
Abstract
D222G mutation of the hemagglutinin (HA) is of special interest because of its close association with the enhanced virulence of 2009 pandemic influenza A (H1N1) virus through the increased binding affinity to α2,3-linked sialylated glycan receptors. However, there is still a lack of detailed understanding about the molecular mechanism of this enhanced virulence. Here, molecular dynamics simulation and binding free energy calculation were performed to explore the altered glycan receptor binding mechanism of HA upon the D222G mutation by studying the interaction of one α2,3-linked sialylglycan (sequence: SIA-GAL-NAG) with the wild type and D222G mutated HA. The binding free energy calculation based on the molecular mechanics generalized Born surface area (MM-GBSA) method indicates that the D222G mutated HA has a much stronger binding affinity with the studied α2,3-linked glycan than the wild type. This is consistent with the experimental result. The increased binding free energy of D222G mutant mainly comes from the increased energy contribution of Gln223. The structural analysis proves that the altered electrostatic potential of receptor binding domain (RBD) and the increased flexibility of 220-loop are the essential reasons leading to the increased affinity of HA to α2,3-linked sialic acid glycans. The obtained results of this study have allowed a deeper understanding of the receptor recognition mechanism and the pathogenicity of influenza virus, which will be valuable to the structure-based inhibitors design targeting influenza virus entry process.

The altered α-2,3 linked glycan binding to hemagglutinin upon D222G mutation
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since the first detection of the H1N1 influenza A virus in human in April 2009, this virus spreads rapidly around the world and has given serious threat to the life of the population all over the world. In most cases, infection with the 2009 pandemic influenza A (H1N1) virus (pdmH1N1) causes mild disease [1] and the case-fatality rate during the start of the pandemic is influenza virus has high evolutionary rate and tendency to acquire point mutation in their genome. Some of these mutations can result in amino acid substitutions at key position in the proteins like hemagglutinin (HA) which is important for the virus replication and thus can alter the properties such as those associated with the virus antigenicity or pathogenecity [2]. From July to December of 2009, in Norway, 11 (18 %) of 61 cases infected by 2009 pandemic influenza A(H1N1) virus with severe disease outcome have been reported to harbor the D222G substitution of HA in clinical specimens [3]. Such mutant was not observed in any of the 205 mild cases investigated [3–5]. In addition to Norway, this mutation has also been detected in Hong Kong, Brazil, Japan, Mexico, Ukraine, and the United States [3]. Because of its close correlation with a severe clinical outcome, D222G mutation arouses great interest. Furthermore, D222G mutation has been described as the single change in HA between two strains of the “Spanish” 1918 H1N1 virus [6, 7].
Why does this single amino acid substitution increase the pathogenicity of the 2009 pandemic influenza A (H1N1) virus? As we know, the influenza virus infection is initiated by virus attachment to cell-surface sialoside receptors via influenza HA. Thus, HA plays a key role for the attachment of the virus to the host cell membrane surface glycoproteins or glycolipids via multivalent interactions to the sialoglycans. Thus, the point mutation in the receptor binding domain of HA may influence the interaction between virus and cell-surface sialoside receptors of the host and then change the pathogenicity of influenza virus. Recently, Chutinimitkul et al. confirmed this hypothesis and they found that the most remarkable difference in phenotype between wild type and D222G 2009 pandemic influenza A(H1N1) lay in the altered sialoside receptor binding [8]. Such differences in attachment are likely explained by an increased affinity for α2,3-linked sialic acids (α2,3-SAs), while maintaining specificity for α2,6-linked sialic acids (α2,6-SAs). However, there is still a lack of understanding in molecular mechanism about how the D222G mutation increases the binding affinity of HA with α2,3-SAs, which is very important to the rational design of HA inhibitors aiming at the severe influenza disease caused by D222G mutation.
Recent biotechnologies can test the affinity of miscellaneous HA binding to various carbohydrate receptors, but they cannot provide intrinsic and detailed information about their interaction [9]. A better understanding of the mechanism of glycan receptor recognition requires new approaches. As we know, during the glycan receptor recognition process of HA, the conformational dynamic factors and binding free energy should be important and responsible for the specific recognition. Molecular dynamics (MD) simulation combined with binding free energy calculation offer a complementary approach to investigate biological phenomena difficult to assess experimentally [10–13]. This method can provide not only plentiful dynamic structural information on protein complex structures in solution but also a wealth of energetic information, including the binding free energy between protein and ligand. In recent years, a combination of molecular dynamics (MD) simulation and binding free energy calculation has been used widely and successfully to study protein-protein, protein-nucleic acid and protein-ligand interactions [14–17]. In the study of glycan receptor recognition mechanism for influenza virus HA, there are also several successful applications of MD combined with binding free energy calculation [10, 11].
