
Abstract
The innate immune system constitutes the first line of defence against microorganisms and plays a primordial role in the activation and regulation of adaptive immunity. The innate immune system is composed of a cellular arm and a humoral arm. Components of the humoral arm include members of the complement cascade and soluble pattern recognition molecules (PRMs). These fluid-phase PRMs represent the functional ancestors of antibodies and play a crucial role in the discrimination between self, non-self and modified-self. Moreover, evidence has been presented that these soluble PRMs participate in the regulation of inflammatory responses and interact with the cellular arm of the innate immune system. Pentraxins consist of a set of multimeric soluble proteins and represent the prototypic components of humoral innate immunity. Based on the primary structure of the protomer, pentraxins are divided into two groups: short pentraxins and long pentraxins. The short pentraxins C-reactive protein and serum amyloid P-component are produced by the liver and represent the main acute phase proteins in human and mouse, respectively. The long pentraxin PTX3 is produced by innate immunity cells (e.g. PMN, macrophages, dendritic cells), interacts with several ligands and plays an essential role in innate immunity, tuning inflammation and matrix deposition. PTX3 provides a paradigm for the mode of action of humoral innate immunity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The mammalian immune system is organized around two components: innate and adaptive immunity. The innate immune system is older in terms of evolution and constitutes the first line of defence against microorganisms. In vertebrates, this system is complemented by adaptive immunity, a more recent feature in terms of evolution; adaptive immunity provides the basis of immunological memory and involves the use of specific receptors.
Organisms are constantly in contact with microbes and our ability to induce a protective immune response resides in our competence to identify potential pathogens. The molecules involved in this recognition are germline-encoded receptors called pattern recognition molecules (PRMs). In essence, these receptors specifically recognize highly conserved motifs called pathogen-associated molecular patterns (PAMPs) expressed by microorganisms (Iwasaki and Medzhitov 2010). Based on their localization, PRMs are divided into the cell-associated receptors, such as the scavenger receptors (Murphy et al. 2005) and the toll-like receptors (TLR; Akira et al. 2006), and the fluid-phase molecules (Bottazzi et al. 2010). Fluid-phase molecules (collectins, ficolins and pentraxins) constitute the humoral arm of the immune system and represent the functional ancestors of antibodies (Bottazzi et al. 2010; Endo et al. 2007; Holmskov et al. 2003).
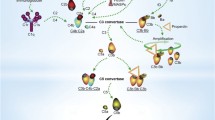
Pentraxins are a pivotal component of the innate immune system (Fig. 1). During the 1930s, C-reactive protein (CRP) was the first PRM to be identified and purified from the serum of infected patients (Abernethy and Avery 1941; Tillett and Francis 1930). Here, we review the key interactions and functions of pentraxins in innate immunity. In particular, we focus our attention on the prototypic long pentraxin 3 (PTX3), whose regulation has been conserved in evolution.

Schematic overview of pentraxins in innate immunity (IL interleukin, TLR toll-like receptor, TNF tumour necrosis factor, CRP C-reactive protein, SAP serum amyloid P-component, PTX pentraxin)
The pentraxin superfamily
Pentraxins constitute a superfamily of multifunctional multimeric proteins that are phylogenetically conserved from arachnids to mammals. All members of the family contain, in their carboxy-terminal, the “pentraxin domain”, which is characterized by a conserved 8-amino-acid long sequence (HxCxS/TWxS, where x is any amino acid) called the “pentraxin signature” (Garlanda et al. 2005). Based on the primary structure of the protomer, pentraxins are divided into two groups: short pentraxins and long pentraxins.
CRP and serum amyloid P-component (SAP) are prototypic short pentraxins. Following the identification of CRP, human SAP was identified as its relative on the basis of amino acid sequence identity (51%). Short pentraxins are 25-kDa proteins characterized by a common structural organization in five or ten identical subunits arranged with pentameric radial symmetry (Emsley et al. 1994; Rubio et al. 1993). CRP and SAP orthologues in various mammal species share substantial sequence similarity but present notable differences in serum basal levels and changes during the acute phase response (e.g. CRP and SAP are the main acute-phase reactants in human and mouse, respectively). Circulating CRP is produced by hepatocytes, mainly in response to the proinflammatory cytokine, interleukin-6 (IL-6). IL-1 might also contribute as an additional CRP-inducing signal acting synergistically with IL-6 (Pepys and Hirschfield 2003). SAP is produced exclusively by hepatocytes and is the main acute-phase protein in mice, whereas in human serum, it is constitutively present at 30–50 mg/l (Emsley et al. 1994; Fig. 1).
During the early 1990s, PTX3, a new pentraxin-domain-containing secreted protein, was identified and classified as a prototypic long pentraxin (Breviario et al. 1992; Lee et al. 1993). Components of the subfamily of long pentraxins differ from short pentraxins in their gene organization, chromosomal localization, cellular sources, inducing stimuli and ligand-recognition ability. The long pentraxins identified after PTX3, as inducible genes or molecules expressed in specific tissues (e.g. neurons, spermatozoa), include guinea pig apexin, neuronal pentraxin (NPTX) 1 or NP1, NPTX2 (also called Narp or NP2) and neuronal pentraxin receptor (NPTXR), a transmembrane molecule (for a review, see Bottazzi et al. 2010). Orthologue molecules of long pentraxins have also been found in lower vertebrates such as zebrafish and puffer-fish (Martinez de la Torre et al. 2010). Long pentraxins have additionally been identified in Xenopus laevis (XL-PXN1; Seery et al. 1993). In an attempt to find new pentraxin-domain-containing proteins, we have recently identified a new long pentraxin, which we have named PTX4. As described for the long pentraxins, PTX4 is characterized by an unrelated N-terminal domain coupled to a C-terminal pentraxin domain. Analysis of PTX4 in silico and by transcript expression shows that the gene is well conserved from mammals to lower vertebrates. Moreover, PTX4 has a unique pattern of mRNA expression, distinct from that of other members of the family (Martinez de la Torre et al. 2010).
Short pentraxins in innate immunity
The first ligand described for CRP was the C-polysaccharide of Streptococcus pneumoniae: this interaction is attributable to the direct binding of CRP to phosphorylcholine, a major constituent of C-type capsule polysaccharides (Tillett and Francis 1930). Moreover, CRP binds various microbes, including fungi, yeasts, bacteria and parasites through phosphorylcholine and carbohydrate structures, promoting phagocytosis and resistance to infections (Szalai 2002). However, binding to bacteria is not always necessary for protection, because CRP also protects mice from infection with Salmonella typhimurium, a pathogen to which CRP does not bind (Szalai et al. 2000). Like CRP, SAP binds various pathogens but these interactions have contrasting consequences on innate immunity (Noursadeghi et al. 2000; Yuste et al. 2007). SAP has been reported to bind to phosphorylethanolamine and to lipopolysaccharide (LPS) of several bacteria and prevents LPS-mediated complement activation and LPS toxicity (de Haas et al. 2000; Schwalbe et al. 1992). In addition, SAP binds S. pneumoniae and plays an important role in the complement-mediated immune response to this pathogen (Yuste et al. 2007). However, for certain organisms to which SAP binds, such as Streptococcus pyogenes and rough strains of Escherichia coli, SAP enhances virulence by protecting the bacteria against phagocytosis, whereas it is protective in infection with organisms to which it does not bind, for instance Listeria monocytogenes (Noursadeghi et al. 2000; Table 1). The antimicrobial activity of the short pentraxins has been thoroughly reviewed elsewhere (Garlanda et al. 2005; Szalai 2002). In the arthropod Limulus polyphemus, diverse forms of CRP and SAP have been identified as abundant constituents of the haemolymph involved in recognizing and destroying pathogens (Armstrong et al. 1996; Robey and Liu 1981; Shrive et al. 1999).
