significantly, while up-regulation of miR-503 reduced SHOX2 expression dramatically compared with the control group (p < 0.01, Fig. 6h–j). Overexpression of IGF2-AS and miR-503 together made also no change in SHOX2 expression compared with the control group. Taken together, these observations indicated that SHOX2 is a target of miR-503, and IGF2-AS acted as a ceRNA for miR-503 to up-regulate SHOX2 and then affects the progression of GC.
IGF2-AS affects GC cell proliferation and motility by modulating miR-503/SHOX2 axis as a ceRNA
To determine whether IGF2-AS exerts roles via regulating miR-503/SHOX2, we performed rescue assays. The results revealed that down-regulation of miR-503 or overexpression of SHOX2 mitigated IGF2-AS silencing induced suppression on viability, migration of BGC823 and SGC7901 cells (Fig. 7a–f). In AGS and MGC803 cells, we observed that up-regulation of miR-503 or knockdown of SHOX2 countermanded overexpression of IGF2-AS induced enhancement in cell viability, migration and invasion (Fig. 7g–l).

IGF2-AS affects GAC cell proliferation and motility by modulating miR-503/SHOX2 axis as a ceRNA. a, b Down-regulation of miR-503 or overexpression of SHOX2 rescued the proliferation of BGC823 and SGC7901 cells with depleted IGF2-AS. c, d Down-regulation of miR-503 or overexpression of SHOX2 rescued the migration ability of BGC823 and SGC7901 cells with depleted IGF2-AS. e, f Down-regulation of miR-503 or overexpression of SHOX2 rescued the invasion of BGC823 and SGC7901 cells with depleted IGF2-AS. g, h Up-regulation of miR-503 or knockdown of SHOX2 reduced cell viability to normal level in AGS and MGC803 cells with overexpressed IGF2-AS. i, j Up-regulation of miR-503 or knockdown of SHOX2 reduced cell migration ability to normal level in AGS and MGC803 cells with overexpressed IGF2-AS. k, l Up-regulation of miR-503 or knockdown of SHOX2 reduced cell invasion ability to normal level in AGS and MGC803 cells with overexpressed IGF2-AS. N = 6, *p < 0.05, **p < 0.01 vs. control group; ##p < 0.01 vs. si-IGF2-AS 2# or pcDNA-IGF2-AS group
On the purpose of investigating the function of IGF2-AS in vivo, we carried out xenograft experiments. Knockdown of IGF2-AS significantly reduced the tumor volume 10 days after injection compared with the scrambled group, suggesting a suppressive effect on tumor growth (Fig. 8a, b). In summary, IGF2-AS affects GAC tumor development by modulating miR-503/SHOX2.

Knockdown of IGF2-AS inhibit tumor growth in vivo.a Images of tumors that taken out from the mice after 21 days injection. Bar = 10 mm. b Tumor growth curve was plotted according to tumor volumes. N = 3, **p < 0.01 vs. scrambled group
Discussion
Significant attention has been paid to lncRNAs in these years, since the vital regulatory role of lncRNAs in tumor progression has been demonstrated by increasing evidences [26, 27]. The main findings of our present work was that lncRNA IGF2-AS was overexpressed in GAC tissues and affects GAC tumor progression possibly by regulating miR-503/SHOX2 axis.
In 2000, Tomohisa Okutsu et al. first identified that PEG8/IGF2-AS was significantly overexpressed in Wilms' tumor and suggested it as a marker for Wilms' tumor [13]. Later, researchers discovered the abnormal expression of IGF2-AS in several other tumors. In hepatocellular carcinoma tissues and cells, the expression level of IGF2-AS was also up-regulated [18]. However, in prostate cancer tissues [16] and non-small cell lung cancer tissues [17], the level of IGF2AS expression was found to be decreased. These contradictory results suggest that IGF2AS may play a diametrically opposite role in the development of different tumors. In our present study, we demonstrated that the level of IGF2AS was significantly higher in GAC tissues and cell lines than that in the para-cancer tissues or the normal gastric epithelial cells, respectively. The overlapping of IGF2AS expression between tumor and nontumor tissues possibly caused by the difference among samples. Univariate and multivariate analyses unearthed that IGF2AS were a potential independent prognosis factor for patients with GAC. However, whether this lncRNA could be used as a robust prognostic marker in the clinical settings need our future verification. Biological function experiments found that knockdown of IGF2AS in GAC cells suppressed cell proliferation, migration and invasion, while the overexpression of IGF2AS in GAC cells obtained the opposite results, suggesting IGF2AS acted as a tumor promoter in GAC progression.