In this study, to explore the mechanism that D222G mutation increases the virulence of 2009 pandemic influenza A (H1N1) virus, we combined molecular dynamics simulation with binding free energy calculation to analyze the difference of the interaction of one α2,3-linked glycan (sequence: SIA-GAL-NAG) with the wild type and mutated HA. The large scale simulation can complement experiments for better understanding of the molecular recognition mechanism by providing atomic details and conformational dynamics that are often inaccessible by experiment. The binding free energy calculations based on the molecular mechanics generalized Born surface area (MM-GBSA) can provide the detailed interaction profile as well as characterize the binding hot spots and identify the difference between the wild type and D222G mutated HA. These specific interaction profiles will give an interesting clue to understand the mechanism of glycan receptor recognition and species’ specificity switch, which will help us get a deeper understanding about the pathogenicity depending on HA strains of influenza virus and will be very valuable to structure-based design of anti-influenza drugs targeting the HA.
Materials and methods
Preparation of the studied systems
During our study, there was no crystal structure of pdmH1N1 HA with α2,3-SA. Thus, the first step is to construct the structure of the studied complexes. The 3D structure of HA of pdmH1N1 used in this study was taken from the Protein Data Bank (pdb code: 3M6S) [18]. In this crystal structure, the HA exists in trimer with the asparagine-linked glycosylation at position 87. In the receptor binding site, there was no glycan receptor crystallized. Here, one α-2,3 linked sialylglycan with the sequence SIA-GAL-NAG (Fig. 1a), which has been confirmed to be with increased affinity for HA D222G mutant from glycan microarray analysis [18], was used in this study. To construct the complex of the studied α-2,3 linked glycan with the wild type HA, firstly, three HA monomers with the corresponding α-2,3 linked glycan were extracted from 1934 HA (PDB ID: 1RVX) [19], respectively. Similarly, each monomer structure of HA of 2009 pandemic influenza A (H1N1) virus were extracted from PDB ID 3M6S. Then, by aligning each monomer, the corresponding α-2,3 linked glycan was fitted to HA of 2009 pandemic influenza A and the complex of the studied α-2,3 linked glycan with the wild type HA was obtained. The mutated HA complex with glycan was obtained by using the wild type complex as the initial structure and changing the specific residue with the program Pymol [20]. When we finished our study, the wild type H1N1 HA-LSTa crystal structure was obtained (pdb code 3UBJ) [21]. By comparing our structure and the reported crystal structure, we found that the RMSD between Cα atoms of the two structures is 0.684, indicating our constructed structure is reasonable.

The studied complex of glycan and HA: (a) the studied α-2,3 linked glycan topology; (b) structure overview of trimer HA; (c) the receptor-binding site of HA
The complexes of wild type and mutated HA were further treated and used as the initial structures for molecular dynamics simulation. The main procedure for the structure treatment included the addition of hydrogen atoms and six disulfide bonds for protein using the leap module together with the parameterization of receptor and ligands in AMBER 10 software package [22]. The standard AMBER force field for bio-organic systems (FF99SB) [23] and for glycans (GLYCAM06) [24] was used to describe the HA and glycan parameters, respectively. For each system, the counter ions (Na+) were added to neutralize each ligand-bound system. Then, the corresponding systems were solvated using atomistic TIP3P water [25] in an octahedron box with at least 10 Å distance around the complex.
Molecular dynamics simulation
The molecular dynamics package AMBER10 [22] was used throughout the whole simulation process as well as the minimization and equilibration protocols. The energy minimization was firstly conducted with steepest descent method switched to conjugate gradient every 2500 steps totally for 5000 steps with 0.1 kcal mol−1 Å−2 restraints on all atoms in the complexes. Following this step, another two rounds of energy minimization were performed by only restraining protein and further releasing all the restraints for 5000 steps of each round. Long-range Coulombic interactions were handled using the particle mesh Ewald (PME) summation [26]. For the equilibration and subsequent production runs, the SHAKE algorithm [27] was employed on all atoms covalently bonded to a hydrogen atom, allowing for an integration time step of 2 fs. The system was gently annealed from 0 to 310 K over a period of 50 ps using a Langevin thermostat with a coupling coefficient of 1.0/ps and a force constant 2.0 kcal mol−1 Å−2 on the complex. All subsequent stages were carried out in the isothermal isobaric (NPT) ensemble using a Berendsen barostat [28] with a target pressure of 1 bar and a pressure coupling constant of 2.0 ps. An additional five rounds of MD (100 ps each at 310 K) were performed with decreasing restraint weights from 2.0, 1.5, 1.0, 0.5 to 0.1 kcal mol−1 Å−2. By releasing all the restraints, the system was again equilibrated for 500 ps. Following the last equilibration step, the production phase of the simulations was run without any restraints for a total of 15 ns.