CRP and SAP, aggregated or attached to most of their ligands, interact with the globular head modules of complement component C1q (Roumenina et al. 2006). Complement activation by short pentraxins may be one of the mechanisms leading to the removal of cellular debris (Nauta et al. 2003b). When bound to self surfaces (e.g. apoptotic cells, damaged tissue), CRP activates the classical pathway of complement through interaction with C1q but this activation is restricted to the initial stages with only little consumption of C5-C9 (Gershov et al. 2000). Surface-bound CRP has been shown to decrease the alternative pathway C3-convertase activity, to inhibit the alternative pathway amplification loop and to reduce the deposition of C3b and lysis by the lectin pathway (Mold and Gewurz 1981; Suankratay et al. 1998). The inhibition of the alternative pathway amplification loop is, at least in part, a consequence of the interaction of CRP with Factor H (Jarva et al. 1999; Mihlan et al. 2009; Okemefuna et al. 2010). In addition to its roles in the activation of classical and alternative complement pathways, CRP has been shown to interact with Ficolin-2 (L-ficolin). Immobilized CRP recruits Ficolin-2, leading to MASP-2 activation and deposition of C4. Likewise, immobilized Ficolin-2 can recruit CRP, suggesting that Ficolin-2 widens the range of bacteria opsonized by CRP (Ng et al. 2007). Indeed, a recent study has shown that infection-induced local inflammatory conditions trigger a strong interaction between CRP and Ficolin-2; this elicits complement amplification and enhances antimicrobial activity of the classical and lectin pathways (Zhang et al. 2009; Table 1). The interaction of CRP with Ficolin-1 (M-ficolin) has been recently reported but the functional significance of this interaction has not been investigated (Tanio et al. 2009).
A specific and saturable binding to all three classes of Fcγ receptors (FcγR) has been demonstrated for both CRP and SAP, the interaction with FcγR being able to mediate not only the phagocytosis of apoptotic cells and microbes, but also protective immune responses (Bharadwaj et al. 1999; Mold et al. 2001). However, the interpretation of these data has been questioned, suggesting that binding may be influenced by CRP contamination with traces of IgG (Saeland et al. 2001). The crystal structure of human SAP in complex with the extracellular domain of FcγRIIa has recently been solved. The structural mechanism for pentraxin binding to FcγRs followed by the functional activation of FcγR-mediated phagocytosis and cytokine secretion has been described; this study suggests that pentraxins and IgG share the binding site and that pentraxins inhibit immune-complex-mediated phagocytosis (Lu et al. 2008). Thus, pentraxins possess similar functions to those of antibodies that activate both the complement and FcγR pathways (Table 1).
The long pentraxin PTX3
Structure and expression
The human PTX3 gene, which is organized in three exons and localized on the chromosome 3q25, encodes for a transcript of 1861 bp. The first two exons encode for the signal peptide and the N-terminal domain (amino acids 18-179), respectively, whereas the third exon encodes for the C-terminal domain containing the pentraxin signature (amino acids 179-381). The murine gene presents the same organization and is located on chromosome 3 (Bottazzi et al. 2010). The primary structure of PTX3 indicates the presence of a unique N-linked glycosylation site in the C-terminal domain at position Asn220. Indeed, investigations have demonstrated the presence of a heterogeneous glycosidic moiety with bi-, tri- and tetra-antennary sugar structures (Inforzato et al. 2006). PTX3 is composed of eight identical protomers associated via disulphide bonds (Inforzato et al. 2008), forming an elongated asymmetric molecule with two differently sized domains interconnected by a stalk. The N-terminal domain provides the main structural determinant underlying this quaternary organization, supporting formation of a disulphide-linked tetramer and a dimer of dimers (a non-covalent tetramer) giving rise to the asymmetry of the molecule (Inforzato et al. 2010).
High homology is found between the human and murine PTX3 promoters in which several potential enhancer-binding elements are found, i.e. activator protein-1 (AP-1), nuclear factor kappa B (NF-κB), selective promoter factor 1 (SP1; Altmeyer et al. 1995; Basile et al. 1997). The NF-κB binding site is involved in the response to the proinflammatory cytokines tumour necrosis factor-α (TNF-α) and IL-1β, whereas AP-1 promotes the basal transcription of PTX3 (Altmeyer et al. 1995; Basile et al. 1997). Other pathways might be involved in the production of PTX3. Indeed, in lung epithelial cells, the production of PTX3 induced by TNF-α does not require NF-κB but relies on the c-Jun N-terminal kinase pathway (Han et al. 2005).
A recent study reports that glucocorticoid hormones (GC) can modulate PTX3 expression in a cell-dependent manner. Indeed, whereas GC induce and amplify the production of PTX3 in fibroblasts and endothelial cells, GC treatment suppresses its production by haematopoietic cells (dendritic cells and macrophages). Moreover, the in vivo administration of GC increases the blood level of PTX3 and patients with Cushing’s syndrome exhibit increased levels of circulating PTX3 (Doni et al. 2008).
PTX3 was originally identified as a cytokine-inducible gene in vascular endothelial cells and fibroblasts (Breviario et al. 1992; Lee et al. 1993). A variety of cell types express PTX3 upon exposure to inflammatory signals, such as cytokines (e.g. IL-1β, TNF-α), TLR agonists, microbial moieties (e.g. LPS, OmpA, lipoarabinomannans) or microorganisms (Bottazzi et al. 2009; Fig. 1). Whereas myeloid dendritic cells are a major source of PTX3, this molecule is also expressed by fibroblasts, endothelial cells, monocytes, macrophages, smooth muscle cells, kidney epithelial cells, synovial cells, chondrocytes, adipocytes and alveolar epithelial cells (Abderrahim-Ferkoune et al. 2003; Breviario et al. 1992; Doni et al. 2003; dos Santos et al. 2004; Goodman et al. 2000; Han et al. 2005; Introna et al. 1996; Nauta et al. 2005). In contrast, the expression of PTX3 is undetectable in T and B lymphocytes, natural killer cells or plasmacytoid dendritic cells (Alles et al. 1994; Doni et al. 2003). Interestingly, interferon-γ (IFNγ) reduces the production of PTX3 by dendritic cells, monocytes and macrophages through the inhibition of its transcription and the reduction of the transcript stability (Doni et al. 2006; Goodman et al. 2000; Polentarutti et al. 1998). On the other hand, IL-10 amplifies LPS-induced PTX3 production (Doni et al. 2006). Given the role that IL-10 plays in the resolution of inflammation, this result suggests that PTX3 participates in tissue repair and remodelling (the function of PTX3 in extracellular matrix has been reviewed elsewhere; Deban et al. 2009). Although the expression of PTX3 mRNA is absent in neutrophils (Alles et al. 1994), we have found that PTX3 is stored in specific granules in a ready-to-use form and undergoes release in response to microorganisms or TLR agonists (Jaillon et al. 2007). PTX3 mRNA expression is confined to immature myeloid cells. Moreover, PTX3 is found associated with neutrophil extracellular traps generated upon neutrophil stimulation and neutrophil-associated-PTX3 promotes the in vivo control of Aspergillus fumigatus infection (Jaillon et al. 2007).
Pathogen recognition by PTX3
Among the first functions reported for PTX3 was its ability to bind certain pathogens. Indeed, PTX3 binds conidia of A. fumigatus, selected gram-positive and gram-negative bacteria (Staphylococcus aureus, Pseudomonas aeruginosa, Klebsiella pneumoniae, S. typhimurium), Paracoccidioides brasiliensis and several viral strains (Bozza et al. 2006; Diniz et al. 2004; Garlanda et al. 2002; Jeannin et al. 2005; Reading et al. 2008). In contrast, PTX3 does not bind Candida albicans or Burkholderia cepacia (Garlanda et al. 2002). Genetically modified mice have allowed the exploration of the in vivo functions of PTX3. Ptx3 -/- mice are highly susceptible to invasive pulmonary aspergillosis (Garlanda et al. 2002). This susceptibility is associated with a low protective T helper cell type 1 (Th1) antifungal response coupled with an inappropriate Th2 response. Treatment with recombinant PTX3 restores the protective Th1 response demonstrating that PTX3 can participate in the tuning of immune responses (Garlanda et al. 2002; Gaziano et al. 2004). PTX3-deficient neutrophils, dendritic cells and alveolar macrophages exhibit defective recognition and killing of conidia. Treatment with recombinant PTX3 or neutrophil-associated PTX3 reverses this phenotype (Garlanda et al. 2002; Jaillon et al. 2007). Moreover, in an experimental model of chronic granulomatous disease (p47phox-/- mice), PTX3 limits the Th17 response and the pathogenic inflammation induced by A. fumigatus infection. In both p47phox-/- and p47phox+/+ mice, PTX3 reduces neutrophil counts and mononuclear cell recruitment to the lung parenchyma and bronchoalveolar lavage fluid. Furthermore, in p47phox-/- mice infected by A. fumigatus, PTX3 modulates cytokine production and adaptive immunity through the enhancement of a Th1/Treg response associated with the restriction of a Th2/Th17 response (D'Angelo et al. 2009). Recent investigations strongly suggest that PTX3 acts as an opsonin, enhancing the recognition and phagocytosis of conidia by neutrophils through a Fcγ receptor II (FcγRII)- and complement-dependent mechanism. Indeed, through its binding to FcγRIIA/CD32, PTX3 promotes the activation of CD11b and thus the phagocytosis of C3b-opsonized A. fumigatus conidia (F. Moalli, unpublished data).