Growing evidences demonstrated that lncRNA possibly functioned as a ceRNA for particular miRNAs to modulate the target genes of the miRNAs [28]. However, there is no reports concerning that IGF2AS acted as a ceRNA of miRNAs to date. RNA-FISH assay and qRT-PCR found that IGF2AS is mainly distributed in the cytoplasm, suggesting that IGF2AS is involved in a ceRNA mechanism. This speculation was further verified by the RIP assay. By predicting the possible miRNAs related to IGF2AS via miRcode website, literatures reading, and miRNA expression and prognosis analysis based on the public data, miR-503 was finally screened out and supposed to be one of the miRNAs associated with IGF2AS. miR-503 has been previously found to be down-regulated in GC and the down-regulation of miR-503 is correlated with more aggressive tumor [29, 30]. Our further biological experiments demonstrated that IGF2AS could direct bind to miR-503 and the level of miR-503 was negatively correlated with the level of IGF2AS.
Afterwards, we predicted the targets of miR-503 using miRWalk. After prognosis analysis of the predicted targets and searching literatures, we selected SHOX2 for further investigation. Luciferase reporter assay confirmed that SHOX2 is a direct target of miR-503. SHOX2 belongs to the homeobox family which encode proteins containing a 60-amino acid residue motif that represents a DNA binding domain [31]. Previously research concerning SHOX2 is mainly focused on its transcriptional regulatory role in several genetic disorders [32, 33]. Recently, increasing evidence demonstrated that SHOX2 expression was disordered in tumors, suggesting its involvement in cancer progression. In esophageal squamous cell carcinoma (ESCC), SHOX2 exhibited an enhanced expression and presented promoting effects on ESCC cell proliferation and invasion [21]. High expression of SHOX2 was indicated to be correlated with the recurrence of hepatocellular carcinoma [22] and related to worse survival of breast cancer patients [23]. Promoter methylation of SHOX2 has been reported as diagnostic and prognostic biomarker in lung cancer [24, 25, 34]. DIMO DIETRICH et al. have previously developed and characterized a CE marked in vitro diagnostic test kit for SHOX2 DNA methylation quantification in bronchia aspirates, and revealed that this test is robust and reliable for the diagnosis of lung cancer [34]. Through Cox Proportional Hazard analysis, DNA methylation of PITX2 and SHOX2 is identified as an independent prognostic biomarker in patients with non–small-cell lung cancer [25]. In our study, we found that higher SHOX2 expression is correlated with poor outcomes of patients with GAC and it could be considered as an independent predictor for GAC. How will SHOX2 expression be used as a prognostic marker in GAC in the clinical settings is worth our future investigation. Biological experiments found that SHOX2 expression is positively correlated with the expression of IGF2-AS and negatively associated with the expression of miR-503. Collectively, the above observations illustrated that knockdown of IGF2AS increased miR-503 expression and then decreased the expression of SHOX2 in a ceRNA-dependent manner, which was further proofed by rescue assay. Moreover, in vivo assays demonstrated that knockdown of IGF2-AS significantly reduced tumor growth, suggesting that IGF2-AS maybe a promising therapeutic target for GAC.
Recent studies identified that IGF2-AS is up-regulated by signal transducer and activator of transcription 3 (STAT3) in human hepatoma cells and involved in hepatitis C virus replication via modulating phosphatidylinositol 4-phosphate (PI4P) [35]. STAT3 is known as a transcription factor that contribute to the growth and migration of cancer cells and its constitutive activation is found in several tumor types, including GC [36, 37]. It has been clearly illustrated that enhanced p-STAT3 activity is closely correlated with worse prognosis of GC patients as well as with undifferentiated type and lymph node metastasis [38]. On the basis of these data, we supposed that the up-regulation of IGF2-AS observed in our study was possibly modulated by STAT3. However, this hypothesis need further verification and how STAT3 regulate IGF2-AS will be an interesting and attractive topic.
To our best knowledge, this is the first study demonstrated that IGF2-AS affects the growth and metastasis of GAC via function as a ceRNA of miR-503 to regulate SHOX2. Our results illuminate the light for a deeper understanding of the mechanisms of GAC. In the future works, we will quest the mechanisms of upregulation of IGF2AS in GAC, the correlation between IGF2-AS and other miRNAs, and the regulatory relationship between miR-503 and other targets, which will further deepen our understanding of the mechanism of GAC.
References
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86.
Network CGA. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513(7517):202–9.
Du C, Li D, Li N, Chen L, Li S, Yang Y, Hou M, Xie M, Zheng Z. DDX5 promotes gastric cancer cell proliferation in vitro and in vivo through mTOR signaling pathway. Sci Rep. 2017;7:42876.
Katoh H, Ishikawa S. Genomic pathobiology of gastric carcinoma. Pathol Int. 2017;67(2):63.
Li H, Ma SQ, Huang J, Chen XP, Zhou HH. Roles of long noncoding RNAs in colorectal cancer metastasis. Oncotarget. 2017;8(24):39859–76.