Binding free energy calculation
MM-GBSA calculation was performed using AMBER10 [22]. The first step of MM-GBSA method is the generation of multiple snapshots from an MD trajectory of the protein-ligand complex, stripped of water molecules and counter ions. Snapshots, equally spaced at 10 ps intervals, were extracted from the equilibration section of MD trajectory. For each snapshot, the free energy is calculated for each molecular species (complex, protein, and ligand). The binding free energy is computed as the difference:
The free energy, G, for each species can be calculated by the following scheme using the MM-GBSA method [29]:
Here, E gas is the gas-phase energy; E int is the internal energy; E bond , E angle , and E torsion are the bond, angle, and torsion energies, respectively; and E ele and E vdw are the Coulomb and van der Waals energies, respectively. E gas was calculated using the AMBER molecular mechanics force field. G sol is the solvation free energy and can be decomposed into polar and nonpolar contributions. G GB is the polar solvation contribution calculated by solving the GB equation [29, 30]. The dielectric constant of solvent was set to 80. The dielectric constant of solute was shown to be an important factor in influencing the calculation of solvation free energy [31]. Here, in order to select the proper dielectric constant of solute, different values were tried. G nonpolar is the nonpolar solvation contribution and was estimated by the solvent accessible surface area (SAS) determined using a water probe radius of 1.4 Å. The surface tension constant γ was set to 0.0072 kcal mol−1 Å−2 [32]. T and S are the temperature and the total solute entropy, respectively. Vibrational entropy contributions were estimated by normal mode analysis [33]. Because of the high computational demand, only 50 snapshots for ligand and the receptor binding region (residues 54–259) of HA were used in the normal mode analysis and each snapshot was optimized for 100,000 steps using a distance-dependent dielectric of 4rij (rij is the distance between atoms i and j) until the root-mean-square of the gradient vector was less than 0.0001 kcal mol−1 Å−2.
To obtain the contribution of each residue to the binding energy, MM-GBSA was used to decompose the interaction energies to each residue involved in the interaction by only considering molecular mechanics and solvation energies without the contribution of entropies.
Cluster analysis
Molecular dynamics simulations can provide a representation of the sampling of a given molecule’s energetically accessible conformational ensemble. Such trajectories contain large amounts of structural information. To group and make sense of this information in the trajectory, clustering analysis is a useful tool. In this study, the self-organization map (SOM) method from the AMBER ptraj module was used to cluster the MD trajectories based on the pairwise similarity measured by the root-mean square deviation (RMSD) [34]. The SOM algorithm proposed by Kohonen has been used successfully in the trajectory analysis [34]. In the SOM-based clustering analysis, each cluster is seeded with a random point, and the clusters are set up in a simple topology where each cluster has some “neighbor” clusters. Then, the system is run through several training cycles on the input data. To process a data point, the most similar cluster is chosen. Here, during the clustering analysis, for each monomer both in the wild type and mutated HA-glycan complexes, only the ligand and residues of the receptor binding site in the 5 Å range of glycan were considered and the corresponding snapshots every 1 ps were extracted from the equilibration section of MD trajectory and selected for cluster analysis. The cluster number was set as 3.
Results and discussion
System stability and structural changes
Molecular dynamics simulation was performed for 15 ns for each system. Here, the equilibration of MD trajectories was monitored from the convergence of the root-mean square deviation (RMSD) of Cα atoms of protein from the original starting coordinates (Fig. 2a). From Fig. 2a, it can be seen that this system is stable in the last 5 ns. In addition, we also monitored the RMSD of Cα atoms for the residues in 5 Å around ligand as well as RMSD of heavy atoms for ligand for each monomer in the wild type and mutated HA complexes (Fig. 2b and c) since our aim here is to study the interaction of HA and glycan. From Fig. 2b, we can see the receptor binding site is very stable and has a small fluctuation after 5 ns. From Fig. 2c, except for the monomer-2 of D222G mutant, the α-2,3 linked glycan has a single binding mode with HA. In the monomer-2 of D222G mutant, it seems that there are two different states for glycan. In order to give a clear description, we extracted two representative snapshots at 10 ns and 15 ns (shown in Fig. 3). By aligning these two snapshots, we find that the glycan at 15 ns is somewhat translated relative to that at 10 ns.