Ptx3-overexpressing macrophages exhibit greater phagocytosis of zymosan and P. brasiliensis than macrophages from wild-type mice (Diniz et al. 2004). Interestingly, in response to zymosan, ptx3-transgenic macrophages have an enhanced expression of dectin-1, the cellular receptor primarily involved in the interaction between macrophages and zymosan. Moreover, since zymosan induces PTX3 expression, dectin-1 up-regulation promotes a positive feedback for this phagocytosis (Diniz et al. 2004).
The recognition of a microbial component by PTX3 can also amplify the inflammatory response, as reported for the interaction between PTX3 and the outer membrane protein A of K. pneumoniae (KpOmpA). KpOmpA is recognized by scavenger receptors lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) and scavenger receptor expressed by endothelial cell-I (SREC-1) and induces a pro-inflammatory response through the collaboration between these receptors and TLR2. KpOmpA induces the expression of PTX3 by dendritic cells and monocytes, which, in turn, bind this microbial component (Jeannin et al. 2005). In contrast to zymosan, PTX3 does not enhance the in vitro recognition of KpOmpA or the cell activation induced by KpOmpA. However, in vivo PTX3 improves the local inflammation induced by KpOmpA, in terms of cell recruitment and proinflammatory cytokine production (Jeannin et al. 2005; Table 1). This effect is complement-dependent and is abrogated after treatment with complement inhibitors. Furthermore, this amplification loop is not a general mechanism, since PTX3 does not modify the inflammatory response induced by LPS, a microbial moiety not recognized by PTX3 (Cotena et al. 2007).
PTX3 is also involved in resistance against some viral infections. Indeed, PTX3 binds both human and murine cytomegalovirus (HCMV and MCMV, respectively) and reduces the viral infection of dendritic cells in vitro. Accordingly, ptx3 -/- mice present a higher susceptibility to infections than wild-type mice and the viral titre is reduced upon treatment with recombinant PTX3. Moreover, PTX3 protects MCMV-infected mice from an A. fumigatus superinfection and enhances the production of IL-12 and IFN-γ by dendritic cells and T cells, respectively. The protective effect of PTX3 is lost in TLR3-/ -, TLR2-/ - or TLR4-/ - mice but is still efficient in TLR9-/ - and MyD88-/ - mice and reflects the activation of interferon regulatory factor 3 (IRF3) mediated by TLR9/MyD88-independent viral recognition sensing (Bozza et al. 2006).
Finally, human and murine PTX3 binds influenza virus (H3N2) through the interaction between the viral haemagglutinin glycoprotein and the sialic acid residue present on PTX3. PTX3 inhibits virus-induced haemagglutination and viral neuramidase activity and neutralizes the virus infectivity. Consistently, ptx3 -/- mice are more susceptible than wild-type mice to influenza virus infection. Treatment with recombinant PTX3 reduces mortality and the viral load (Reading et al. 2008).
Interaction of PTX3 with complement components
The first described and best characterized ligand of PTX3 is the complement component C1q. Unlike classical pentraxins, PTX3 interacts with C1q in a calcium-independent manner, without previous aggregation of the protein. Interaction of PTX3 with plastic-immobilized C1q induces the activation of the classical complement pathway, as demonstrated by an increased deposition of C3 and C4. In contrast, fluid-phase PTX3 binding to C1q inhibits complement activation via competitive blocking of relevant interaction sites. These data indicate that PTX3 exerts a dual role in classical pathway activation, depending on the way that C1q is presented (Bottazzi et al. 1997; Nauta et al. 2003a). This hypothesis is supported by the observation that PTX3 enhances the deposition of both C1q and C3 on the cell surface when incubated with apoptotic cells. On the contrary, in the fluid phase, PTX3 reduces C1q and C3 deposition on apoptotic cells and C1q-mediated phagocytosis of apoptotic cells by dendritic cells (Baruah et al. 2006a; Table 1). Furthermore, PTX3 desialylation enhances the activation of the classical complement pathway, suggesting the involvement of the PTX3 sugar moiety in C1q recognition and complement activation (Inforzato et al. 2006).
Recently, PTX3 has been shown to interact with Ficolin-2, similarly to CRP. Indeed, Ficolin-2 could be affinity-isolated from human plasma on immobilized PTX3. In binding studies, Ficolin-1 and particularly Ficolin-2 interact with PTX3 in a calcium-independent manner. Ficolin-2, but not Ficolin-1 and Ficolin-3, binds A. fumigatus directly and this binding is enhanced by PTX3 and vice versa. Likewise, the Ficolin-2-dependent deposition of complement components on the surface of A. fumigatus is enhanced by PTX3. This effect is alleviated by a common amino acid change in the fibrinogen-like domain of Ficolin-2. Moreover, a polymorphism in the FCN2 gene causing a T236M amino acid change in the fibrinogen-like binding domain, which affects binding to GlcNAc, reduces Ficolin-2 binding to PTX3 (Ma et al. 2009). PTX3 and Ficolin-2 may recruit each other to the surface of recognized microbes and amplify synergistically complement-mediated innate response (Table 1). Thus, two distinct components of the humoral innate immune system (pentraxins and ficolins), which activate different complement pathways, cooperate and amplify microbial recognition and antimicrobial effector functions.
In addition to the interaction with the classical and lectin complement cascades, PTX3 has recently been observed to be able to interact with Factor H, the main soluble regulator of the alternative pathway of complement activation. PTX3-bound Factor H remains functionally active and PTX3 enhances Factor H and iC3b deposition on apoptotic cells. These observations suggest that the interaction of PTX3 with Factor H modulates alternative pathway activation by promoting Factor H deposition on PTX3-coated surfaces and preventing exaggerated complement activation (Deban et al. 2008; Table 1). Together, the above observations suggest a dual role for PTX3 in the regulation of complement-mediated immune responses. Interactions with key components of all three complement pathways (C1q, Ficolin-2, Factor H) identify PTX3 as an important component of the complex network controlling complement functions.
Self and modified-self discrimination
In addition to PAMP recognition, PRMs bind apoptotic cells and modulate their clearance by phagocytes (Jeannin et al. 2008). Both short pentraxins and PTX3 are involved in this process but promote two opposite effects. Indeed, whereas CRP and SAP recognize dying cells and enhance their elimination (Gillmore et al. 2004; Pepys and Hirschfield 2003), the opsonization of apoptotic cells by PTX3 inhibits their clearance (Gershov et al. 2000; van Rossum et al. 2004). Moreover, PTX3 inhibits the cross-presentation of apoptotic-cell-derived antigens to CD8+ T cells (Baruah et al. 2006a; Table 1).
Interestingly, PTX3 prevents the binding of C1q to apoptotic cells and thus inhibits their clearance mediated by C1q (Baruah et al. 2006a). In addition, PTX3 interacts with Factor H, favouring its deposition on apoptotic cells and thus playing a role as the modulator of the alternative pathway of complement activation in the context of injured tissues (Deban et al. 2008). Given that the binding of Factor H to dying cells can protect them against complement-mediated lysis (Trouw et al. 2007), PTX3 might promote the clearance of apoptotic cells in an anti-inflammatory context. Notably, this mechanism has also been described for CRP (Gershov et al. 2000; Jarva et al. 1999; Mihlan et al. 2009; Okemefuna et al. 2010).