Wang P, Li J, Zhao W, Shang C, Jiang X, Wang Y, Zhou B, Bao F, Qiao H (2018) A novel LncRNA-miRNA-mRNA triple network identifies LncRNA RP11-363E7.4 as an important regulator of miRNA and gene expression in gastric cancer. Cell Physiol Biochem. 47(3):1025.
Lei K, Liang X, Gao Y, Xu B, Xu Y, Li Y, Tao Y, Shi W, Liu J. Lnc-ATB contributes to gastric cancer growth through a MiR-141-3p/TGFβ2 feedback loop. Biochem Biophys Res Commun. 2017;484(3):514–21.
Zhen L, Yun-hui L, Hong-yu D, Jun M, Yi-long Y. Long noncoding RNA NEAT1 promotes glioma pathogenesis by regulating miR-449b-5p/c-Met axis. Tumour Biol. 2016;37(1):673.
Xu TP, Huang MD, Xia R, Liu XX, Sun M, Yin L, Chen WM, Han L, Zhang EB, Kong R. Decreased expression of the long non-coding RNA FENDRR is associated with poor prognosis in gastric cancer and FENDRR regulates gastric cancer cell metastasis by affecting fibronectin1 expression. J Hematol Oncol. 2014;7(1):63.
Li H, Yu B, Li J, Su L, Yan M, Zhu Z, Liu B. Overexpression of lncRNA H19 enhances carcinogenesis and metastasis of gastric cancer. Oncotarget. 2014;5(8):2318–29.
Hu Y, Wang J, Qian J, Kong X, Tang J, Wang Y, Chen H, Hong J, Zou W, Chen Y, Xu J. Long noncoding RNA GAPLINC regulates CD44-dependent cell invasiveness and associates with poor prognosis of gastric cancer. Can Res. 2014;74(23):6890–902.
Gu Y, Chen T, Li G, Yu X, Lu Y, Wang H, Teng L. LncRNAs: emerging biomarkers in gastric cancer. Future Oncol (London, England). 2015;11(17):2427–41.
Okutsu T, Kuroiwa Y, Kagitani F, Kai M, Aisaka K, Tsutsumi O, Kaneko Y, Yokomori K, Surani MA, Kohda T. Expression and imprinting status of human PEG8/IGF2AS, a paternally expressed antisense transcript from the IGF2 locus Wilms’ tumors. J Biochem. 2000;127(3):475–83.
Song C, Song C, Chen K, Zhang X. Inhibition of long non-coding RNA IGF2AS protects apoptosis and neuronal loss in anesthetic-damaged mouse neural stem cell derived neurons. Biomed Pharmacother. 2017;85:218–24.
Zhao Z, Liu B, Li B, Song C, Diao H, Guo Z, Li Z, Zhang J. Inhibition of long noncoding RNA IGF2AS promotes angiogenesis in type 2 diabetes. Biomed Pharmacother. 2017;92:445–50.
Chen Q, Sun T, Wang F, Gong B, Xie W, Ma M, Yang X. Long noncoding RNA IGF2AS is acting as an epigenetic tumor suppressor in human prostate cancer. Urology. 2019;124:310.e1–e8.
Zhang X, Zhang X, Hu R, Hao L. Prognostic implication and functional role of long noncoding RNA IGF2AS in human non-small cell lung cancer. J Cell Biochem. 2019;120:12067.
Bao H, Guo CG, Qiu PC, Zhang XL, Dong Q, Wang YK. Long non-coding RNA Igf2as controls hepatocellular carcinoma progression through the ERK/MAPK signaling pathway. Oncol Lett. 2017;14(3):2831–7.
Wu D, Cao G, Huang Z, Jin K, Hu H, Yu J, Zeng Y. Decreased miR-503 expression in gastric cancer is inversely correlated with serum carcinoembryonic antigen and acts as a potential prognostic and diagnostic biomarker. Oncotargets Ther. 2017;10:129–35.
Wang Y, Cao Z, Wang L, Liu S, Cai J. Downregulation of microRNA‑142‑3p and its tumor suppressor role in gastric cancer. Oncol Lett. 2018;15(5):8172–80.
Yi J, Jin L, Chen J, Feng B, He Z, Chen L, Song H. MiR-375 suppresses invasion and metastasis by direct targeting of SHOX2 in esophageal squamous cell carcinoma. Acta Biochim Biophys Sin. 2017;49(2):159–69.
Yang T, Zhang H, Cai SY, Shen YN, Yuan SX, Yang GS, Wu MC, Lu JH, Shen F. Elevated SHOX2 expression is associated with tumor recurrence of hepatocellular carcinoma. Ann Surg Oncol. 2013;20(3):S644–49.