The monitoring of the equilibration for the MD trajectories: (a) The time series of the RMSD of Cα atoms from the initial structure; (b) Time evolution of RMSD of Cα atoms for the residues in 5 Å around of glycan; (c) Time evolution of the RMSD of heavy atoms for the α-2,3 linked glycan

The alignment of two snapshots at 10 ns (colored as white) and 15 ns (colored as yellow) in the monomer-2 of D222G mutated HA
To identify if D222G mutation will affect the conformational flexibility, we also calculated the RMS fluctuations of backbone atoms averaged for each residue in the wild type and mutated HA. Since there are many residues in the studied system, to give a clear display, we just showed the comparison of RMSF of the residues around the glycan binding region (Fig. 4). In pdmH1N1, the receptor binding region (RBD) mainly includes the 130-loop, 190-helix and 220-loop. From Fig. 4, it can be seen that the flexibility of 130-loop and 220-loop in D222G mutant obviously increased compared to that in wild type HA. The increased flexibility in the receptor binding region may be responsible for the increased ability in the identification of more kinds of glycan receptors, such as α2,3-SAs. However, this conclusion is not consistent with that from the work by Yang et al. [18]. In the work of Yang et al., by mutating D222 to G222 based on the crystal structure of wild type HA, they deduced that the D222G mutation may not drastically change the properties of H1N1 HA, since the overall structure of the receptor binding site may be generally preserved. We think this inconsistency may arise given that Yang et al. [18] used static structural information, while our results are obtained through dynamics simulations.

The comparison of RMSF of the residues of the receptor binding region in the wild type and mutated HA
The comparison of the binding free energy of the studied glycan with wild type and D222G mutated HA
To explore the difference of interaction profile from the energy perspective for the studied α-2,3 linked glycan with the wild type and mutated HA, the binding free energy calculation and pair interaction energy analysis were performed by using MM-GBSA method. Five hundred snapshots were extracted from the last 5 ns equilibrated section of molecular dynamics (MD) trajectory and used for the enthalpy calculation. Because of the high computational demand, it is difficult to calculate the entropy for such a large system including more than 23,000 atoms excluding the water molecules. However, entropy, as an important component, is critical to the accurate evaluation of binding free energy. To make the calculation of entropy feasible here, the residues (residues 54–259) in 20 Å around the glycan and only 50 snapshots extracted from the last 5 ns of molecular dynamics (MD) trajectory were used to evaluate the entropy change.
Furthermore, the solute dielectric constant is shown to be a very important factor to affect the accurate calculation of the electrostatic interaction contribution and polar solvation free energy during the MM-GBSA calculation process. Here, the ligand is a glycan with large polarity. Thus, the binding surface exhibits the hydrophilicity to some degree. To select the proper solute dielectric constant, we tried 1, 2, 4, 5 and 6. By comparing the binding free energy results in the different dielectric constants of solute, we found more stable enthalpy for each monomer in the same trimer could be obtained with the increasing of solute dielectric constant. When the solute dielectric constant was 6, each monomer in the same trimer had very similar enthalpy with glycan and the reasonable binding free energy results were obtained. The calculated average binding free energies when the solute dielectric constant was 6 and the detailed contribution of various energy components are shown in Table 1. The calculated results when the solute dielectric constant was set as other values are given in the supporting information (Tables S1–S4).
From Table 1, it can be seen that the wild type HA has a positive binding free energy for the studied α-2,3 linked glycan, indicating that the HA cannot bind α2,3-SAs. After D222G mutation, the average binding free energy between the studied glycan and HA is −1.49 kcal mol−1. Although it is difficult to give an accurate estimation of absolute binding free energy, generally, the MM-GBSA can give a good rank of binding free energy for different systems [31, 35, 36]. Here, the 4 kcal mol−1 lower binding free energy of D222 mutated HA with the glycan relative to that of wild type system indicates that the HA with D222G mutant has a much stronger binding ability to α2,3-SAs. Such calculation result is completely consistent with the experimental results that the D222G mutation do increase the binding of the α2,3-SAs [8], indicating that our calculated results are reliable. By decomposing the free energy into identifiable contributions, we can understand the origin to determine the binding of the α-2,3 linked glycan and HA. From the contribution of individual energy term, the averaged nonpolar or hydrophobic interaction (ΔGnonpolar = ΔEvdw + ΔGsol-np) with −31.13 kcal mol−1 and −32.36 kcal mol−1 contribution in wild type and mutated HA, respectively, provides the main driving force for the glycan binding. Relative to the hydrophobic interaction, the electrostatic interaction (ΔGpolar = ΔEele + ΔGsol-ele) has an even smaller unfavorable contribution. Basically, the direct intermolecular electrostatic interactions (ΔEele) are highly favorable to the binding both for the wild type and mutated HA but their contributions are compensated by the large desolvation penalties (ΔGsol-ele) associated with the binding process.