Recently, we have reported that the sequential fusion of neutrophil granules with the cell membrane occurs during the apoptotic process. This fusion leads to the translocation of PTX3 from granules to the surface of apoptotic neutrophils. PTX3 accumulation on the apoptotic blebs is a result of an active process and acts as a late “eat me” signal for the recognition of apoptotic neutrophils by macrophages (Jaillon et al. 2009). Collectively, these results underline the dichotomy between soluble PTX3, which inhibits the clearance of dying cells, and membrane-associated PTX3, which promotes the elimination of apoptotic neutrophils. This difference might have physiological significance. Indeed, soluble PTX3 secreted following a proinflammatory signal may lead to an avoidance of the capture of dying cells in an inflammatory context and thus prevent the initiation of an immune response against self-antigens (Jeannin et al. 2008). In contrast, membrane-associated PTX3 promotes the phagocytosis of late apoptotic neutrophils and thus enhances their elimination before the loss of their cell-membrane permeability and the release of self-antigens and alarmins such as high-mobility group box 1 (Jaillon et al. 2009; Poon et al. 2010).
PTX3 in inflammation and leucocyte recruitment
PTX3 behaves as an acute phase response protein since its blood levels, which are low under normal conditions (about 25 ng/ml in the mouse, <2 ng/ml in man), increase rapidly (peak at 6-8 h) and dramatically (200-800 ng/ml in human and mouse) during endotoxic shock, sepsis and other inflammatory and infectious conditions, correlating with the severity of the disease (Mantovani et al. 2008). PTX3 transgenic mice show increased resistance to LPS toxicity and to cecal ligation and puncture (Dias et al. 2001). In a model of kainate-induced seizures, PTX3-deficient mice have more widespread and severe IL-1-induced neuronal damage. In this model, PTX3 confers resistance to neurodegeneration, possibly by binding to dying neurons and rescuing them from otherwise irreversible damage (Ravizza et al. 2001). In a model of acute myocardial infarction caused by coronary artery ligation and reperfusion, ptx3−/− mice display increased myocardial damage associated with a greater no-reflow area, an increased neutrophil infiltration, a decreased number of capillaries, and an increased number of apoptotic cardiomyocytes, suggesting that PTX3 plays a non-redundant regulatory cardioprotective role in acute myocardial infarction in mice (Salio et al. 2008). Whether PTX3 plays a role in the progression of post-infarction left ventricular dysfunction and failure remains to be assessed. Similarly, in an atherosclerosis model, Norata et al. (2009) have recently found that PTX3 has atheroprotective effects in mice. Ptx3 expression increases in the vascular wall of apolipoprotein E (APO-E)-knockout mice from 3 to 18 months of age. Aortic lesions are significantly increased in double-knockout mice lacking both ptx3 and APO-E and in mice heterozygous for ptx3 compared with APO-E-deficient mice. In addition, mice lacking ptx3 show a more pronounced inflammatory profile in the vascular wall and increased macrophage accumulation within the plaque (Norata et al. 2009). Together, the above observations suggest a cardiovascular protective function of PTX3 through the modulation of the immuno-inflammatory balance in the cardiovascular system. In contrast to the protective role in cardiac ischaemia-reperfusion injury, PTX3 has divergent effects in the cascade of events leading to tissue inflammation and injury following intestinal ischaemia-reperfusion injury. Indeed, in this model, the absence of ptx3 is accompanied by greatly diminished tissue inflammation and decreased lethality after reperfusion (Souza et al. 2009). Thus, PTX3 has different roles in mediating reperfusion injury in the heart (protective, localized response) versus the intestine (deleterious, systemic response).
Recently, our group has shown that PTX3 selectively binds P-selectin via its N-linked glycosidic moiety (Table 1). Using three in vivo models of P-selectin-dependent inflammation (pleurisy, acid-induced acute respiratory distress syndrome and intravital microscopy analysis of mesenteric inflammation), we have demonstrated that exogenously administered PTX3 and endogenous PTX3 released from haematopoietic cells act as a negative feedback loop, preventing excessive P-selectin-dependent recruitment of neutrophils. PTX3 released from activated leucocytes function locally to dampen neutrophil recruitment and regulate inflammation (Deban et al. 2010). This PTX3 activity is reminiscent of the recently characterized glycosylation-dependent regulatory effect of antibodies on inflammation (Kaneko et al. 2006). Therefore, PTX3, which is an essential component of humoral innate immunity, and immunoglobulins, the humoral arm of adaptive immunity, share functional outputs, including complement activation, opsonization and the glycosylation-dependent regulation of inflammation.
PTX3 as a marker in human pathology
CRP has been extensively used clinically for over 75 years as a nonspecific systemic marker of infection, inflammation and tissue damage (Verma et al. 2005). Plasma levels of CRP are independently associated with risk of coronary heart disease, with an open question as to whether CRP is causally associated with this pathology or merely a marker of underlying atherosclerosis. Two recent genome-wide association studies have reported data that argue against a causal association of CRP with coronary heart disease. Indeed, CRP gene polymorphisms associated with a marked increase in CRP levels are not in themselves associated with an increased risk of ischaemic vascular disease (Elliott et al. 2009; Zacho et al. 2008).
The structural and functional similarity of PTX3 to the classic diagnostic pentraxin CRP has given rise to efforts aimed at assessing the usefulness of PTX3 as a marker in diverse human pathological conditions. The hypothesis driving such work is that PTX3, unlike CRP, might represent a rapid marker for the primary local activation of innate immunity and inflammation.
Increased levels of PTX3 have been observed in diverse infectious disorders including sepsis and septic shock, meningococcal disease, tuberculosis and dengue infection (Azzurri et al. 2005; Mairuhu et al. 2005; Mauri et al. 2010; Sprong et al. 2009). In all these conditions, PTX3 levels are correlated with disease severity and have a prognostic value. Furthermore, evidence has linked PTX3 levels to ischaemic heart disorders. PTX3 levels increase rapidly in acute myocardial infarction (AMI), reaching a peak at around 7 h after the onset of symptoms (Peri et al. 2000). In a series of 748 patients with ST-elevation AMI, PTX3, measured together with established markers including CRP, has emerged as the only independent predictor of mortality (Latini et al. 2004). Patients with arterial inflammation, who are eligible for coronary intervention, exhibit high concentrations of plasma PTX3; in particular, patients with unstable angina pectoris have PTX3 levels three times higher than the normal range (Inoue et al. 2007). Thus, PTX3 is a new candidate for being a prognostic marker in ischaemic heart disorders including AMI (Brugger-Andersen et al. 2009; Jenny et al. 2009; Latini et al. 2004; Matsui et al. 2010). Likewise, PTX3 has been suggested as a useful inflammatory biomarker for predicting cognitive impairment in elderly hypertensive patients (Yano et al. 2010). Conversely, the assessment of PTX3 as a marker of metabolic syndrome characterized by increased cardiovascular risk has produced controversial data and further studies with larger number of patients are required before definitive conclusions can be made (Miyaki et al. 2010; Ogawa et al. 2010; Zanetti et al. 2009). Increased levels of PTX3 have been observed in a restricted set of autoimmune disorders (e.g. in blood in small vessel vasculitis, in synovial fluid in rheumatoid arthritis), but not in others (e.g. systemic lupus erythematosus; Fazzini et al. 2001; Luchetti et al. 2000). In small vessel vasculitis, PTX3 levels are correlated with the clinical activity of the disease and represent a candidate marker for monitoring the disease (Fazzini et al. 2001). The presence of anti-PTX3 autoantibodies has been determined in patients with systemic lupus erythematosus. Correlation of anti-PTX3 antibody titre and disease activity remains to be confirmed (Augusto et al. 2009). Recent results show that pregnancy, a condition associated with the relevant involvement of inflammatory molecules at the implantation site, is associated with a slight increase in maternal circulating PTX3 levels compared with the non-pregnant condition. Higher maternal PTX3 levels have been observed in pregnancies complicated by preeclampsia, which represents the clinical manifestation of an endothelial dysfunction as part of an excessive maternal inflammatory response to pregnancy (Cetin et al. 2006; Rovere-Querini et al. 2006). PTX3 plasma and vaginal levels are increased during pregnancy complicated by spontaneous preterm delivery and, in particular, in the cases of placenta vasculopathy (Assi et al. 2007).
The general characteristic emerging from studies of PTX3 blood levels in human pathology is the rapidity of its increase compared with that of CRP, consistent with its original identification as an immediate early gene, together with a lack of correlation between levels of CRP and PTX3. The data collected so far in cases of diverse pathology indicate a correlation between PTX3 plasma levels and severity of disease, suggesting a possible role of PTX3 as a marker of pathology. Whether the impressive correlation with outcome and severity actually reflects a role in the pathogenesis of damage remains to be elucidated.