Hong S, Noh H, Teng Y, Shao J, Rehmani H, Ding HF, Dong Z, Su SB, Shi H, Kim J. SHOX2 is a direct miR-375 target and a novel epithelial-to-mesenchymal transition inducer in breast cancer cells. Neoplasia. 2014;16(4):279–90.
Schmidt B, Liebenberg V, Dietrich D, Schlegel T, Kneip C, Seegebarth A, Flemming N, Seemann S, Distler J, Lewin J. SHOX2 DNA Methylation is a Biomarker for the diagnosis of lung cancer based on bronchial aspirates. BMC Cancer. 2010;10(1):600.
Dietrich D, Hasinger O, Liebenberg V, Field JK, Kristiansen G, Soltermann A. DNA methylation of the homeobox genes PITX2 and SHOX2 predicts outcome in non-small-cell lung cancer patients. Diagn Mol Pathol. 2012;21(2):93.
Tony G, Sven D. The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol. 2012;9(6):703–19.
Fu Z, Chen C, Zhou Q, Wang Y, Zhao Y, Zhao X, Li W, Zheng S, Ye H, Wang L, He Z. LncRNA HOTTIP modulates cancer stem cell properties in human pancreatic cancer by regulating HOXA9. Cancer Lett. 2017;410:68.
Chao Y, Di W, Lin G, Xi L, Jin Y, Dong W, Wang T, Li X. Competing endogenous RNA networks in human cancer: hypothesis, validation, and perspectives. Oncotarget. 2016;7(12):13479–90.
Wu D, Cao G, Huang Z, Jin K, Hu H, Yu J, Zeng Y. Decreased miR-503 expression in gastric cancer is inversely correlated with serum carcinoembryonic antigen and acts as a potential prognostic and diagnostic biomarker. OncoTargets Ther. 2017;10:129–35.
Peng Y, Liu YM, Li LC, Wang LL, Wu XL. microRNA-503 inhibits gastric cancer cell growth and epithelial-to-mesenchymal transition. Oncol Lett. 2014;7(4):1233–8.
Hongbing L, Espinoza-Lewis RA, Chaohui C, Xuefeng H, Yanding Z, Yiping C. The role of Shox2 in SAN development and function. Pediatr Cardiol. 2012;33(6):882–9.
Blaschke RJ, Hahurij ND, Sanne K, Steffen J, Wisse LJ, Kirsten D, Tina M, Konstantinos A, Jessica S, Hardt SE. Targeted mutation reveals essential functions of the homeodomain transcription factor Shox2 in sinoatrial and pacemaking development. Circulation. 2007;115(14):1830–8.
Hongbing L, Chao-Hui C, Espinoza-Lewis RA, Zhen J, Ivana S, Xuefeng H, Minkui L, Yanding Z, Yiping C. Functional redundancy between human SHOX and mouse Shox2 genes in the regulation of sinoatrial node formation and pacemaking function. J Biol Chem. 2011;286(19):17029–38.
Dimo D, Christoph K, Olaide R, Triantafillos L, Anke S, Thomas S, Nadja F, Sebastian R, Jürgen D, Michael F. Performance evaluation of the DNA methylation biomarker SHOX2 for the aid in diagnosis of lung cancer based on the analysis of bronchial aspirates. Int J Oncol. 2012;40(3):825–32.
Yulin X, Ming J, Jing Y, Changjiang Z, Yan Z, Xuemei K, Lin L, Xiaohong W. STAT3-regulated long non-coding RNAs lnc-7SK and lnc-IGF2-AS promote hepatitis C virus replication. Mol Med Rep. 2015;12(5):6738.
Koseki T, Suehiro N, Masuda Y, Miyoshi N, Muraoka D, Ogo N, Asai A. Discovery of a new STAT3 inhibitor acting on the linker domain. Biol Pharm Bull. 2019;42(5):792–800.
Koh JS, Joo MK, Park JJ, Yoo HS, Choi BI, Lee BJ, Chun HJ, Lee SW. Inhibition of STAT3 in gastric cancer: role of pantoprazole as SHP-1 inducer. Cell Biosci. 2018;8:50.
Ji K, Zhang L, Zhang M, Chu Q, Li X, Wang W. Prognostic value and clinicopathological significance of p-stat3 among gastric carcinoma patients: a systematic review and meta-analysis. Medicine. 2016;95(5):e2641.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
Our study was approved by the ethics committee of the Affiliated Yantai Yuhuangding Hospital of Qingdao University.
Consent to participate
Informed consent was signed by all patients.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Huang, J., Chen, Yx. & Zhang, B. IGF2-AS affects the prognosis and metastasis of gastric adenocarcinoma via acting as a ceRNA of miR-503 to regulate SHOX2. Gastric Cancer 23, 23–38 (2020). https://doi.org/10.1007/s10120-019-00976-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10120-019-00976-2