By comparing the difference in the contribution of individual energy term, both the polar and nonpolar terms have an increased contribution for mutated HA relative to wild type HA. In order to seek which residues are responsible for the increased binding affinity for the D222G mutated HA, pair interaction energy analysis was employed. As for the wild type HA, residue contribution profile of each monomer was given in Fig. 5. By comparing Figs. 5a-5c, it can be seen that the energy profile for each monomer is very similar. In all three monomers, five residues have more than 1.0 kcal mol−1 contribution to the binding of glycan commonly. They include Val132, Thr133, Trp150, Leu191 and Gln223. Lys142 also has an obvious contribution in monomer-2 and monomer-3. From the perspective of enthalpy (Table 1), monomer-3 both in the wild type and mutated HA has the strongest binding with the studied α-2,3 linked glycan. In the following analysis, we will focus on monomer-3. The comparison of residue contribution profile in monomer 3 of the wild type and mutated HA indicates that the increased contribution of Gln223 should be responsible for the increased binding affinity of the D222G mutated HA with glycan (shown in Fig. 5d). To search for the reason of the varied contribution of the residues in wild type and mutated HA, deeper structural analysis is necessary.

The residue contributions for the glycan binding: (a) monomer 1 in WT HA; (b) monomer 2 in WT HA; (c) monomer 3 in WT HA; (d) The energy difference of each residue contribution in the mutated HA relative to wild type one in monomer 3
As for the glycan receptor, to explore which section is responsible for its interaction with HA, we further decompose the binding free energy to each glycan component including SIA, GAL and ANG. The corresponding results were given in Table 2. From Table 2, it can be seen that SIA is a main section to interact with HA while GAL and ANG have a relatively small contribution for this α-2,3 linked glycan binding, which is consistent with the glycan binding mode.
Structural analysis of wild type and D222G mutated HA
To explore the reason for the binding affinity change of HA to α2,3-SAs upon the D222G mutation, further structural analysis was performed. Generally, the change of hydrogen bond mode will have a large impact on the protein-ligand interaction and the feature of the binding pocket. To identify the reason for the changed contribution of the residues upon the D222G mutation, the hydrogen bond analysis was performed. The default geometric criterion to define the hydrogen bond is used: the donor-acceptor heavy atom distance should be less than 3.5 Å, and the donor-hydrogen-acceptor angle should be larger than 120°. Figure 6 shows the comparison of percentage occupation of hydrogen bonds between the SIA section of the α-2,3 linked glycan and the wild type as well as mutated HA. From this figure, it can be seen that the hydrogen bond interaction mode between the glycan and HA changed largely upon the D222G mutation. The occupation of the hydrogen bonds between the carboxyl group of SIA and the nitrogen atom of side chain of Gln223 reduced greatly in the D222G mutated HA. In addition, the hydrogen bond modes between the 1,2,3-trihydroxy propyl group of the glycan and the mutated HA have a large differencefrom that in wild type system. The hydrogen bonds between the 1,2,3-trihydroxy propyl group and Tyr91, Gln223 disappear almost completely in the D222G mutated HA. Instead, the His180 and Glu187 as hydrogen acceptors, form the stronger hydrogen bonds with 1,2,3-trihydroxy propyl group in the mutated HA compared with the wild type HA. From the hydrogen bond analysis, D222G mutation changes the hydrogen bond mode of Gln223 on a large scale, which is consistent with the RMSF and binding free energy decomposition analysis. The RMSF analysis indicates the flexibility of 220-loop in the mutated HA increased compared with the wild type system. Furthermore, the results from the binding free energy calculation disclose that the residue Gln223 in D222G mutated HA has a clearly increased contribution to the binding of the α-2,3 linked glycan. From the above hydrogen bond (HB) analysis, the number of HB of the residue 223 in D222G mutated HA was reduced compared to that in wild type HA, which may suggest that the direct electrostatic interaction should decrease. However, the direct favorable electrostatic interaction can be compensated by the unfavorable polar solvation contribution generally. As a result, the polar contribution of Q223 is almost the same in the wild type (−0.50 kcal mol−1) and mutated HA (−0.51 kcal mol−1). However, D222G mutation makes the nonpolar contribution of Q223 increase obviously (−1.67 kcal mol−1 in the mutated HA and −0.86 kcal mol−1 in wild type HA), leading to the increased contribution of Q223 on the whole. All the above analyses show the importance of 220-loop. The importance of the 220-loop brings our attention to its structural changes. By monitoring the distance between the oxygen atom from main chain of Arg221 and the terminal nitrogen atom from side chain of Gln223 (shown in Fig. 7), we find an interesting change upon the D222G mutation. In the wild type HA, one hydrogen bond between the oxygen atom from the main chain of Arg221 and the terminal nitrogen atom from the side chain of Gln223 is always stably formed. However, the D222G mutation makes this hydrogen bond almost disappear. Based on this phenomenon, this hydrogen bond locks the 220-loop in a special and stable conformation in the wild type HA like a lock. When a small and flexible residue glycine replaces aspartic acid, the hydrogen bond lock disappears and the 220-loop becomes more flexible. This change means the RBD may identify many more kinds of glycans such as α2,3-SAs.