Concluding remarks
Pentraxins are components of the complex and complementary network of cellular and humoral pattern recognition receptors involved in the recognition and response to microbial elements and damaged tissues (Fig. 1). Whereas the regulation of expression of short pentraxins has diverged from mouse to man, the gene targeting of the prototypic, evolutionarily conserved, long pentraxin PTX3 has unequivocally defined the role of this molecule in innate immunity and inflammation. Recent progress has further defined the structure, regulation, microbial recognition and in vivo functions of PTX3. As a component of the humoral arm of innate immunity, PTX3 plays a role similar to that played by antibodies as the humoral arm of the adaptive immune system, including complement activation, opsonization and glycosylation-dependent regulation of inflammation. The available evidence suggests that PTX3 not only acts as a PRM, but can also function as a tuner of inflammatory reactions and may contribute to the discrimination between self, infectious non-self and modified-self. In addition, PTX3 may represent a useful marker of infectious and inflammatory (particularly cardiovascular) pathology.
References
Abderrahim-Ferkoune A, Bezy O, Chiellini C, Maffei M, Grimaldi P, Bonino F, Moustaid-Moussa N, Pasqualini F, Mantovani A, Ailhaud G, Amri EZ (2003) Characterization of the long pentraxin PTX3 as a TNFalpha-induced secreted protein of adipose cells. J Lipid Res 44:994–1000
Abernethy TJ, Avery OT (1941) The occurrence during acute infections of a protein not normally present in the blood. I. Distribution of the reactive protein in patients' sera and the effect of calcium on the flocculation reaction with C polysaccharide of pneumococcus. J Exp Med 73:173–182
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124:783–801
Alles VV, Bottazzi B, Peri G, Golay J, Introna M, Mantovani A (1994) Inducible expression of PTX3, a new member of the pentraxin family, in human mononuclear phagocytes. Blood 84:3483–3493
Altmeyer A, Klampfer L, Goodman AR, Vilcek J (1995) Promoter structure and transcriptional activation of the murine TSG-14 gene encoding a tumor necrosis factor/interleukin-1-inducible pentraxin protein. J Biol Chem 270:25584–25590
Armstrong PB, Swarnakar S, Srimal S, Misquith S, Hahn EA, Aimes RT, Quigley JP (1996) A cytolytic function for a sialic acid-binding lectin that is a member of the pentraxin family of proteins. J Biol Chem 271:14717–14721
Assi F, Fruscio R, Bonardi C, Ghidini A, Allavena P, Mantovani A, Locatelli A (2007) Pentraxin 3 in plasma and vaginal fluid in women with preterm delivery. Br J Obstet Gynaecol 114:143–147
Augusto JF, Onno C, Blanchard S, Dubuquoi S, Mantovani A, Chevailler A, Jeannin P, Subra JF (2009) Detection of anti-PTX3 autoantibodies in systemic lupus erythematosus. Rheumatology (Oxford) 48:442–444
Azzurri A, Sow OY, Amedei A, Bah B, Diallo S, Peri G, Benagiano M, D'Elios MM, Mantovani A, Del Prete G (2005) IFN-gamma-inducible protein 10 and pentraxin 3 plasma levels are tools for monitoring inflammation and disease activity in Mycobacterium tuberculosis infection. Microbes Infect 7:1–8
Baruah P, Dumitriu IE, Peri G, Russo V, Mantovani A, Manfredi AA, Rovere-Querini P (2006a) The tissue pentraxin PTX3 limits C1q-mediated complement activation and phagocytosis of apoptotic cells by dendritic cells. J Leukoc Biol 80:87–95
Baruah P, Propato A, Dumitriu IE, Rovere-Querini P, Russo V, Fontana R, Accapezzato D, Peri G, Mantovani A, Barnaba V, Manfredi AA (2006b) The pattern recognition receptor PTX3 is recruited at the synapse between dying and dendritic cells, and edits the cross-presentation of self, viral, and tumor antigens. Blood 107:151–158
Basile A, Sica A, d'Aniello E, Breviario F, Garrido G, Castellano M, Mantovani A, Introna M (1997) Characterization of the promoter for the human long pentraxin PTX3. Role of NF-kappaB in tumor necrosis factor-alpha and interleukin-1beta regulation. J Biol Chem 272:8172–8178
Bharadwaj D, Stein MP, Volzer M, Mold C, Du Clos TW (1999) The major receptor for C-reactive protein on leukocytes is Fcgamma receptor II. J Exp Med 190:585–590
Bickerstaff MC, Botto M, Hutchinson WL, Herbert J, Tennent GA, Bybee A, Mitchell DA, Cook HT, Butler PJ, Walport MJ, Pepys MB (1999) Serum amyloid P component controls chromatin degradation and prevents antinuclear autoimmunity. Nat Med 5:694–697
Bottazzi B, Vouret-Craviari V, Bastone A, De Gioia L, Matteucci C, Peri G, Spreafico F, Pausa M, D'Ettorre C, Gianazza E, Tagliabue A, Salmona M, Tedesco F, Introna M, Mantovani A (1997) Multimer formation and ligand recognition by the long pentraxin PTX3. Similarities and differences with the short pentraxins C-reactive protein and serum amyloid P component. J Biol Chem 272:32817–32823
Bottazzi B, Garlanda C, Cotena A, Moalli F, Jaillon S, Deban L, Mantovani A (2009) The long pentraxin PTX3 as a prototypic humoral pattern recognition receptor: interplay with cellular innate immunity. Immunol Rev 227:9–18
Bottazzi B, Doni A, Garlanda C, Mantovani A (2010) An integrated view of humoral innate immunity: pentraxins as a paradigm. Annu Rev Immunol 28:157–183
Botto M, Hawkins PN, Bickerstaff MC, Herbert J, Bygrave AE, McBride A, Hutchinson WL, Tennent GA, Walport MJ, Pepys MB (1997) Amyloid deposition is delayed in mice with targeted deletion of the serum amyloid P component gene. Nat Med 3:855–859
Bozza S, Bistoni F, Gaziano R, Pitzurra L, Zelante T, Bonifazi P, Perruccio K, Bellocchio S, Neri M, Iorio AM, Salvatori G, De Santis R, Calvitti M, Doni A, Garlanda C, Mantovani A, Romani L (2006) Pentraxin 3 protects from MCMV infection and reactivation through TLR sensing pathways leading to IRF3 activation. Blood 108:3387–3396
Breviario F, d'Aniello EM, Golay J, Peri G, Bottazzi B, Bairoch A, Saccone S, Marzella R, Predazzi V, Rocchi M, Della Valle G, Dejana E, Mantovani A, Introna M (1992) Interleukin-1-inducible genes in endothelial cells. Cloning of a new gene related to C-reactive protein and serum amyloid P component. J Biol Chem 267:22190–22197
Brugger-Andersen T, Ponitz V, Kontny F, Staines H, Grundt H, Sagara M, Nilsen DW (2009) The long pentraxin 3 (PTX3): a novel prognostic inflammatory marker for mortality in acute chest pain. Thromb Haemost 102:555–563
Cetin I, Cozzi V, Pasqualini F, Nebuloni M, Garlanda C, Vago L, Pardi G, Mantovani A (2006) Elevated maternal levels of the long pentraxin 3 (PTX3) in preeclampsia and intrauterine growth restriction. Am J Obstet Gynecol 194:1347–1353
Cotena A, Maina V, Sironi M, Bottazzi B, Jeannin P, Vecchi A, Corvaia N, Daha MR, Mantovani A, Garlanda C (2007) Complement dependent amplification of the innate response to a cognate microbial ligand by the long pentraxin PTX3. J Immunol 179:6311–6317
D'Angelo C, De Luca A, Zelante T, Bonifazi P, Moretti S, Giovannini G, Iannitti RG, Zagarella S, Bozza S, Campo S, Salvatori G, Romani L (2009) Exogenous pentraxin 3 restores antifungal resistance and restrains inflammation in murine chronic granulomatous disease. J Immunol 183:4609–4618
Deban L, Jarva H, Lehtinen MJ, Bottazzi B, Bastone A, Doni A, Jokiranta TS, Mantovani A, Meri S (2008) Binding of the long pentraxin PTX3 to Factor H: interacting domains and function in the regulation of complement activation. J Immunol 181:8433–8440
Deban L, Bottazzi B, Garlanda C, Torre YM de la, Mantovani A (2009) Pentraxins: multifunctional proteins at the interface of innate immunity and inflammation. Biofactors 35:138–145
Deban L, Russo RC, Sironi M, Moalli F, Scanziani M, Zambelli V, Cuccovillo I, Bastone A, Gobbi M, Valentino S, Doni A, Garlanda C, Danese S, Salvatori G, Sassano M, Evangelista V, Rossi B, Zenaro E, Constantin G, Laudanna C, Bottazzi B, Mantovani A (2010) Regulation of leukocyte recruitment by the long pentraxin PTX3. Nat Immunol 11:328–334
Dias AA, Goodman AR, Dos Santos JL, Gomes RN, Altmeyer A, Bozza PT, Horta MF, Vilcek J, Reis LF (2001) TSG-14 transgenic mice have improved survival to endotoxemia and to CLP-induced sepsis. J Leukoc Biol 69:928–936
Diniz SN, Nomizo R, Cisalpino PS, Teixeira MM, Brown GD, Mantovani A, Gordon S, Reis LF, Dias AA (2004) PTX3 function as an opsonin for the dectin-1-dependent internalization of zymosan by macrophages. J Leukoc Biol 75:649–656
Doni A, Peri G, Chieppa M, Allavena P, Pasqualini F, Vago L, Romani L, Garlanda C, Mantovani A (2003) Production of the soluble pattern recognition receptor PTX3 by myeloid, but not plasmacytoid, dendritic cells. Eur J Immunol 33:2886–2893
Doni A, Michela M, Bottazzi B, Peri G, Valentino S, Polentarutti N, Garlanda C, Mantovani A (2006) Regulation of PTX3, a key component of humoral innate immunity in human dendritic cells: stimulation by IL-10 and inhibition by IFN-gamma. J Leukoc Biol 79:797–802
Doni A, Mantovani G, Porta C, Tuckermann J, Reichardt HM, Kleiman A, Sironi M, Rubino L, Pasqualini F, Nebuloni M, Signorini S, Peri G, Sica A, Beck-Peccoz P, Bottazzi B, Mantovani A (2008) Cell-specific regulation of ptx3 by glucocorticoid hormones in hematopoietic and nonhematopoietic cells. J Biol Chem 283:29983–29992
Elliott P, Chambers JC, Zhang W, Clarke R, Hopewell JC, Peden JF, Erdmann J, Braund P, Engert JC, Bennett D, Coin L, Ashby D, Tzoulaki I, Brown IJ, Mt-Isa S, McCarthy MI, Peltonen L, Freimer NB, Farrall M, Ruokonen A, Hamsten A, Lim N, Froguel P, Waterworth DM, Vollenweider P, Waeber G, Jarvelin MR, Mooser V, Scott J, Hall AS, Schunkert H, Anand SS, Collins R, Samani NJ, Watkins H, Kooner JS (2009) Genetic loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA 302:37–48
Emsley J, White HE, O'Hara BP, Oliva G, Srinivasan N, Tickle IJ, Blundell TL, Pepys MB, Wood SP (1994) Structure of pentameric human serum amyloid P component. Nature 367:338–345
Endo Y, Matsushita M, Fujita T (2007) Role of ficolin in innate immunity and its molecular basis. Immunobiology 212:371–379
Fazzini F, Peri G, Doni A, Dell'Antonio G, Dal Cin E, Bozzolo E, D'Auria F, Praderio L, Ciboddo G, Sabbadini MG, Manfredi AA, Mantovani A, Querini PR (2001) PTX3 in small-vessel vasculitides: an independent indicator of disease activity produced at sites of inflammation. Arthritis Rheum 44:2841–2850
Garlanda C, Hirsch E, Bozza S, Salustri A, De Acetis M, Nota R, Maccagno A, Riva F, Bottazzi B, Peri G, Doni A, Vago L, Botto M, De Santis R, Carminati P, Siracusa G, Altruda F, Vecchi A, Romani L, Mantovani A (2002) Non-redundant role of the long pentraxin PTX3 in anti-fungal innate immune response. Nature 420:182–186
Garlanda C, Bottazzi B, Bastone A, Mantovani A (2005) Pentraxins at the crossroads between innate immunity, inflammation, matrix deposition, and female fertility. Annu Rev Immunol 23:337–366
Gaziano R, Bozza S, Bellocchio S, Perruccio K, Montagnoli C, Pitzurra L, Salvatori G, De Santis R, Carminati P, Mantovani A, Romani L (2004) Anti-Aspergillus fumigatus efficacy of pentraxin 3 alone and in combination with antifungals. Antimicrob Agents Chemother 48:4414–4421
Gershov D, Kim S, Brot N, Elkon KB (2000) C-reactive protein binds to apoptotic cells, protects the cells from assembly of the terminal complement components, and sustains an antiinflammatory innate immune response: implications for systemic autoimmunity. J Exp Med 192:1353–1364
Gillmore JD, Hutchinson WL, Herbert J, Bybee A, Mitchell DA, Hasserjian RP, Yamamura K, Suzuki M, Sabin CA, Pepys MB (2004) Autoimmunity and glomerulonephritis in mice with targeted deletion of the serum amyloid P component gene: SAP deficiency or strain combination? Immunology 112:255–264
Goodman AR, Levy DE, Reis LF, Vilcek J (2000) Differential regulation of TSG-14 expression in murine fibroblasts and peritoneal macrophages. J Leukoc Biol 67:387–395
Haas CJ de, Leeuwen EM van, Bommel T van, Verhoef J, Kessel KP van, Strijp JA van (2000) Serum amyloid P component bound to gram-negative bacteria prevents lipopolysaccharide-mediated classical pathway complement activation. Infect Immun 68:1753–1759
Han B, Mura M, Andrade CF, Okutani D, Lodyga M, Santos CC dos, Keshavjee S, Matthay M, Liu M (2005) TNFalpha-induced long pentraxin PTX3 expression in human lung epithelial cells via JNK. J Immunol 175:8303–8311
Holmskov U, Thiel S, Jensenius JC (2003) Collectins and ficolins: humoral lectins of the innate immune defense. Annu Rev Immunol 21:547–578
Inforzato A, Peri G, Doni A, Garlanda C, Mantovani A, Bastone A, Carpentieri A, Amoresano A, Pucci P, Roos A, Daha MR, Vincenti S, Gallo G, Carminati P, De Santis R, Salvatori G (2006) Structure and function of the long pentraxin PTX3 glycosidic moiety: fine-tuning of the interaction with C1q and complement activation. Biochemistry 45:11540–11551
Inforzato A, Rivieccio V, Morreale AP, Bastone A, Salustri A, Scarchilli L, Verdoliva A, Vincenti S, Gallo G, Chiapparino C, Pacello L, Nucera E, Serlupi-Crescenzi O, Day AJ, Bottazzi B, Mantovani A, De Santis R, Salvatori G (2008) Structural characterization of PTX3 disulfide bond network and its multimeric status in cumulus matrix organization. J Biol Chem 283:10147–10161
Inforzato A, Baldock C, Jowitt TA, Holmes DF, Lindstedt R, Marcellini M, Rivieccio V, Briggs DC, Kadler KE, Verdoliva A, Bottazzi B, Mantovani A, Salvatori G, Day AJ (2010) The angiogenic inhibitor long pentraxin PTX3 forms an asymmetric octamer with two binding sites for FGF2. J Biol Chem 285:17681–17692
Inoue K, Sugiyama A, Reid PC, Ito Y, Miyauchi K, Mukai S, Sagara M, Miyamoto K, Satoh H, Kohno I, Kurata T, Ota H, Mantovani A, Hamakubo T, Daida H, Kodama T (2007) Establishment of a high sensitivity plasma assay for human pentraxin3 as a marker for unstable angina pectoris. Arterioscler Thromb Vasc Biol 27:161–167
Introna M, Alles VV, Castellano M, Picardi G, De Gioia L, Bottazzai B, Peri G, Breviario F, Salmona M, De Gregorio L, Dragani TA, Srinivasan N, Blundell TL, Hamilton TA, Mantovani A (1996) Cloning of mouse ptx3, a new member of the pentraxin gene family expressed at extrahepatic sites. Blood 87:1862–1872
Iwasaki A, Medzhitov R (2010) Regulation of adaptive immunity by the innate immune system. Science 327:291–295