Percentage occupation of hydrogen bonds between the glycan and HA in the wild type and D222G mutated system

The monitoring of the distance between the oxygen atom from main chain of Arg221 and the terminal nitrogen atom from side chain of Gln223
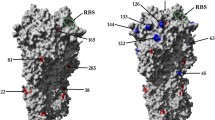
Furthermore, we performed structural cluster analysis for the RBD of monomer-3 to seek the structural difference between the wild type and mutated HA. We selected a three cluster solution using the self-organization map method based on pairwise similarity measured by RMSD. The percentage occupation of different clusters and the electrostatic surface of the largest cluster in the wild type and mutated HA was shown in Fig. 8. For the wild type HA, the percentage of each cluster is similar where cluster 3 is the largest cluster. As for the D222G mutated HA, the largest cluster is cluster 1 accounting for 46.8 % conformations. Since the electrostatic force has been shown to be very important to the recognition and association between the protein and ligand [37], from a quick glance at the electrostatic potential surfaces of different clusters, it can be seen that the electrostatic surface of each cluster both for the wild type or mutated HA is similar. However, the comparison of the electrostatic surface between the wild type and D222G mutated HA shows a large difference. The RBD of D222G mutant is becoming more positive. By checking this change in detail, we find that in the wild type HA, the carboxyl group of SIA lies in the region with neural, even negative charge, which is unfavorable to its binding. After D222G mutation, the corresponding region has positive charge and easily forms the favorable electrostatic interaction with the carboxyl group of SIA, leading to increased interaction of the α-2,3 linked glycan with HA. The electrostatic potential analysis can explain further why the D222G mutation increases the binding ability of HA to α2,3-SAs.

(a) Percentage occupation of different clusters in the wild type HA; (b) Percentage occupation of different clusters in D222G mutated HA; (c) electrostatic surface of largest cluster in WT HA; (d) electrostatic surface of largest cluster in WT HA (blue region with positive change and red region with negative charge)
Since explicit water molecules can affect the interaction of protein and ligand [38], the solvent effects of the explicit water molecules in the binding site were further explored. As shown in Fig. 9, the water molecules were monitored at 5 ns, 10 ns and 15 ns in the monomer-3 of wild type and mutated HA. From this figure, it can be seen that there are more water molecules in the binding pocket in the wild type HA, indicating that the binding pocket of wild type HA is more hydrophilic that that of D222G mutant. Furthermore, in the wild type HA, residue 222 can form a weak hydrogen bond with glycan using a water molecule as a bridge for a long time. However, the D222G mutation makes this hydrogen bond disappear completely. Although the hydrogen bond interaction decreased, the increased hydrophobicity makes the mutated HA form stronger hydrophobic interactions with the glycan, leading to the increased overall binding affinity of mutated HA relative to wild type.

Themonitoring ofwatermoleculesof binding pocket in the monomer-3 ofwild type HA and D222G mutant: (a)7WT HA at 5 ns; (b) MT HA at 5 ns;(c) WT HA at 10 ns; (d) MT HA at10 ns; (e) WT HA at 15 ns; (f) MT HAat 15 ns. (red sphere are water)
Conclusions
In this study, to explore the altered receptor binding mechanism of HA, molecular dynamics simulation and binding free energy calculations were performed to study the interaction of the α-2,3 linked glycan with the wild type and D222G mutated HA. To make the simulation reflect the real state, a large trimer of HA was used and 15 ns molecular dynamics simulation for each system was performed. The MM-GBSA calculations based on the molecular dynamics simulation trajectories indicate that the D222G mutated HA has a −1.46 kcal mol−1 binding free energy with the studied α-2,3 linked glycan while the binding free energy of the wild type HA with glycan is 2.79 kcal mol−1. This result is consistent with the experimental result that the D222G mutated HA is able to bind the α2,3-linked sialic acid while the wild type HA can not. By comparing the residue contribution profile, we find that the increased binding ability of D222G mutant mainly comes from the increased contribution of Gln223. Furthermore structural analysis suggests the altered electrostatic potential of receptor binding domain (RBD) and the flexibility of 220-loop should be responsible for the increased affinity of HA to α2,3-linked sialic acid. The obtained results of this study have allowed a deeper understanding of the receptor recognition mechanism and the pathogenicity of influenza virus, which will be valuable to the structure-based inhibitors design targeting influenza virus entry process.