Jaillon S, Peri G, Delneste Y, Fremaux I, Doni A, Moalli F, Garlanda C, Romani L, Gascan H, Bellocchio S, Bozza S, Cassatella MA, Jeannin P, Mantovani A (2007) The humoral pattern recognition receptor PTX3 is stored in neutrophil granules and localizes in extracellular traps. J Exp Med 204:793–804
Jaillon S, Jeannin P, Hamon Y, Fremaux I, Doni A, Bottazzi B, Blanchard S, Subra JF, Chevailler A, Mantovani A, Delneste Y (2009) Endogenous PTX3 translocates at the membrane of late apoptotic human neutrophils and is involved in their engulfment by macrophages. Cell Death Differ 16:465–474
Jarva H, Jokiranta TS, Hellwage J, Zipfel PF, Meri S (1999) Regulation of complement activation by C-reactive protein: targeting the complement inhibitory activity of Factor H by an interaction with short consensus repeat domains 7 and 8-11. J Immunol 163:3957–3962
Jeannin P, Bottazzi B, Sironi M, Doni A, Rusnati M, Presta M, Maina V, Magistrelli G, Haeuw JF, Hoeffel G, Thieblemont N, Corvaia N, Garlanda C, Delneste Y, Mantovani A (2005) Complexity and complementarity of outer membrane protein A recognition by cellular and humoral innate immunity receptors. Immunity 22:551–560
Jeannin P, Jaillon S, Delneste Y (2008) Pattern recognition receptors in the immune response against dying cells. Curr Opin Immunol 20:530–537
Jenny NS, Arnold AM, Kuller LH, Tracy RP, Psaty BM (2009) Associations of pentraxin 3 with cardiovascular disease and all-cause death: the Cardiovascular Health Study. Arterioscler Thromb Vasc Biol 29:594–599
Kaneko Y, Nimmerjahn F, Ravetch JV (2006) Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 313:670–673
Latini R, Maggioni AP, Peri G, Gonzini L, Lucci D, Mocarelli P, Vago L, Pasqualini F, Signorini S, Soldateschi D, Tarli L, Schweiger C, Fresco C, Cecere R, Tognoni G, Mantovani A (2004) Prognostic significance of the long pentraxin PTX3 in acute myocardial infarction. Circulation 110:2349–2354
Lee GW, Lee TH, Vilcek J (1993) TSG-14, a tumor necrosis factor- and IL-1-inducible protein, is a novel member of the pentaxin family of acute phase proteins. J Immunol 150:1804–1812
Lu J, Marnell LL, Marjon KD, Mold C, Du Clos TW, Sun PD (2008) Structural recognition and functional activation of FcgammaR by innate pentraxins. Nature 456:989–992
Luchetti MM, Piccinini G, Mantovani A, Peri G, Matteucci C, Pomponio G, Fratini M, Fraticelli P, Sambo P, Di Loreto C, Doni A, Introna M, Gabrielli A (2000) Expression and production of the long pentraxin PTX3 in rheumatoid arthritis (RA). Clin Exp Immunol 119:196–202
Ma YJ, Doni A, Hummelshoj T, Honore C, Bastone A, Mantovani A, Thielens NM, Garred P (2009) Synergy between ficolin-2 and pentraxin 3 boosts innate immune recognition and complement deposition. J Biol Chem 284:28263–28275
Mairuhu AT, Peri G, Setiati TE, Hack CE, Koraka P, Soemantri A, Osterhaus AD, Brandjes DP, Meer JW van der, Mantovani A, Gorp EC van (2005) Elevated plasma levels of the long pentraxin, pentraxin 3, in severe dengue virus infections. J Med Virol 76:547–552
Mantovani A, Garlanda C, Doni A, Bottazzi B (2008) Pentraxins in innate immunity: from C-reactive protein to the long pentraxin PTX3. J Clin Immunol 28:1–13
Martinez de la Torre Y, Fabbri M, Jaillon S, Bastone A, Nebuloni M, Vecchi A, Mantovani A, Garlanda C (2010) Evolution of the pentraxin family: the new entry PTX4. J Immunol 184:5055–5064
Matsui S, Ishii J, Kitagawa F, Kuno A, Hattori K, Ishikawa M, Okumura M, Kan S, Nakano T, Naruse H, Tanaka I, Nomura M, Hishida H, Ozaki Y (2010) Pentraxin 3 in unstable angina and non-ST-segment elevation myocardial infarction. Atherosclerosis 210:220–225
Mauri T, Bellani G, Patroniti N, Coppadoro A, Peri G, Cuccovillo I, Cugno M, Iapichino G, Gattinoni L, Pesenti A, Mantovani A (2010) Persisting high levels of plasma pentraxin 3 over the first days after severe sepsis and septic shock onset are associated with mortality. Intensive Care Med 36:621–629
Mihlan M, Stippa S, Jozsi M, Zipfel PF (2009) Monomeric CRP contributes to complement control in fluid phase and on cellular surfaces and increases phagocytosis by recruiting Factor H. Cell Death Differ 16:1630–1640
Miyaki A, Maeda S, Yoshizawa M, Misono M, Sasai H, Shimojo N, Tanaka K, Ajisaka R (2010) Is pentraxin 3 involved in obesity-induced decrease in arterial distensibility? J Atheroscler Thromb 17:278–284
Mold C, Gewurz H (1981) Inhibitory effect of C-reactive protein on alternative C pathway activation by liposomes and Streptococcus pneumoniae. J Immunol 127:2089–2092
Mold C, Gresham HD, Du Clos TW (2001) Serum amyloid P component and C-reactive protein mediate phagocytosis through murine Fc gamma Rs. J Immunol 166:1200–1205
Murphy JE, Tedbury PR, Homer-Vanniasinkam S, Walker JH, Ponnambalam S (2005) Biochemistry and cell biology of mammalian scavenger receptors. Atherosclerosis 182:1–15
Nauta AJ, Bottazzi B, Mantovani A, Salvatori G, Kishore U, Schwaeble WJ, Gingras AR, Tzima S, Vivanco F, Egido J, Tijsma O, Hack EC, Daha MR, Roos A (2003a) Biochemical and functional characterization of the interaction between pentraxin 3 and C1q. Eur J Immunol 33:465–473
Nauta AJ, Daha MR, Kooten C van, Roos A (2003b) Recognition and clearance of apoptotic cells: a role for complement and pentraxins. Trends Immunol 24:148–154
Nauta AJ, Haij S de, Bottazzi B, Mantovani A, Borrias MC, Aten J, Rastaldi MP, Daha MR, Kooten C van, Roos A (2005) Human renal epithelial cells produce the long pentraxin PTX3. Kidney Int 67:543–553
Ng PM, Le Saux A, Lee CM, Tan NS, Lu J, Thiel S, Ho B, Ding JL (2007) C-reactive protein collaborates with plasma lectins to boost immune response against bacteria. EMBO J 26:3431–3440
Norata GD, Marchesi P, Pulakazhi Venu VK, Pasqualini F, Anselmo A, Moalli F, Pizzitola I, Garlanda C, Mantovani A, Catapano AL (2009) Deficiency of the long pentraxin PTX3 promotes vascular inflammation and atherosclerosis. Circulation 120:699–708
Noursadeghi M, Bickerstaff MC, Gallimore JR, Herbert J, Cohen J, Pepys MB (2000) Role of serum amyloid P component in bacterial infection: protection of the host or protection of the pathogen. Proc Natl Acad Sci USA 97:14584–14589
Ogawa T, Kawano Y, Imamura T, Kawakita K, Sagara M, Matsuo T, Kakitsubata Y, Ishikawa T, Kitamura K, Hatakeyama K, Asada Y, Kodama T (2010) Reciprocal contribution of pentraxin 3 and C-reactive protein to obesity and metabolic syndrome. Obesity (Silver Spring) (in press)
Okemefuna AI, Nan R, Miller A, Gor J, Perkins SJ (2010) Complement Factor H binds at two independent sites to C-reactive protein in acute phase concentrations. J Biol Chem 285:1053–1065
Pepys MB, Hirschfield GM (2003) C-reactive protein: a critical update. J Clin Invest 111:1805–1812
Peri G, Introna M, Corradi D, Iacuitti G, Signorini S, Avanzini F, Pizzetti F, Maggioni AP, Moccetti T, Metra M, Cas LD, Ghezzi P, Sipe JD, Re G, Olivetti G, Mantovani A, Latini R (2000) PTX3, a prototypical long pentraxin, is an early indicator of acute myocardial infarction in humans. Circulation 102:636–641