References
Perez-Padilla R, de la Rosa-Zamboni D, Ponce de Leon S, Hernandez M, Quinones-Falconi F, Bautista E, Ramirez-Venegas A, Rojas-Serrano J, Ormsby CE, Corrales A, Higuera A, Mondragon E, Cordova-Villalobos JA (2009) Pneumonia and respiratory failure from swine-origin influenza A (H1N1) in Mexico. N Engl J Med 361(7):680–689
Rogers GN, Paulson JC, Daniels RS, Skehel JJ, Wilson IA, Wiley DC (1983) Single amino acid substitutions in influenza haemagglutinin change receptor binding specificity. Nature 304(5921):76–78
Preliminary review of D222G amino acid substitution in the haemagglutinin of pandemic influenza A (H1N1) 2009 viruses (2010). Wkly Epidemiol Rec 85 (4):21–22
Puzelli S, Facchini M, De Marco MA, Palmieri A, Spagnolo D, Boros S, Corcioli F, Trotta D, Bagnarelli P, Azzi A, Cassone A, Rezza G, Pompa MG, Oleari F, Donatelli I (2010) Molecular surveillance of pandemic influenza A(H1N1) viruses circulating in Italy from May 2009 to February 2010: association between haemagglutinin mutations and clinical outcome. Euro Surveill 15 (43):pii = 19696
Kilander A, Rykkvin R, Dudman SG, Hungnes O (2010) Observed association between the HA1 mutation D222G in the 2009 pandemic influenza A(H1N1) virus and severe clinical outcome, Norway 2009–2010. Euro Surveill 15 (9):pii = 19498
Stevens J, Blixt O, Glaser L, Taubenberger JK, Palese P, Paulson JC, Wilson IA (2006) Glycan microarray analysis of the hemagglutinins from modern and pandemic influenza viruses reveals different receptor specificities. J Mol Biol 355(5):1143–1155
Chan PK, Lee N, Joynt GM, Choi KW, Cheung JL, Yeung AC, Lam P, Wong R, Leung BW, So HY, Lam WY, Hui DC (2011) Clinical and virological course of infection with haemagglutinin D222G mutant strain of 2009 pandemic influenza A (H1N1) virus. J Clin Virol 50(4):320–324
Chutinimitkul S, Herfst S, Steel J, Lowen AC, Ye J, van Riel D, Schrauwen EJ, Bestebroer TM, Koel B, Burke DF, Sutherland-Cash KH, Whittleston CS, Russell CA, Wales DJ, Smith DJ, Jonges M, Meijer A, Koopmans M, Rimmelzwaan GF, Kuiken T, Osterhaus AD, Garcia-Sastre A, Perez DR, Fouchier RA (2010) Virulence-associated substitution D222G in the hemagglutinin of 2009 pandemic influenza A(H1N1) virus affects receptor binding. J Virol 84(22):11802–11813
Liao HY, Hsu CH, Wang SC, Liang CH, Yen HY, Su CY, Chen CH, Jan JT, Ren CT, Cheng TJ, Wu CY, Wong CH (2010) Differential receptor binding affinities of influenza hemagglutinins on glycan arrays. J Am Chem Soc 132(42):14849–14856
Newhouse EI, Xu D, Markwick PR, Amaro RE, Pao HC, Wu KJ, Alam M, McCammon JA, Li WW (2009) Mechanism of glycan receptor recognition and specificity switch for avian, swine, and human adapted influenza virus hemagglutinins: a molecular dynamics perspective. J Am Chem Soc 131(47):17430–17442
Das P, Li J, Royyuru AK, Zhou R (2009) Free energy simulations reveal a double mutant avian H5N1 virus hemagglutinin with altered receptor binding specificity. J Comput Chem 30(11):1654–1663
Nunthaboot N, Rungrotmongkol T, Malaisree M, Kaiyawet N, Decha P, Sompornpisut P, Poovorawan Y, Hannongbua S (2010) Evolution of human receptor binding affinity of H1N1 hemagglutinins from 1918 to 2009 pandemic influenza A virus. J Chem Inf Model 50(8):1410–1417
Xu D, Newhouse EI, Amaro RE, Pao HC, Cheng LS, Markwick PR, McCammon JA, Li WW, Arzberger PW (2009) Distinct glycan topology for avian and human sialopentasaccharide receptor analogues upon binding different hemagglutinins: a molecular dynamics perspective. J Mol Biol 387(2):465–491
Liu H, Yao X (2010) Molecular basis of the interaction for an essential subunit PA-PB1 in influenza virus RNA polymerase: insights from molecular dynamics simulation and free energy calculation. Mol Pharmaceutics 7(1):75–85
Liu H, Yao X, Wang C, Han J (2010) In silico identification of the potential drug resistance sites over 2009 influenza A (H1N1) virus neuraminidase. Mol Pharmaceutics 7(3):894–904
Dirauf P, Meiselbach H, Sticht H (2010) Effects of the V82A and I54V mutations on the dynamics and ligand binding properties of HIV-1 protease. J Mol Model 16(10):1577–1583
Pan D, Sun H, Bai C, Shen Y, Jin N, Liu H, Yao X (2011) Prediction of zanamivir efficiency over the possible 2009 influenza A (H1N1) mutants by multiple molecular dynamics simulations and free energy calculations. J Mol Model 17(10):2465–2473
Yang H, Carney P, Stevens J (2010) Structure and Receptor binding properties of a pandemic H1N1 virus hemagglutinin. PLoS Curr 2:RRN1152
Gamblin SJ, Haire LF, Russell RJ, Stevens DJ, Xiao B, Ha Y, Vasisht N, Steinhauer DA, Daniels RS, Elliot A, Wiley DC, Skehel JJ (2004) The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science 303(5665):1838–1842
DeLano WL (2002) The PyMOL Molecular Graphics System, DeLano Scientific. Palo Alto, CA. http://www.pymol.org
Xu R, McBride R, Nycholat CM, Paulson JC, Wilson IA (2011) Structural characterization of the hemagglutinin receptor specificity from the 2009 H1N1 influenza pandemic. J Virol doi:JVI.06322-11
Case DA, Cheatham TE 3rd, Darden T, Gohlke H, Luo R, Merz KM Jr, Onufriev A, Simmerling C, Wang B, Woods RJ (2005) The Amber biomolecular simulation programs. J Comput Chem 26(16):1668–1688
Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65(3):712–725
Kirschner KN, Yongye AB, Tschampel SM, Gonzalez-Outeirino J, Daniels CR, Foley BL, Woods RJ (2008) GLYCAM06: a generalizable biomolecular force field. Carbohydrates J Comput Chem 29(4):622–655
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79(2):926–935
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG (1995) A smooth particle mesh Ewald method. J Chem Phys 103(19):8577–8593
Ryckaert J-P, Ciccotti G, Berendsen HJC (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23(3):327–341
Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR (1984) Molecular dynamics with coupling to an external bath. The J Chem Phys 81(8):3684–3690
Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE 3rd (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 33(12):889–897
Srinivasan J, Cheatham TE, Cieplak P, Kollman PA, Case DA (1998) Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate − DNA helices. J Am Chem Soc 120(37):9401–9409
Hou T, Wang J, Li Y, Wang W (2011) Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model 51(1):69–82
Sitkoff D, Sharp KA, Honig B (1994) Accurate calculation of hydration free energies using macroscopic solvent models. J Phys Chem 98(7):1978–1988
Chong LT, Duan Y, Wang L, Massova I, Kollman PA (1999) Molecular dynamics and free-energy calculations applied to affinity maturation in antibody 48 G7. Proc Natl Acad Sci USA 96(25):14330–14335
Shao J, Tanner SW, Thompson N, Cheatham TE (2007) Clustering molecular dynamics trajectories: 1. Characterizing the performance of different clustering algorithms. J Chem Theor Comput 3(6):2312–2334
Strockbine B, Rizzo RC (2007) Binding of antifusion peptides with HIVgp41 from molecular dynamics simulations: quantitative correlation with experiment. Proteins 67(3):630–642
Raju RK, Burton NA, Hillier IH (2010) Modelling the binding of HIV-reverse transcriptase and nevirapine: an assessment of quantum mechanical and force field approaches and predictions of the effect of mutations on binding. Phys Chem Chem Phys 12(26):7117–7125
McCammon JA (2009) Darwinian biophysics: electrostatics and evolution in the kinetics of molecular binding. Proc Natl Acad Sci USA 106(19):7683–7684
García-Sosa AT, Mancera RL (2010) Free energy calculations of mutations involving a tightly bound water molecule and ligand substitutions in a ligand-protein complex. Mol Inform 29(8–9):589–600
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant Nos: 20905033 and 21103075). The authors also would like to thank the Super-computing Center of Gansu Province for providing the computing resources.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Pan, D., Xue, W., Wang, X. et al. Molecular mechanism of the enhanced virulence of 2009 pandemic Influenza A (H1N1) virus from D222G mutation in the hemagglutinin: a molecular modeling study. J Mol Model 18, 4355–4366 (2012). https://doi.org/10.1007/s00894-012-1423-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-012-1423-2