Polentarutti N, Picardi G, Basile A, Cenzuales S, Rivolta A, Matteucci C, Peri G, Mantovani A, Introna M (1998) Interferon-gamma inhibits expression of the long pentraxin PTX3 in human monocytes. Eur J Immunol 28:496–501
Poon IK, Hulett MD, Parish CR (2010) Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell Death Differ 17:381–397
Ravizza T, Moneta D, Bottazzi B, Peri G, Garlanda C, Hirsch E, Richards GJ, Mantovani A, Vezzani A (2001) Dynamic induction of the long pentraxin PTX3 in the CNS after limbic seizures: evidence for a protective role in seizure-induced neurodegeneration. Neuroscience 105:43–53
Reading PC, Bozza S, Gilbertson B, Tate M, Moretti S, Job ER, Crouch EC, Brooks AG, Brown LE, Bottazzi B, Romani L, Mantovani A (2008) Antiviral activity of the long chain pentraxin PTX3 against influenza viruses. J Immunol 180:3391–3398
Robey FA, Liu TY (1981) Limulin: a C-reactive protein from Limulus polyphemus. J Biol Chem 256:969–975
Rossum AP van, Fazzini F, Limburg PC, Manfredi AA, Rovere-Querini P, Mantovani A, Kallenberg CG (2004) The prototypic tissue pentraxin PTX3, in contrast to the short pentraxin serum amyloid P, inhibits phagocytosis of late apoptotic neutrophils by macrophages. Arthritis Rheum 50:2667–2674
Roumenina LT, Ruseva MM, Zlatarova A, Ghai R, Kolev M, Olova N, Gadjeva M, Agrawal A, Bottazzi B, Mantovani A, Reid KB, Kishore U, Kojouharova MS (2006) Interaction of C1q with IgG1, C-reactive protein and pentraxin 3: mutational studies using recombinant globular head modules of human C1q A, B, and C chains. Biochemistry 45:4093–4104
Rovere-Querini P, Antonacci S, Dell'Antonio G, Angeli A, Almirante G, Cin ED, Valsecchi L, Lanzani C, Sabbadini MG, Doglioni C, Manfredi AA, Castiglioni MT (2006) Plasma and tissue expression of the long pentraxin 3 during normal pregnancy and preeclampsia. Obstet Gynecol 108:148–155
Rubio N, Sharp PM, Rits M, Zahedi K, Whitehead AS (1993) Structure, expression, and evolution of guinea pig serum amyloid P component and C-reactive protein. J Biochem 113:277–284
Saeland E, Royen A van, Hendriksen K, Vile-Weekhout H, Rijkers GT, Sanders LA, Winkel JG van de (2001) Human C-reactive protein does not bind to FcgammaRIIa on phagocytic cells. J Clin Invest 107:641–643
Salio M, Chimenti S, De Angelis N, Molla F, Maina V, Nebuloni M, Pasqualini F, Latini R, Garlanda C, Mantovani A (2008) Cardioprotective function of the long pentraxin PTX3 in acute myocardial infarction. Circulation 117:1055–1064
Santos CC dos, Han B, Andrade CF, Bai X, Uhlig S, Hubmayr R, Tsang M, Lodyga M, Keshavjee S, Slutsky AS, Liu M (2004) DNA microarray analysis of gene expression in alveolar epithelial cells in response to TNFalpha, LPS, and cyclic stretch. Physiol Genomics 19:331–342
Schwalbe RA, Dahlback B, Coe JE, Nelsestuen GL (1992) Pentraxin family of proteins interact specifically with phosphorylcholine and/or phosphorylethanolamine. Biochemistry 31:4907–4915
Seery LT, Schoenberg DR, Barbaux S, Sharp PM, Whitehead AS (1993) Identification of a novel member of the pentraxin family in Xenopus laevis. Proc Biol Sci 253:263–270
Shrive AK, Metcalfe AM, Cartwright JR, Greenhough TJ (1999) C-reactive protein and SAP-like pentraxin are both present in Limulus polyphemus haemolymph: crystal structure of Limulus SAP. J Mol Biol 290:997–1008
Souza DG, Amaral FA, Fagundes CT, Coelho FM, Arantes RM, Sousa LP, Matzuk MM, Garlanda C, Mantovani A, Dias AA, Teixeira MM (2009) The long pentraxin PTX3 is crucial for tissue inflammation after intestinal ischemia and reperfusion in mice. Am J Pathol. 174:1309-1318
Sprong T, Peri G, Neeleman C, Mantovani A, Signorini S, Meer JW van der, Deuren M van (2009) Pentraxin 3 and C-reactive protein in severe meningococcal disease. Shock 31:28–32
Suankratay C, Mold C, Zhang Y, Potempa LA, Lint TF, Gewurz H (1998) Complement regulation in innate immunity and the acute-phase response: inhibition of mannan-binding lectin-initiated complement cytolysis by C-reactive protein (CRP). Clin Exp Immunol 113:353–359
Szalai AJ (2002) The antimicrobial activity of C-reactive protein. Microbes Infect 4:201–205
Szalai AJ, VanCott JL, McGhee JR, Volanakis JE, Benjamin WH Jr (2000) Human C-reactive protein is protective against fatal Salmonella enterica serovar typhimurium infection in transgenic mice. Infect Immun 68:5652–5656
Tanio M, Wakamatsu K, Kohno T (2009) Binding site of C-reactive protein on M-ficolin. Mol Immunol 47:215–221
Tennent GA, Lovat LB, Pepys MB (1995) Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer disease and systemic amyloidosis. Proc Natl Acad Sci USA 92:4299–4303
Tillett WS, Francis TJ (1930) Serological reactions in pneumonia with a non protein somatic fraction of pneumococcus. J Exp Med 52:561–585
Trouw LA, Bengtsson AA, Gelderman KA, Dahlback B, Sturfelt G, Blom AM (2007) C4b-binding protein and Factor H compensate for the loss of membrane-bound complement inhibitors to protect apoptotic cells against excessive complement attack. J Biol Chem 282:28540–28548
Verma S, Szmitko PE, Ridker PM (2005) C-reactive protein comes of age. Nat Clin Pract 2:29–36
Yano Y, Matsuda S, Hatakeyama K, Sato Y, Imamura T, Shimada K, Kodama T, Kario K, Asada Y (2010) Plasma pentraxin 3, but not high-sensitivity C-reactive protein, is a useful inflammatory biomarker for predicting cognitive impairment in elderly hypertensive patients. J Gerontol 65:547–552
Yuste J, Botto M, Bottoms SE, Brown JS (2007) Serum amyloid P aids complement-mediated immunity to Streptococcus pneumoniae. PLoS Pathog 3:1208–1219
Zacho J, Tybjaerg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG (2008) Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med 359:1897–1908
Zanetti M, Bosutti A, Ferreira C, Vinci P, Biolo G, Fonda M, Valente M, Cattin L, Guarnieri G, Barazzoni R (2009) Circulating pentraxin 3 levels are higher in metabolic syndrome with subclinical atherosclerosis: evidence for association with atherogenic lipid profile. Clin Exp Med 9:243–248
Zhang J, Koh J, Lu J, Thiel S, Leong BS, Sethi S, He CY, Ho B, Ding JL (2009) Local inflammation induces complement crosstalk which amplifies the antimicrobial response. PLoS Pathog 5:e1000282
Author information
Authors and Affiliations
Corresponding author
Additional information
Financial support from the European Commission (“MUGEN” LSHG-CT-2005-005203, “TOLERAGE” 2008-02156), European Research Council (project HIIS), Ministero dell’Istruzione, Università e della Ricerca (MIUR) (project FIRB), Telethon (Telethon grant no. GGP05095), Fondazione CARIPLO (Project Nobel and Project 2009-2582), Ministero della Salute (Ricerca finalizzata), the Italian Association for Cancer Research (AIRC) and Leem recherche/ARIIS (Alliance pour la Recherche et l’Innovation des Industries de Santé) is gratefully acknowledged.
Livija Deban and Sebastian Jailon contributed equally to this review.
Rights and permissions
About this article
Cite this article
Deban, L., Jaillon, S., Garlanda, C. et al. Pentraxins in innate immunity: lessons from PTX3. Cell Tissue Res 343, 237–249 (2011). https://doi.org/10.1007/s00441-010-1018-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-010-1018-0